Introduction
Lung cancer is the most lethal malignancy worldwide.
Treatment options remain limited for some patients with advanced
lung cancer. Approximately half of limited stage lung cancer
patients relapse despite curative intent surgery/radiation and
adjuvant chemotherapy (1). Thus,
there is an urgent need to develop novel or modified therapeutic
approaches to enhance the efficacy of treatment for this
malignancy.
Epidermal growth factor receptor (EGFR), a 170 kDa
membrane-bound protein encoded by 28 exons on chromosome 7p12, is a
typical member of the tyrosine kinase (TK) family and belongs to a
subfamily that consists of four closely related members: EGFR
(ErbB1), HER-2/neu (ErbB2), HER-3 (ErbB3) and HER-4 (ErbB4). All
members have an extracellular ligand-binding domain, a single
membrane spanning domain and an intracellular domain (2,3); they
are also known to have intrinsic TK activity apart from ErbB3
(4). EGFR has been shown to play a
central role in the occurrence and progression of multiple solid
tumors, including lung cancer (5).
Several anti-EGFR molecules have been reported to induce neoplastic
growth inhibition. Among these, gefitinib (Iressa), an orally
available synthetic anilinoquinazoline agent, selectively binds to
the TK region of the intracellular domain of EGFR, prevents ATP
binding, and blocks EGFR signaling transduction pathways, thereby
inhibiting cancer cell growth (6).
Over the years, non-small cell lung cancer (NSCLC) containing EGFR
mutations has been shown to be sensitive to gefitinib, and this
agent has been successfully used in the treatment of these patients
(7,8). Unfortunately, some patients with EGFR
gene mutations develop resistance to TK inhibitor (TKI) treatment
after a certain period of time, and the mechanisms behind this
resistance remain unknown.
Aberrant hypermethylation in the gene promoter has
become one of the major mechanisms for silencing tumor suppressor
or other cancer-associated genes in many types of human cancer, and
it is an epigenetic modification that plays an important role in
the control of gene expression in mammalian cells (9). Recently, the role it plays in
carcinogenesis has raised considerable interest (10). In many tumor types, CpG island
hypermethylation in the promoter region of several tumor suppressor
genes has been observed and has been shown to correlate closely
with the loss of mRNA and protein expression. While
hypermethylation typically affects tumor suppressor genes, it also
silences oncogenes, such as cyclooxygenase-2 (COX-2) (11) and telomerase reverse transcriptase
(TERT) (12). Since the promoter
region of the EGFR gene contains a CpG island that extends into
exon 1, we hypothesized that EGFR gene promoter methylation may
influence the antitumor effect of gefitinib on NSCLC cells.
Materials and methods
Cell culture and reagents
Three NSCLC cell lines with different EGFR mutation
statuses and levels of sensitivity to EGFR-TKIs, obtained from the
Shanghai Cell Station, Chinese Academy of Sciences, Shanghai, China
were used: H1650 (del E746-A750), H1299 (wild-type EGFR) and PC-9
(del E746-A750). These cells were maintained in RPMI-1640 culture
medium containing 10% heat-inactivated fetal bovine serum (FBS),
100 U/ml penicillin and 100 mg/ml streptomycin, and they were
incubated in humidified air and 5% CO2 at 37°C.
Gefitinib was a generous gift from AstraZeneca (Cheshire, UK).
Stock solutions were prepared in dimethyl sulfoxide (DMSO) and
stored at −20°C. Gefitinib solution was prepared in fresh medium
prior to each experiment and the control cells were treated with
medium containing an equal concentration of DMSO.
5-Aza-deoxycytidine (5-aza-CdR, decitabine), a methylation
inhibitor, was purchased from Sigma (St. Louis, MO, USA) and
prepared as described above.
Methylation-specific PCR (MSP)
For the demethylation experiments, NSCLC cells
(H1650 and H1299) were treated with varying concentrations (1–10
μM) of 5-aza-CdR for up to 72 h. The PC-9 cells were not treated
with 5-aza-CdR due to their unmethylated status. Genomic DNA was
extracted by proteinase K digestion followed by purification with a
series of phenol/chloroform and isopropyl alcohol precipitations.
The extracted DNA samples were stored in TE buffer at −20°C until
use. Bisulfite-based DNA modification, which converted all
unmethylated cytosines to uracil, was performed by using the
Methylcode Bisulfite Conversion kit (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer’s instructions. The modified DNA was
used as a template for MSP with primers specific for either the
modified-methylated or unmethylated EGFR gene promoter sequences.
PCR amplification was performed with the following primer sets that
included the CpG island of EGFR: forward primer,
5′-GGTTGGGTTTGTAAGTTCGC-3′ and reverse primer,
5′-ATAAACAACGATAACCCCCG-3′ for the methylated EGFR sequence (150
bp); and forward primer, 5′-GGTTGGGTTTGTAAGTTTGT-3′ and reverse
primer, 5′-ATAAACAACAATAACCCCCA-3′ for the unmethylated EGFR
sequence (150 bp). The PCR amplification program consisted of 10
min at 95°C, followed by 40 cycles of 30 sec denaturation at 95°C,
30 sec of annealing at 56°C, 30 sec of extension at 72°C, with a
final extension at 72°C for 10 min for both primers using HotStar
Taq DNA polymerase (Qiagen, Valencia, CA, USA). PCR products were
separated on 3% agarose gels with ethidium bromide and visualized
under UV illumination. As the positive control, M.SssI
methylase (New England BioLabs, Ipswich, MA, USA) was used to
methylate normal human peripheral blood.
Cell proliferation assays
Cell proliferation assays were performed using a
Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) according to the manufacturer’s instructions.
Briefly, the H1650, H1299 and PC-9 cells were seeded at a density
of 3,000–5,000 cells/well in 96-well microtiter plates for 24 h.
While the H1650 and H1299 cells were treated with increasing
concentrations of gefitinib (0.01–100 μM), 5-aza-CdR (0.01–100 μM),
or a combination of gefitinib and 5-aza-CdR for up to 72 h, the
PC-9 cells were treated with increasing concentrations of gefitinib
(0.01–100 μM) alone for 72 h. Subsequently, 10 μl of the CCK-8
solution were added to each well in the plate. The absorbance (A)
at 450 nm was measured using a microplate reader, and a calibration
curve was prepared using the data obtained from the wells that
contained known numbers of viable cells. Each experiment was
carried out in five replicate wells at each drug concentration and
repeated at least three times.
Apoptosis measurements
The apoptosis assay was conducted using Annexin V
staining and flow cytometry assays. The H1650 and H1299 cells
seeded in 6-well plates were treated with gefitinib (1 μM),
5-aza-CdR (1 μM), or a combination of gefitinib and 5-aza-CdR based
on their sensitivity to various drugs for 72 h. PC-9 cells were
treated with various concentrations of gefitinib (0.01–100 μM) for
72 h. Subsequently, the treated and untreated cells were collected
and washed with PBS. Annexin V staining was performed following the
manufacturer’s instructions (Trevigen, Inc., Gaithersburg, MD,
USA). Briefly, the cells were incubated for 15 min at room
temperature in the presence of 1 μl of Annexin V-FITC, 1 μl of
propidium iodide, and 98 μl of 1X binding buffer (all reagents were
provided by the manufacturer). Following incubation, 400 μl of 1X
binding buffer were added to each tube, and the cells were analyzed
by flow cytometry.
Western blot analysis
Cells were lysed in a buffer containing 1 mM
protease inhibitor [phenylmethylsulfonyl fluoride (PMSF)] and were
cleared by centrifugation at 12,000 rpm for 10 min. Protein
concentration was determined using the BCA assay (Bio-Rad,
Hercules, CA, USA). Protein (80 μg) was dissolved in loading
buffer, denatured by heating at 100°C for 5 min, and subsequently
separated on 8% polyacrylamide gels by SDS-gel electrophoresis.
After separation, the proteins were transferred onto an immunoblot
polyvinylidene difluoride membrane (Bio-Rad). Overall protein
loading was confirmed by Ponceau S staining. Membranes were blocked
with 5% non-fat milk for 2 h at 37°C and then incubated with
anti-EGFR antibody (Millipore, Billerica, MA, USA) overnight at
4°C. The membranes were washed three times with 1X PBST and
incubated with secondary antibody conjugated with peroxidase (Dako,
Carpinteria, CA, USA) for 1 h at 37°C. After a final wash with
PBST, the membranes were developed using chemiluminescence and
exposed to X-ray film. The expression of EGFR was normalized to
β-actin.
RT-PCR and quantitative real-time
PCR
Total RNA was extracted using TRIzol Reagent
(Invitrogen) according to the manufacturer’s instructions. RNA
concentration was qualitatively assessed using a Nanodrop UV
spectrophotometer. cDNA was generated using an OmniScript RT kit
(Qiagen) according to the manufacturer’s instructions. Quantitative
gene expression analysis by reverse transcriptase-polymerase chain
reaction (RT-PCR) was performed using the Universal TaqMan PCR
protocol (qPCR) and the ABI 7300 sequence detection system.
Briefly, 2 μl of cDNA were used for each RT reaction. The 20 μl PCR
reaction mixture contained 1X primers and probe mixture (Applied
Biosystems, Foster City, CA, USA). The assay IDs were as follows:
Hs01076092-m1 (EGFR) and Hs99999905-m1 (GAPDH); 1X ABsolute qPCR
mix (Roche, USA). The PCR conditions were 95°C for 10 min, followed
by 40 cycles at 95°C for 15 sec, and 60°C for 1 min. Each sample
was assayed in triplicate with commercial RNA as the positive
control and RNase-free water as the negative control. Relative gene
expression quantifications were calculated according to the
comparative Ct method using GAPDH as the internal control and
commercial RNA control as calibrators on each plate. The final
results were determined by the formula, 2−ΔΔCt(13).
Statistical analyses
Data are expressed as the means ± SD. Statistical
analysis was performed using SPSS software version 13.0. Data were
analyzed by one-way analysis of variance (ANOVA). A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
CpG island methylation status
determination in the promoter of the EGFR gene in NSCLC cells
Three NSCLC cell lines with different EGFR gene
mutation statuses were chosen in this study, including a cell line
containing wild-type EGFR (H1299), a cell line containing mutant
EGFR resistant to TKIs (H1650) and a cell line sensitive to TKIs
(PC-9). The histological data of these NSCLC cell lines are shown
in Table I. The methylation status
of the EGFR gene was determined by MSP in the H1650, H1299 and PC-9
cells. As shown in Fig. 1, no
methylation was observed in the PC-9 cells, while hypermethylation
was detected in the H1650 and H1299 cells. We found that the EGFR
gene promoter became unmethylated when the cells were treated with
1–10 μM 5-aza-CdR for 48 and 72 h (Fig.
1).
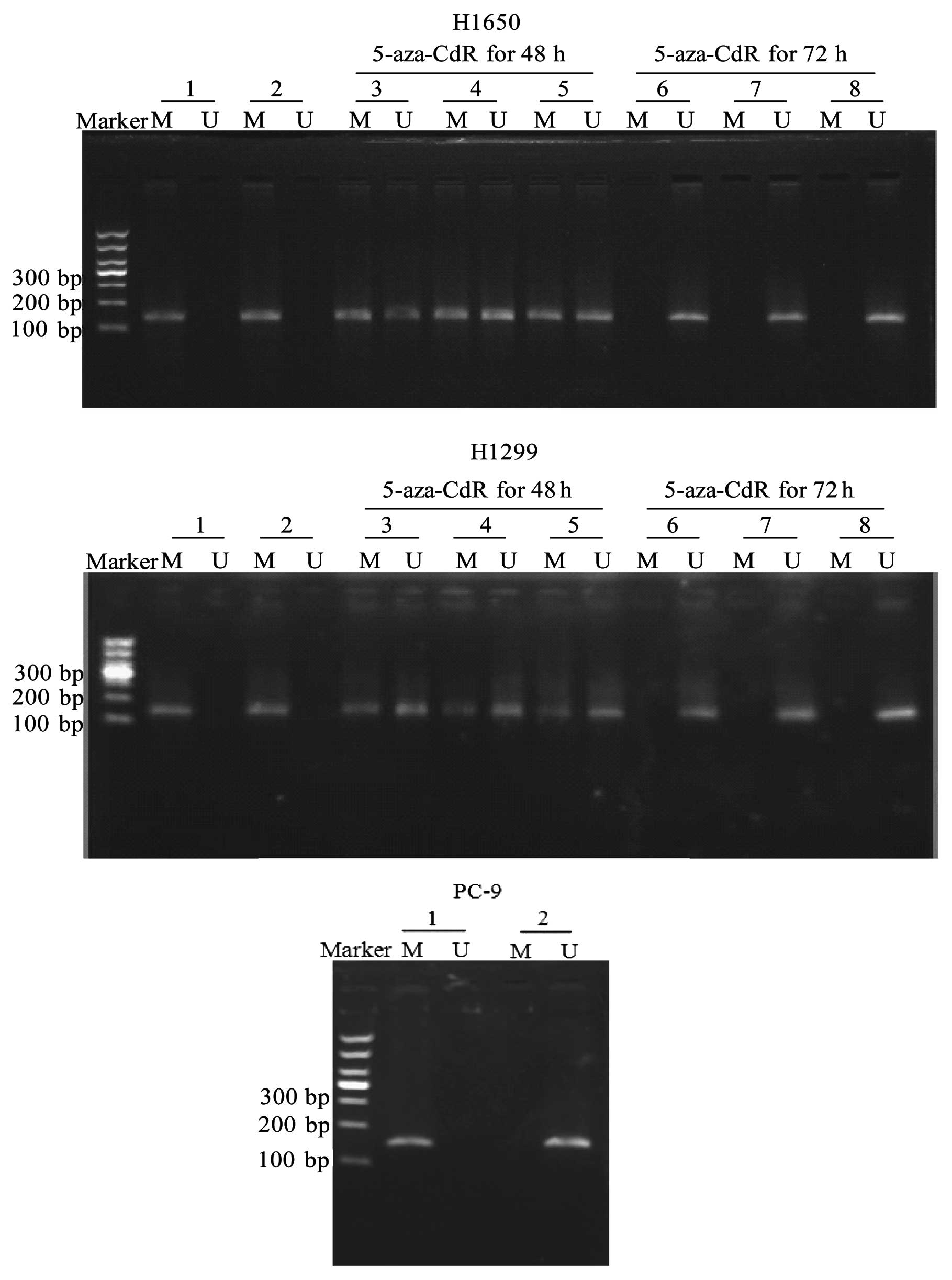 | Figure 1The CpG island methylation status in
the promoter of the EGFR gene in NSCLC cells. NSCLC cells were
treated with various concentrations (1–10 μM) of 5-aza-CdR, a
methylation inhibitor, for up to 72 h. Subsequently, the cells were
harvested. Genomic DNA was extracted by proteinase K digestion
followed by purification with a series of phenol/chloroform and
isopropyl alcohol precipitations. The modified DNA was used as a
template for MSP with primers specific for either the
modified-methylated or the modified-unmethylated EGFR promoter
sequences. The methylation status of the EGFR gene was determined
by MSP. PCR products were separated on 3% agarose gels with
ethidium bromide and visualized under UV illumination. As the
positive control, M.SssI methylase was used to methylate
normal human peripheral blood. Marker, DL1,000 DNA Marker; lane 1,
positive control; lane 2, vehicle control; lane 3, 1 μM 5-aza-CdR;
lane 4, 5 μM 5-aza-CdR; lane 5, 10 μM 5-aza-CdR; lane 6, 1 μM
5-aza-CdR; lane 7, 5 μM 5-aza-CdR; lane 8, 10 μM 5-aza-CdR; M,
product of methylated specific primer (150 bp); U, product of
unmethylated specific primer (150 bp). |
 | Table IThe IC50 values of
gefitinib and 5-aza-CdR in the NSCLC cell lines. |
Table I
The IC50 values of
gefitinib and 5-aza-CdR in the NSCLC cell lines.
| | | | | Gefitinib
IC50 (μM) |
|---|
| NSCLC cell line | Histology | EGFR mutation
status | EGFR mutation
status | 5-aza-CdR
IC50 (μM)a |
|
|---|
| No 5-aza-CdR | 5-aza-CdR |
|---|
| H1650 | AD | mut | Methylated | 15.91±1.42 | 14.53±1.13 | 2.93±0.95 |
| H1299 | LC | wt | Methylated | 16.69±1.64 | 18.64±1.98 | 3.41±1.01 |
| PC-9 | AD | mut | Unmethylated | | 1.42±0.73 | |
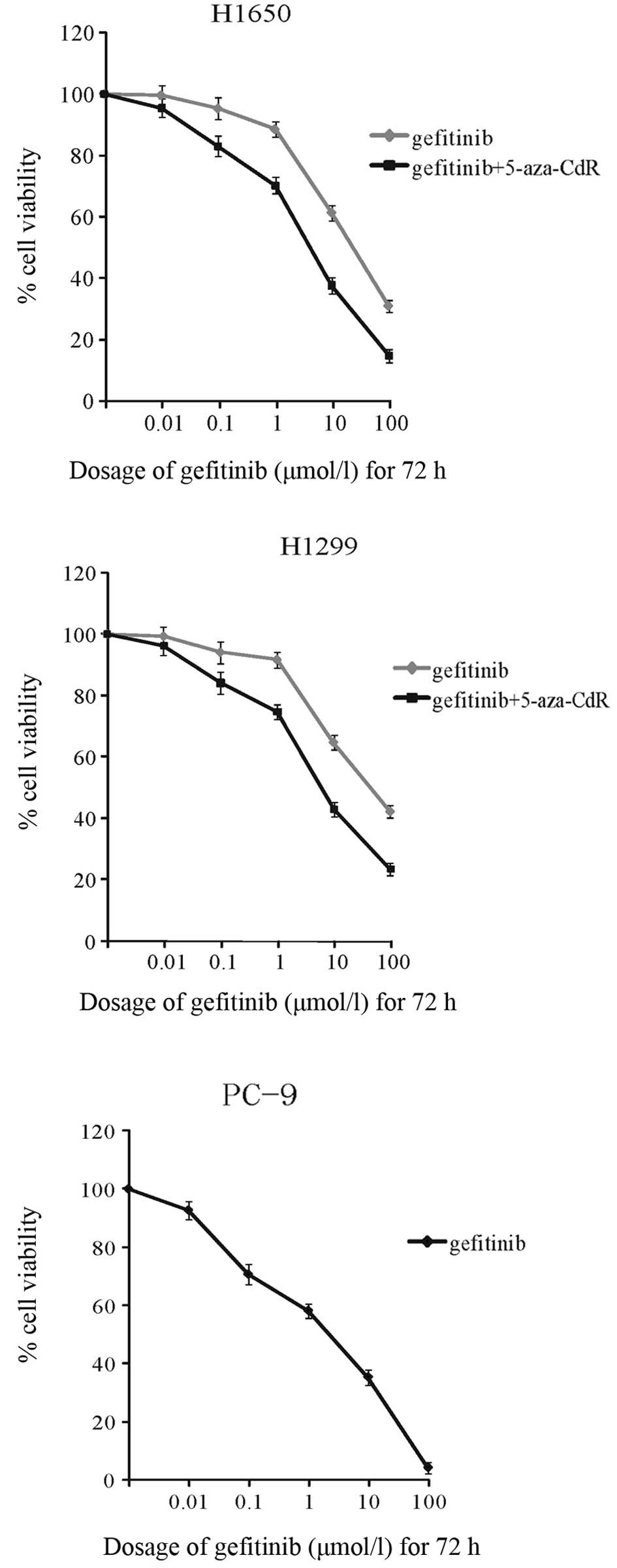
Induction of growth inhibition by
5-aza-CdR and gefitinib
We then examined the effects of 5-aza-CdR and
gefitinib on cell growth. The half maximal inhibitory concentration
(IC50) values of gefitinib in the H1650, H1299 and PC-9
cells were 14.53±1.13, 18.64±1.98 and 1.42±0.73 μM, respectively.
Of note, we found that in the presence of 1 μM 5-aza-CdR, the
IC50 values of gefitinib decreased to 2.93±0.95 and
3.41±1.01 μM in the H1650 and H1299 cells, respectively. For the
combined treatment, H1650 and H1299 cells were treated with varying
concentrations (0.01–100 μM) of gefitinib in the presence of 1 μM
5-aza-CdR; an enhanced inhibition of cell growth was observed
(Fig. 2). These results suggested
that 5-aza-CdR increased the cellular sensitivity of gefitinib as a
cell growth inhibitor. The PC-9 cells, which are unmethylated in
the EGFR gene promoter, were more sensitive to gefitinib than the
H1650 and H1299 cells (Fig. 2).
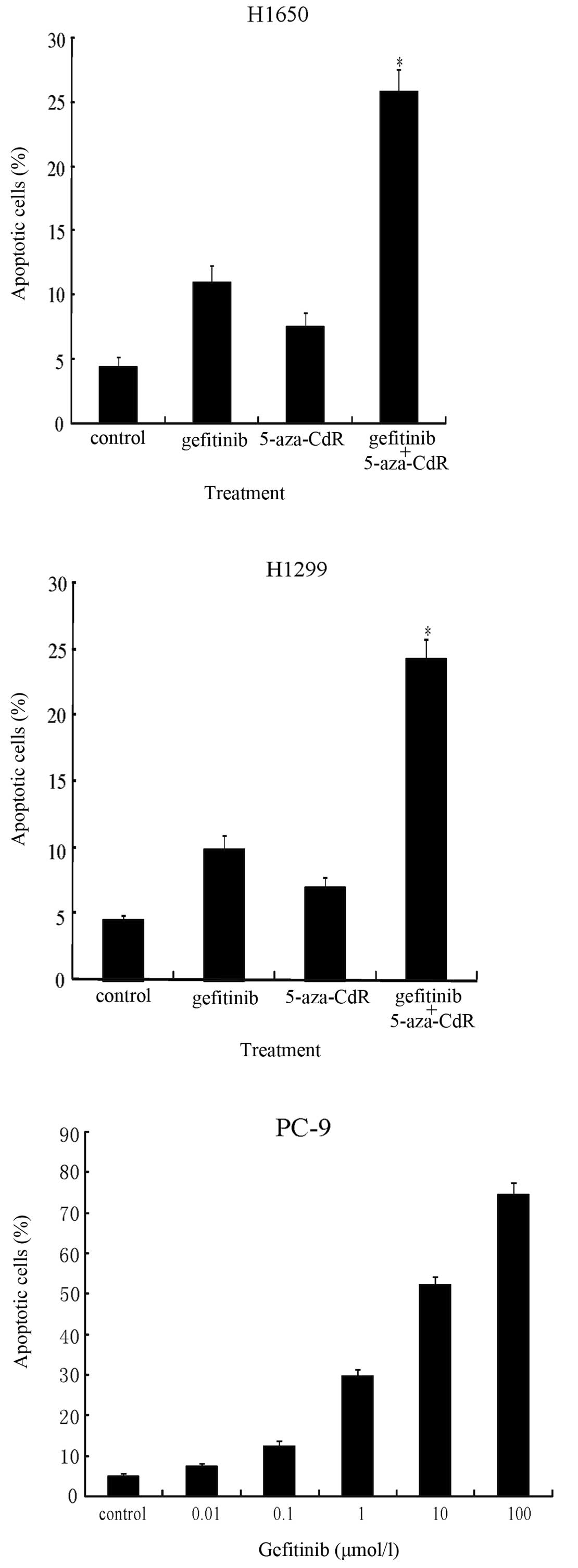
Induction of apoptosis by gefitinib and
5-aza-CdR
We hypothesized that a greater sensitivity to
gefitinib could be achieved by EGFR demethylation in the H1650 and
H1299 cells. We found that the H1650 and H1299 cell lines were
relatively resistant to gefitinib with IC50 values of
14.53±1.13 μM and 18.64±1.98 μM, respectively. Compared to either
5-aza-CdR or gefitinib treatment alone, a significant additional
increase in apoptosis was observed in the H1650 cells (25.73%,
P<0.05) and H1299 cells (24.27%, P<0.05) treated with a
combination of 5-aza-CdR (1 μM), which showed maximal
demethylation, and gefitinib (1 μM) for 72 h as determined by
Annexin V staining (Fig. 3). The
treatment of the unmethylated PC-9 cells with gefitinib also
increased apoptosis in a dose-dependent manner (Fig. 3).
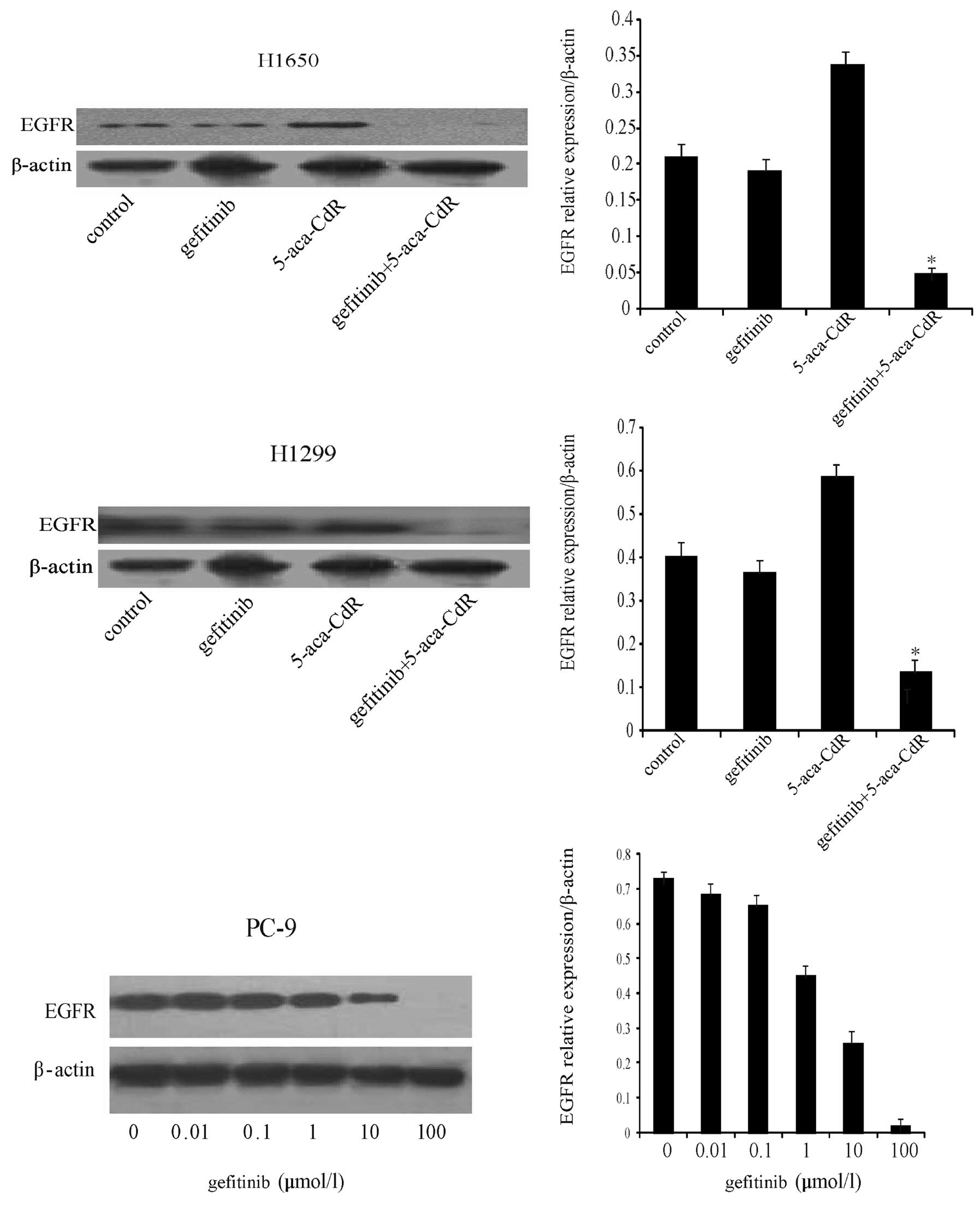
Effects of gefitinib and 5-aza-CdR on
EGFR protein expression
In order to determine whether gene methylation
affects protein expression, the H1650 and H1299 cells were treated
with gefitinib (1 μM), 5-aza-CdR (1 μM), or both. As shown in
Fig. 4, EGFR protein expression was
low in the two cell lines. To confirm that the low expression of
EGFR was due to hypermethylation, the H1650 and H1299 cells were
treated with 5-aza-CdR, which resulted in CpG island demethylation
in the EGFR gene promoter (Fig. 1).
We found that EGFR protein expression was increased following
treatment with 5-aza-CdR. A significant decrease in EGFR protein
expression was observed in the H1650 and H1299 cells after
treatment with a combination of gefitinib and 5-aza-CdR compared to
treatment with either agent alone. A dose-dependent reduction in
EGFR protein expression was observed in the PC-9 cells treated with
gefitinib (Fig. 4).
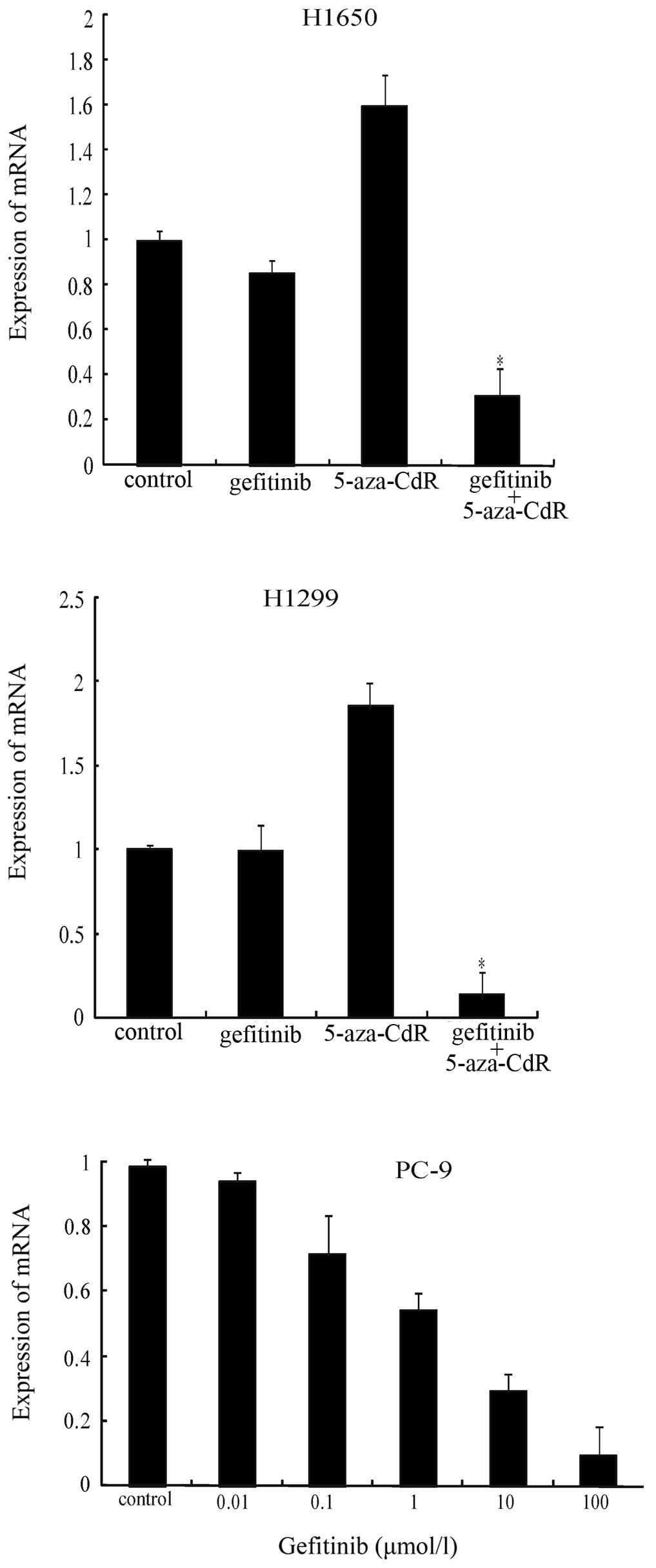
Effect of gefitinib and 5-aza-CdR on EGFR
mRNA expression
We also evaluated EGFR mRNA expression by real-time
PCR (qPCR) in the H1650, H1299 and PC-9 cells. Cells exposed to
5-aza-CdR for 72 h demonstrated an induction of EGFR mRNA
expression (Fig. 5). The effect of
the simultaneous exposure of the NSCLC cells to gefitinib in
combination with 5-aza-CdR at concentrations below the
IC50 value was examined. Similar to the protein
expression, further inhibition of EGFR mRNA expression was observed
with the combination treatment (Fig.
5); these results indicated that the cells became more
sensitive to gefitinib in the presence of 5-aza-CdR and suggested a
clear synergistic effect. As expected, a dose-dependent reduction
in EGFR mRNA expression was observed in the PC-9 cells treated with
gefitinib (Fig. 5).
Discussion
The majority of patients with metastatic NSCLC have
a poor outcome, with a median survival time of less than ten months
(14). The development of EGFR-TKIs
has significantly affected the treatment of NSCLC. EGFR-TKIs, such
as gefitinib and erlotinib, have been found to be efficient in the
treatment of certain NSCLC patients with EGFR mutations (7,8).
However, resistance to TKIs has been found in some NSCLC patients
with EGFR mutations. Furthermore, most primary NSCLC cell lines,
including cell lines containing wild-type EGFR, are resistant to
EGFR-TKI treatment. The exact reasons for this remain unknown.
Further investigation in this area would unveil new information and
may enhance the therapeutic efficacy and survival of patients with
NSCLC. Our study showed that the NSCLC cell line, H1650, which
contains a EGFR mutation (del E746-A750), was less sensitive to
EGFR-TKIs (15). Since lung cancer
cells rapidly develop drug resistance to single TKI therapy,
studies on other potential agents that can complement and enhance
the anti-neoplastic activity of TKIs are required. Previously,
promoter hypermethylation has been recognized as a potential
mechanism by which genes regulating cellular proliferation are
silenced during cancer development (16,17).
Promoter hypermethylation involves DNA methylation of CpG islands
in or near the promoter region of certain genes, rendering them
transcriptionally silent. In this study, we demonstrate that the
expression level of the EGFR gene is low in the H1650 and H1299
cells. To elucidate the mechanism of the downregulation of EGFR
gene expression, we focused on examining the promoter methylation
of the EGFR gene in lung cancer cells.
In this study, we demonstrate that the promoter of
the EGFR gene is methylated in the H1650 and H1299 cells, which
correlates with the downregulation of EGFR gene expression. Of
note, 5-aza-CdR, a demethylating agent, increased EGFR gene
transcription. This suggests that epigenetic regulation may be
responsible for controlling EGF-R gene expression. The
downregulation of expression of important cellular growth control
genes, such as Ras-association domain family member 1 (RASSF1A) and
hypoxia-inducible factor-1 (HIF-1), has been shown to play an
important role in cancer progression and metastasis, resulting in a
poor outcome that is associated with the promoter hypermethylation
of these genes (18,19).
DNA methylation and the associated silencing have
been shown to be involved in the development of drug resistance
(20), which prompted
investigations for the use of a hypomethylation approach to
re-sensitize malignant cells to classical cytotoxic agents.
5-Aza-CdR, a potent DNA methylation inhibitor, has exhibited potent
anti-neoplastic activity in animal models and has shown promising
anticancer activity in hematological malignancies (21), as well as lung cancer (22). Since 5-aza-CdR reactivates genes
through blockade of DNA methylation, it has an important
therapeutic potential in inhibiting tumorigenesis. This led us to
investigate the potential mechanisms of action of the therapeutic
agents in malignant tumors, including lung cancer. Montero et
al(23) showed that 5-aza-CdR
treatment increased the sensitivity to gefitinib, an EGFR-TKI, in
EGFR-methylated breast cancer cells. Other published data has
suggested that folic acid inhibits the constitutive and induced the
activity of the EGFR promoter in colon cancer cells through
methylation, as this effect was reversed by 5-aza-CdR (24,25).
In a study by Momparler and Ayoub (26), it was observed that patients with
stage IV NSCLC who received five cycles of 5-aza-CdR treatment
survived 81 months. Consistent with these data, in this study, by
evaluating the anti-neoplastic effects of 5-aza-CdR and gefitinib,
we showed that a combination of lower doses (<IC50
values) of 5-aza-CdR and gefitinib significantly inhibited NSCLC
cell growth. Similarly, 5-aza-CdR further increased the induction
of apoptosis by gefitinib in NSCLC cells. These results suggest
that NSCLC cells with EGFR methylation are resistant to gefitinib,
and that 5-aza-CdR, a hypomethylating drug, may increase the
cellular sensitivity to gefitinib in controlling NSCLC cell growth
and apoptosis. The PC-9 cells, which did not harbor a CpG island
methylation within the EGFR promoter, were more sensitive to
gefitinib than the H1650 and H1299 cells. Thus, 5-aza-CdR had no
further effect on PC-9 cell proliferation.
Promoter methylation of the EGFR gene is one of the
key mechanisms that affects cancer cell sensitivity to TKIs.
Previous studies have shown that certain somatic mutations within
the TK and ATP-binding domain of the EGFR gene are associated with
a response to EGFR-TKIs in NSCLC (27,28).
The positive association between EGFR mutations and response to
erlotinib or gefitinib in NSCLC patients has been shown in several
clinical studies, with a significant enhancement of patient
survival (29). However, in larger
randomized studies, such as the BR.21 trial, a similar survival
advantage was observed for patients treated with erlotinib,
independent of EGFR mutations or wild-type EGFR gene, indicating
that EGFR mutations were not the only biomarker for predicting
NSCLC survival with small-molecule EGFR-TKI treatment (30). Our results suggested that the CpG
island methylation status in the EGFR promoter influenced the
sensitivity to gefitinib in NSCLC cells.
Our results demonstrated the enhanced effects of
5-aza-CdR on EGFR-TKI-induced cell growth inhibition and the
induction of apoptosis. The reduced expression of EGFR may be due
to hypermethylation. In addition, 5-aza-CdR treatment resulted in
CpG island demethylation in the promoter of the EGFR gene. These
results suggest that demethylation potentially induces EGFR gene
expression. Whether this could affect the sensitivity and
therapeutic efficacy of gefitinib remains unknown. In general,
methylation causes gene silencing and demethylation induces gene
expression. EGFR expression is not considered as a significant
predictive factor for a response to gefitinib (31). There is no clear correlation between
EGFR expression and gefitinib sensitivity. We reasoned that the
demethylation effect of 5-aza-CdR caused the induction of EGFR gene
expression. Thus, the balance between methylation and demethylation
changes may contribute to the synergistic effects of this
combination treatment. On the basis of EGFR demethylation, this
would enhance the therapeutic response of gefitinib in NSCLC cells.
Further mechanistic studies are required to confirm these
results.
Two major EGFR-TKI-resistance mechanisms have been
revealed. Gefitinib is an ATP competitive inhibitor of EGFR-TKI. It
has been found that the T790M secondary mutation increases the
affinity of the oncogenic mutant EGFR for ATP, and this leads to
the reduced efficacy of EGFR-TKIs (31). Almost half of the patients with
acquired resistance appear to have this mutation (32,33).
Met proto-oncogene (MET) amplification is another mechanism that
escapes the antitumor effect of EGFR-TKIs, which allows cancer cell
survival by persistent enhancement of Akt signaling and MET
amplification when the EGFR signal is blocked in the presence of
EGFR-TKIs. MET amplification has been found in approximately 20% of
patients with acquired resistance (34,35).
The link between MET amplification and promoter methylation of the
EGFR gene involving EGFR-TKI resistance has not been reported;
whether 5-aza-CdR has any direct or indirect effect on ATP or MET
amplification needs to be determined. A recent study showed that
5-aza-CdR decreased c-Met protein expression in NSCLC cells
(36), suggesting a potentially
novel mechanism of this agent in controlling cancer cell
growth.
In conclusion, the data presented in this study show
that NSCLC cells with EGFR methylation are more resistant to
gefitinib, and that the combination treatment with 5-aza-CdR, a
demethylating agent, increases the sensitivity to gefitinib. These
results suggest that the combination of a demethylating agent with
an EGFR-TKI may have a synergistic anti-lung cancer effect and that
the blockade of DNA methylation of the EGFR gene promoter region
should be considered as one of the potential mechanisms for
reversing resistance to EGFR-TKIs in NSCLC cells. Further studies
to better define the role of 5-aza-CdR in controlling cancer cell
growth and the potential synergistic antitumor effects of TKIs and
5-aza-CdR in NSCLC are warranted.
References
|
1
|
Ahmed SM and Salgia R: Epidermal growth
factor receptor mutations and susceptibility to targeted therapy in
lung cancer. Respirology. 11:687–692. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hynes NE and Lane HA: ERBB Receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signaling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Citri A, Skaria KB and Yarden Y: The deaf
and the dumb: The biology of ErbB-2 and ErbB-3. Exp Cell Res.
284:54–65. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baselga J: Why the epidermal growth factor
receptor? The rationale for cancer therapy. Oncologist. 7:2–8.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cappuzzo F, Finocchiaro G, Metro G,
Bartolini S, Magrini E, Cancellieri A, Trisolini R, Castaldini L,
Tallini G and Crino L: Clinical experience with gefitinib: An
update. Crit Rev Oncol Hematol. 24:31–45. 2006. View Article : Google Scholar
|
|
7
|
Haber DA, Bell DW, Sordella R, et al:
Molecular targeted therapy of lung cancer: EGFR mutations and
response to EGFR inhibitors. Cold Spring Harb Symp Quant Biol.
70:419–426. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Janne PA and Johnson BE: Effect of
epidermal growth factor receptor tyrosine kinase domain mutations
on the outcome of patients with non-small cell lung cancer treated
with epidermal growth factor receptor tyrosine kinase inhibitors.
Clin Cancer Res. 12:4416–4420. 2006. View Article : Google Scholar
|
|
9
|
Esteller M: Cancer epigenomics: DNA
methylation and histone-modification maps. Nat Rev Gene. 8:286–298.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in Cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar
|
|
11
|
Silva M, Azenha D, Pereira C, et al:
Gastric carcionoma and chronic gastritis: epigenetic regulation of
CDH1 (E-cadherin), CDKN2A (p16INK4A), PTGS2 (COX-2) and EGFR genes
through methylation. Acta Med Port. 23:5–14. 2010.(In
Portuguese).
|
|
12
|
Wang Z, Xu J, Geng X and Zhang W: Analysis
of DNA methylation status of the promoter of human telomerase
reverse transciptase in gastric carcinogenesis. Arch Med Res.
41:1–6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taron M, Rosell R, Felip E, et al: BRCA1
mRNA expression levels as an indicator of chemoresistance in lung
cancer. Hum Mol Genet. 13:2443–2449. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grivaux M, Zureik M, Marsal L, et al:
Five-year survival for lung cancer patients managed in general
hospitals. Rev Mal Respir. 28:e31–e38. 2011.PubMed/NCBI
|
|
15
|
Gadgeel SM, Ali S, Philip PA, Wozniak A
and Sarkar FH: Genistein enhances the effect of epidermal growth
factor receptor tyrosine kinase inhibitors and inhibits nuclear
factor kappa B in non-small cell lung cancer cell lines. Cancer.
115:2165–2176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vucic EA, Brown CJ and Lam WL: Epigenetics
of cancer progression. Pharmacogenomics. 9:215–234. 2008.
View Article : Google Scholar
|
|
17
|
Duffy MJ, Napieralski R, Martens JW, et
al: Methylated genes as new cancer biomarkers. Eur J Cancer.
45:335–346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim YT, Park SJ, Lee SH, et al: Prognostic
implication of aberrant promoter hypermethylation of CpG islands in
adenocarcinoma of the lung. J Thorac Cardiovasc Surg.
130:13782005.PubMed/NCBI
|
|
19
|
Safar AM, Spencer H III, Su X, et al:
Methylation profiling of archived non-small cell lung cancer: a
promising prognostic system. Clin Cancer Res. 11:4400–4405. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Plimack ER, Stewart DJ and Issa JP:
Combining epigenetic and cytotoxic therapy in the treatment of
solid tumors. J Clin Oncol. 25:4519–4521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Daskalakis M, Blaqitko-Dorfs N and
Hackanson B: Decitabine. Recent Results Cancer Res. 184:131–157.
2010. View Article : Google Scholar
|
|
22
|
Chai G, Li L, Zhou W, et al: HDAC
inhibitors act with 5-aza-2′-deoxycytidine to inhibit cell
proliferation by suppressing removal of incorporated abases in lung
cancer cells. PLoS One. 3:e24452008.
|
|
23
|
Montero AJ, Díaz-Montero CM, Mao L, et al:
Epigenetic inactivation of EGFR by CpG island hypermethylation in
cancer. Cancer Biol Ther. 5:1494–1501. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Majumdar AP, Kodali U and Jaszewski R:
Chemopreventive role of folic acid in colorectal cancer. Front
Biosci. 9:2725–2732. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nagothu KK, Rishi AK, Jaszewski R, Kucuk O
and Majumdar AP: Folic acid-mediated inhibition of serum-induced
activation of EGFR promoter in colon cancer cells. Am J Physiol
Gastrointest Liver Physiol. 287:G541–G546. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Momparler RL and Ayoub J: Potential of
5-aza-2′-deoxycytidine (Decitabine) a potent inhibitor of DNA
methylation for therapy of advanced non-small cell lung cancer.
Lung Cancer. 34(Suppl 4): S111–S115. 2001.
|
|
27
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Paez JG, Jänne PA, Lee JC, et al: EGFR
mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsao M-S, Sakurada A, Cutz J-C, et al:
Erlotinib in lung cancer - molecular and clinical predictors of
outcome. N Engl J Med. 353:133–144. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yun CH, Menqwasser KE, Toms AV, et al: The
T790M mutation in EGFR kinase causes drug resistance by increasing
the affinity for ATP. Proc Natl Acad Sci USA. 105:2070–2075. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kosaka T, Yatabe Y, Endoh H, et al:
Analysis of epidermal growth factor receptor gene mutation in
patients with non-small cell lung cancer and acquired resistance to
gefitinib. Clin Cancer Res. 12:5764–5769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo A, Villen J, Kornhauser J, et al:
Signaling networks assembled by oncogenic EGFR and c-met. Proc Natl
Acad Sci USA. 105:692–697. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Watanabe K, Emoto N, Hamano E, et al:
Genome structure-based screening identified epigenetically silenced
microRNA associated with invasiveness in non-small-cell lung
cancer. Int J Cancer. 130:2580–2590. 2012. View Article : Google Scholar
|