Introduction
Cells sense various forms of injury and insult by
biochemical and molecular machinery and execute appropriate
programs of response; they undergo cell cycle arrest, to allow
adequate time for repair of damage, or they die, if the damage is
too severe. In tumor cells surviving cytotoxic/apoptotic stimuli, a
network integrating several pathways may involve different survival
pathways, such as mitogen-activated protein kinase (MAPK) signaling
cascades, the phosphoinositide 3-kinase (PI3K)/Akt pathway, the
nuclear factor (NF)-κB signaling system and the heat-shock response
(1–5). Among them, of particular relevance are
the MAPKs, serine/threonine-protein kinases regulated by dual
tyrosine and threonin phosphorylation, which by participating in
signaling cascades conserved through evolution regulate important
biological activities (6,7). The three major MAPK groups in
mammalian cells include extracellular signal-regulated kinases
(ERKs), c-Jun NH2-terminal protein kinase (JNK) and p38
(8–10). MAPK signaling cascades are activated
by a variety of different cellular stimuli and mediate diverse
responses. ERKs are acutely stimulated by growth and
differentiation factors through activated receptor tyrosine
kinases, heterotrimeric G protein-coupled receptors or cytokine
receptors (8). JNK and p38 MAPKs
are activated in response to a variety of stress signals including
UV irradiation, chemotherapeutics, osmotic stress, hypoxia/anoxia,
hyperthermia, and they are involved in the regulation of apoptosis
(8). Stress-activated MAPKs can
mediate survival signals, or, on the contrary, death signals
(5,11–13).
Monensin plays a role in the exchange of
Na+ and H+, transporting one molecule of
Na+ into the cells for each molecule of H+
transported out, resulting in intracellular alkalinization
(14). This drug has been reported
to cause tumor cell death (15–19).
In the present study, we investigated whether
moderate stress, caused by appropriate doses of monensin, can
promote survival in U937 cells, derived from a human lymphoma. We
thus identified a p38 MAPK/COX-2 dependent mechanism that appears
to counter cell death.
Materials and methods
Materials
RPMI-1640, fetal calf serum, L-glutamine,
penicillin-streptomycin, phosphate-buffered saline (PBS), monensin,
arachidonic acid (AA), NS398, lipopolysaccharide (LPS) and
anti-β-actin antibodies were from Sigma-Aldrich (St. Louis, MO,
USA). SB203580, SP600125, PD98059 and acetylsalicylic acid (ASA)
were from Calbiochem (Inalco, Milan, Italy). Celecoxib was kindly
donated by Pharmacia (Uppsala, Sweden). Anti-phospho-p38 and
anti-p38 antibodies were from Cell Signaling Technology (Beverly,
MA, USA). Anti-COX-1 and anti-COX-2 antibodies were from Cayman
Chemical (Cabru, Milan, Italy). Horseradish peroxidase
(HRP)-conjugated anti-immunoglobulin antibodies, enhanced
chemiluminescence (ECL) reagents and Hyperfilm-ECL film were from
Amersham (Arlington Heights, IL, USA). Protein standards for
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and
nitrocellulose membranes were from Bio-Rad. The membrane permeant
CDCF-DA was from Molecular Probes (SIC, Rome, Italy), and other
reagents were of the highest purity and purchased from Bio-Rad or
Sigma.
Cell viability and growth
U937 cells, derived from the pleural effusion of a
patient with histiocytic lymphoma (20), were grown in complete medium
(RPMI-1640 medium supplemented with 1.0% sodium pyruvate, 5% FCS, 2
mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin) at
37°C, in a fully humidified atmosphere of 95% room air/5%
CO2. Cells were resuspended three times a week in fresh
complete medium as 3×105/ml. Cell growth was evaluated
by hemocytometry counts of cells excluding Trypan blue (0.04%
Trypan blue in PBS, w/v), and viability was assessed by calculating
alive (trypan blue excluding) cells as percentage of all cells
counted. Cells used in every experiment were ≥94% viable and were
obtained from cultures in exponential growth. They were washed once
and resuspended in complete medium (1×106/ml) and
transferred to 24-well microplates. They were then treated with
inhibitors or vehicles, incubated for 30 min, and subsequently
exposed to test agents or, again, to vehicles. At the end of each
experiment, the cells were gently mixed and aliquots were obtained
for cell counting and cell cycle analysis. The vehicles, even when
used in combination, were ≤0.3% (v/v) and did not modify any
investigated parameter in comparison with control culture.
Flow cytometric analysis of cell
death
Nuclear DNA fragmentation was quantified by flow
cytometry of hypodiploid (subG1) DNA after cell fixation and
staining with propidium iodide (PI) (21,22).
Briefly, cells were washed with PBS, pelleted and fixed in ice cold
ethanol/water (70/30, v/v) for 1 h, pelleted again and washed twice
with PBS, and finally resuspended in PBS containing RNAse (20
μg/ml) and PI (100 μg/ml). Events in the different cell cycle
phases were gated manually using an EPICS XL cytofluorimeter
(Beckman Coulter, Hialeah, FL, USA). At least 10,000 events/sample
were acquired. Collected data were analyzed using the Multicycle
software for DNA content and cell cycle analysis (Phoenix Flow
System, San Diego, CA, USA). The subG1 events representative of the
apoptotic cells, and the events in the other cell cycle phases, are
given as a percentage of the total cell population.
Western blot analysis
Whole cell lysates were prepared as previously
described (23,24). Briefly, the cells were kept for 30
min on ice in lysis buffer [NaCl 150 mM, CaCl2 1 mM,
MgCl2 1 mM, NaN3 0.1%, NaF 10 mM, Triton
X-100 1% (v/v), orthovanadate 1 mM, aprotinin 2 μg/ml, leupeptin 2
μg/ml, iodoacetamide 10 mM, PMSF 2 mM, and pepstatin 20 μM]. The
appropriate volumes of 4X SDS-sample buffer and 2-mercaptoethanol
5% (v/v) were then added. Cell lysates were briefly sonicated,
warmed at 95°C for 5 min, and cleared by centrifugation at 14.000 ×
g in a microfuge for 15 min at 4°C. Supernatants were collected and
proteins were quantified by RC DC protein assay. Equal amounts of
proteins were separated from the different samples by SDS-PAGE, and
blotted onto nitrocellulose membranes.
Anisomycin-treated U937 cells were used as positive
control for p38 MAPK detection. Transfer efficiency was checked
with Ponceau staining. The blots were blocked in Tris-buffered
saline (TBS), containing BSA 2% (w/v), probed with specific primary
antibodies, washed with PBS-Tween-20, and then incubated with a
peroxidase-conjugated secondary antibody. Finally, each membrane
was probed to detect β-actin. The final dilutions and incubation
times suggested by the manufacturer were used for each antibody.
Immunodetection was performed using the ECL reagents and
Hyperfilm-ECL film. In the case of COX-1 and COX-2 detection, U937
cells were lysed in RIPA buffer and, as positive control for COX-2,
the lysates of peripheral blood mononuclear cells were used,
separated by Ficoll-Hypaque density-gradient centrifugation,
cultured at 37°C for 16 h in the presence of 10 μg/ml LPS. Proteins
were resolved by 10% SDS-PAGE, blotted and processed as indicated
above. Immunodetection was performed with monoclonal antibodies
anti-COX-2 (1:1,000) or anti-COX-1 (1:1,000) and specific
peroxidase-conjugated anti-immunoglobulin antibodies. Proteins were
detected by ECL.
Assay of COX-2 and COX-1 activity
To distinguish between the peroxidative role of
COX-1 and COX-2, U937 cells resuspended in complete medium
(1×106/ml) were pretreated with either the COXs
inhibitors (ASA, celecoxib and NS398) or the vehicle, or were left
untreated, and, thereafter, with monensin or, again, with the
vehicle for 6 h. After washing with HBSS, incubating for 10 min
with CDCF-DA (2 μM) in HBSS and another washing with the same
medium, the cells (≥10,000) were analyzed using an EPICS XL
cytofluorimeter, with excitation and emission settings at 495 and
525 nm, respectively. Aliquots of CDCF-DA loaded cells were
incubated with exogenous AA (50 μM) for 5 min, immediately prior to
cytofluorimetry analysis.
CDCF-DA is an oxidation sensitive fluorescent probe,
which is first deacetylated inside the cells to the nonfluorescent
compound 2′,7′-CDCFH and can subsequently be oxidized to the
fluorescent compound 2′,7′-CDCF by a variety of peroxides. This
probe allows to investigate the activity of the COXs, as these
enzymes catalyze a) a cyclooxygenase reaction in which AA is
converted to PGG2 and b) a peroxidase reaction in which PGG2 is
reduced to PGH2 (25). Thus, CDCFH
may be oxidized through this peroxidase reaction to CDCF and become
fluorescent. Inhibition of COX-2 with either celecoxib or NS398, or
inhibition of COX-1 with ASA, reveals the relative contribution of
either COX-1 or COX-2 to AA transformation. Fluorescent cells were
analyzed by an EPICS XL cytofluorimeter on a log scale (FL2) and
recorded as mean fluorescence intensity (MFI) of the whole cell
population. A minimum of 10,000 events were examined for each
sample.
Statistical analysis
Results are expressed as the means ± standard
deviation (SD) of repeated experiments. Statistical differences
were evaluated using the paired two-tailed Student’s t-test. P≤0.05
was considered to indicate statistically significant
differences.
Results
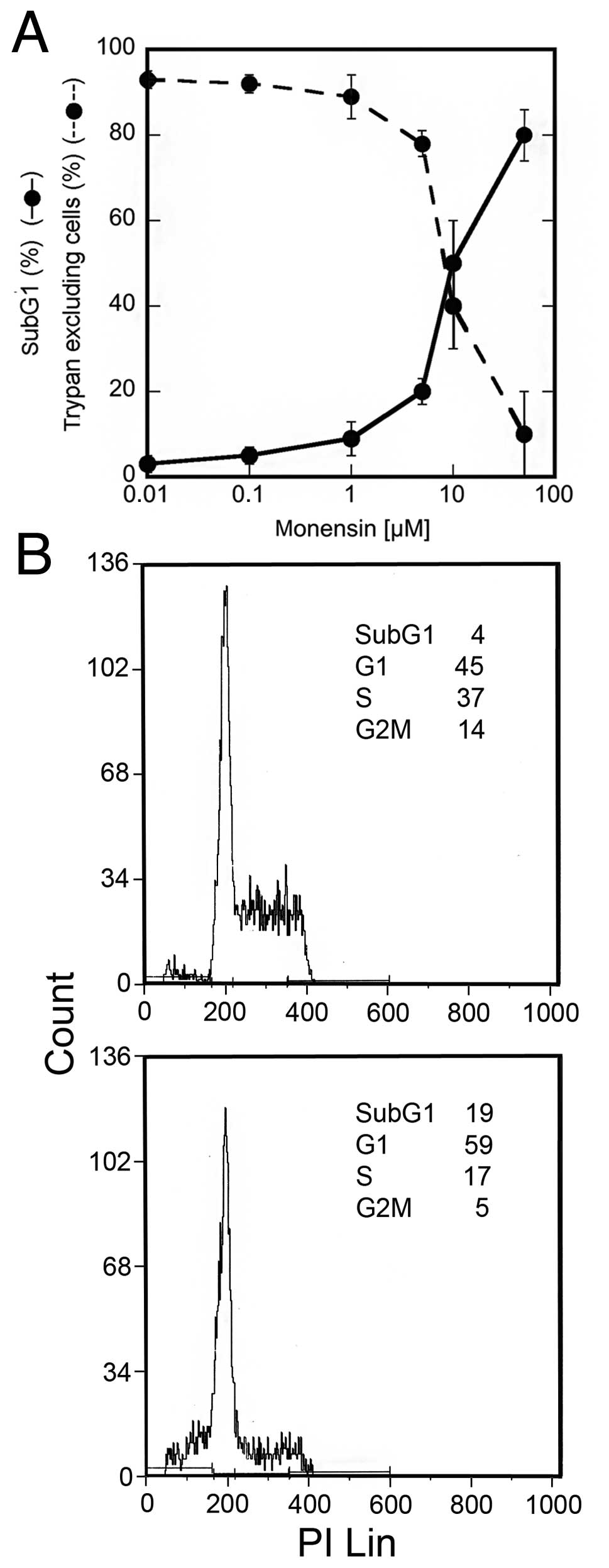
Monensin and cell survival
The Na+ ionophore monensin causes cell
death in a dose-dependent manner. A 24-h treatment with high
concentrations of this drug were cytotoxic for a large proportion
of U937 cells, while lower concentrations were less effective,
suggesting the activation of a survival pathway (Fig. 1A). In particular, 5 μM monensin
caused a slight decrease in trypan blue excluding cells (78±3%) in
comparison with untreated cultures (93±2%), in addition to the
appearance of 20±3% of subG1 events. SubG1 events were studied by
cytofluorimetry of cell cycle phases of cells fixed and stained
with PI; hypodiploid DNA events are easily discernable from the
narrow peak of cells with diploid DNA content, and are considered
to be indicative of apoptotic nuclei (21,22).
Furthermore, analysis of events in the different cell cycle phases
showed that monensin (5 μM) caused a decrease in S and G2M phases,
while the percentage of G1 events increased (Fig. 1B). Cell counts indicated that at
this concentration monensin did not allow cell growth (data not
shown). These results suggest that monensin, at this concentration,
causes activation of a survival pathway in most U937 cells,
increasing the time spent in the G1 cell cycle phase. In order to
investigate this survival pathway, the experiments were performed
using 5 μM monensin and viability parameters were investigated
after 24 h.
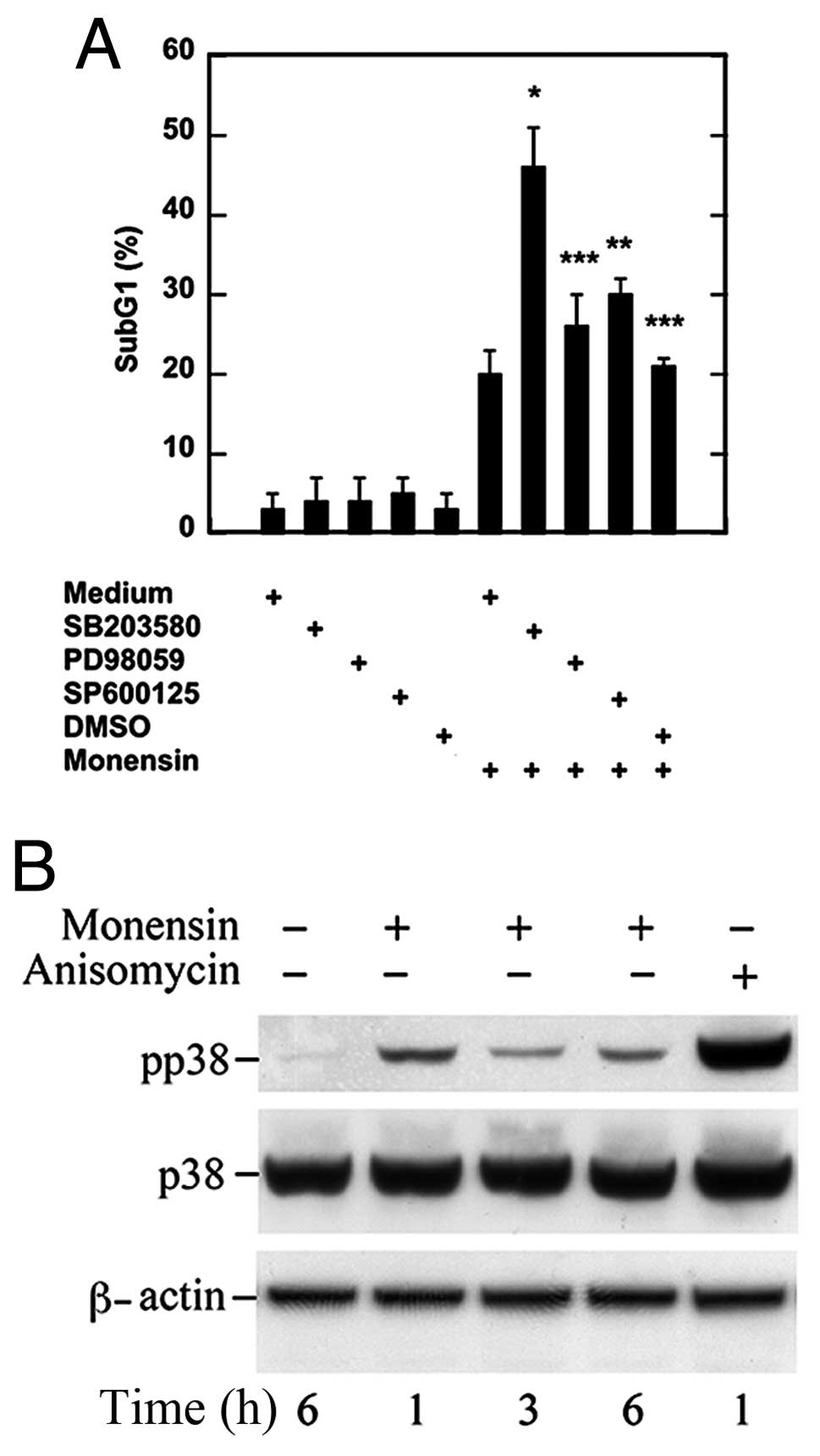
p38 MAPK is activated and promotes the
survival signal
MAPKs are central mediators of cellular survival and
death pathways (11–13). To investigate their involvement in
the previously detected survival pathway activated by monensin, we
pretreated the cells with pharmacological inhibitors at
concentrations affecting specifically one MAPK (26) and then analyzed cell viability.
p38 inhibition with SB203580, but not ERK inhibition
with PD98059, caused a significant cell death increase (Fig. 2A). Also SP600125, a JNK inhibitor,
caused an increase in cell death (Fig.
2A) even if less significant than that caused by SB203580
(P=0.05 vs. P=0.009). Under the same conditions, neither inhibitor
nor DMSO without monensin affected cell viability (Fig. 2A).
To confirm p38 MAPK involvement in the survival
pathway activated by monensin, we performed time-kinetic studies in
which phosphorylated p38 and then total p38 were analyzed by
western blotting with specific antibodies. In the lysate of
untreated U937 cells a faint band relative to phospho-p38 was
detected, which increased after 1 h and was still present after 6 h
of monensin treatment (Fig. 2B).
When probed with antibodies against total p38, a 38 kDa band was
also present in the lysate of untreated cells and showed no change
at the investigated time points (Fig.
2B). Thus, monensin activates p38 MAPK which is involved in a
survival pathway in U937 cells.
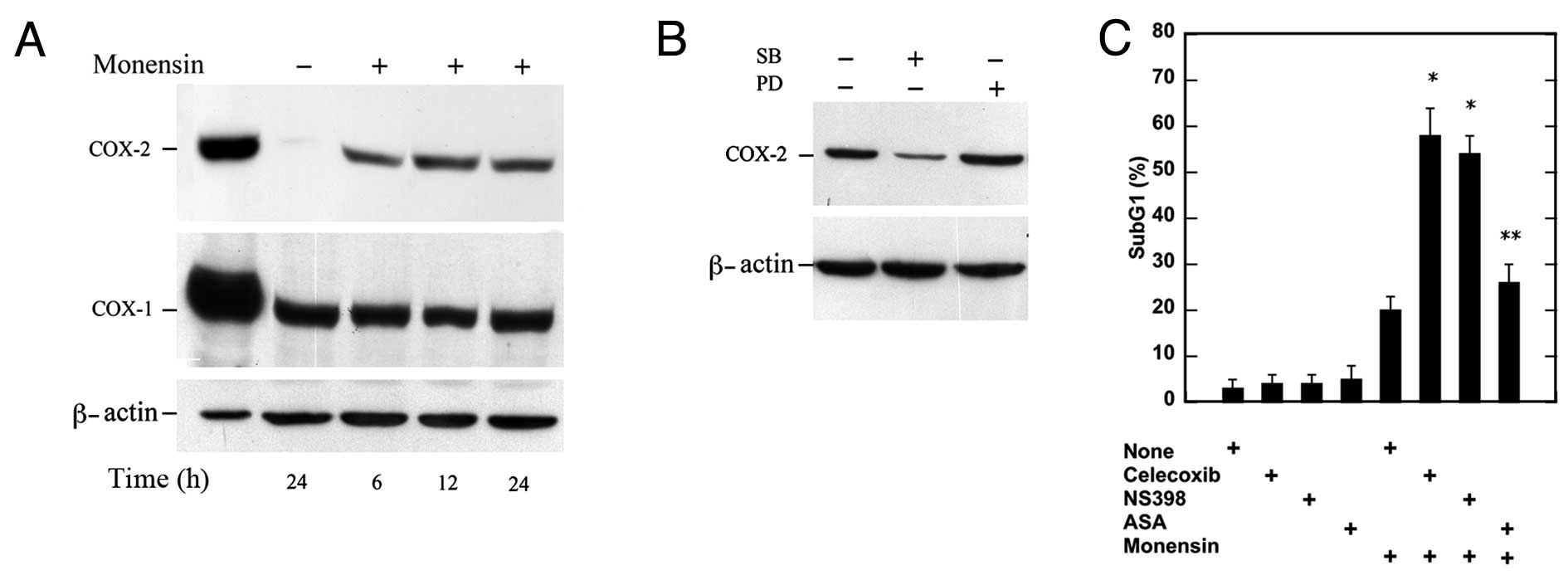
COX-2 is induced by p38 and contributes
to the survival pathway
COX enzymes form prostaglandins (PGs) and
thromboxane from AA and, thus far, two COX isoenzymes (COX-1 and
COX-2) have been detected. They are differently regulated: COX-1 is
constitutively expressed by the majority of cell types, while COX-2
is almost undetectable in normal tissues, is regulated at the
transcriptional and at the post-transcriptional levels (27) and its expression can be regulated by
MAPK (28,29). Therefore, we examined the expression
of this protein by western blotting in the lysates of U937 cells.
Untreated cells did not express COX-2 in a considerable amount
(30) (Fig. 3A), whereas monensin caused an
increase in this protein which became evident after 6 h and was
still present after 24 h. The same lysates probed with anti-COX-1
antibodies showed that the expression of this enzyme is
constitutive in U937 cells and is apparently not affected by
monensin (Fig. 3A). We therefore
examined the expression of COX-2 after p38 MAPK inhibition by
SB203580 or ERK inhibition by PD98059. p38 MAPK inhibition, but not
ERK inhibition, caused a large decrease in COX-2 expression
(Fig. 3B).
In comparison to monensin-treated cells, cell death
was significantly increased after COX-2 inhibition with celecoxib
(1 μM) or NS398 (5 μM) and not after COX-1 inhibition by ASA (100
μM) (Fig. 3C). These results
indicate that COX-2 expression is induced by phosphorylated p38 and
plays a role in the survival pathway activated by monensin in U937
cells.
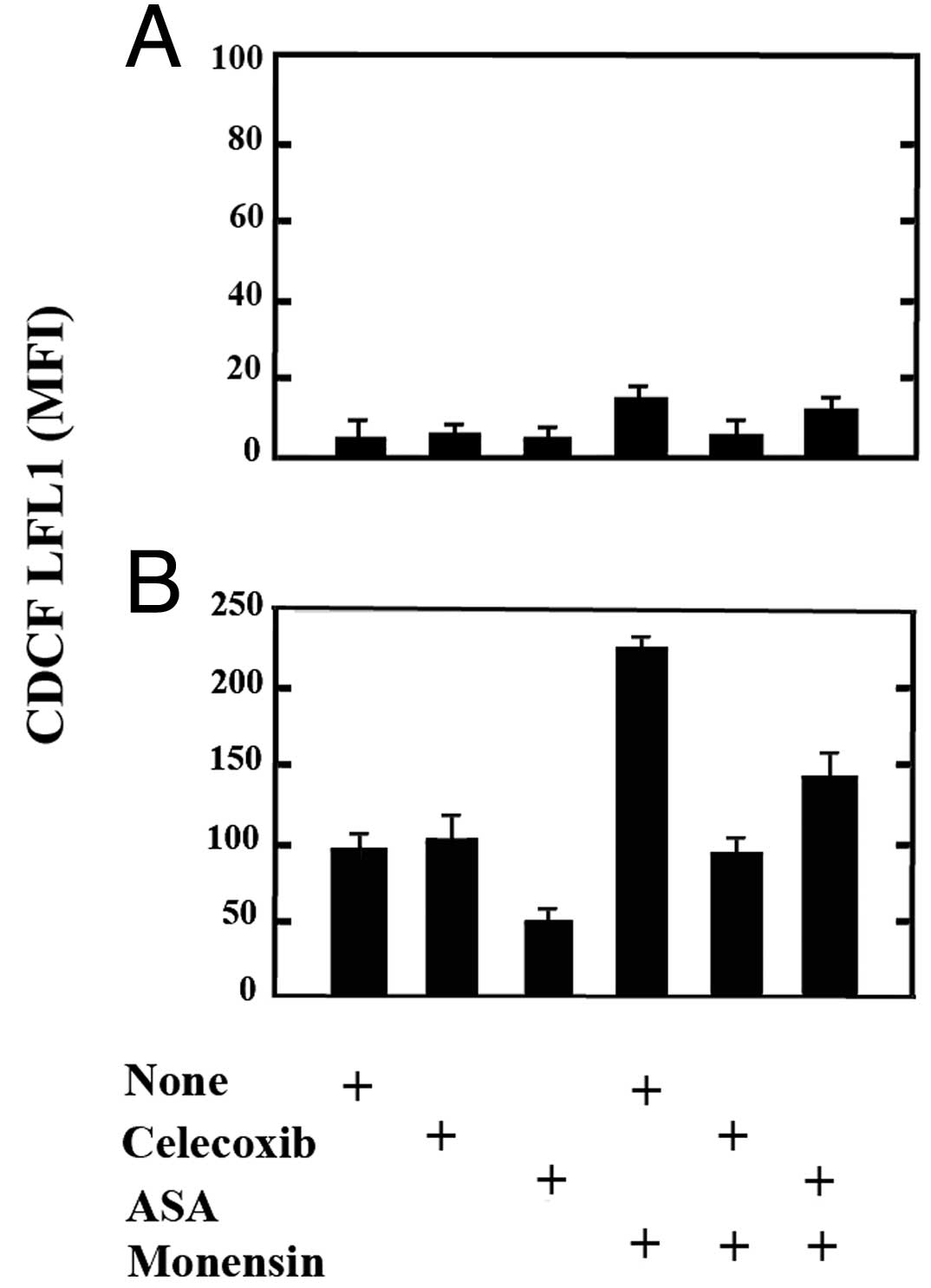
COX-2 efficiently transforms AA
The increased expression of COX-2 suggests that
COX-1, although constitutively expressed in U937 cells, may
function less efficiently than COX-2 and/or that monensin-stressed
cells may have an increased need to transform AA or to avoid its
accumulation. To examine the function of the COXs in living cells
we used CDCF-DA, a probe which becomes fluorescent upon oxidation
as a result of the peroxidase activities of either COX-1 or COX-2
(25).
U937 cells untreated or exposed to monensin for 6 h,
suspended in a medium containing CDCF-DA, were washed and analyzed.
The fluorescence of monensin-treated cells was 3 to 4-fold that of
untreated cells and the responsible enzyme was COX-2, as CDCF
fluorescence was prevented by celecoxib, but not by ASA (Fig. 4A). By this assay we also analyzed
the activity of either COXs towards exogenous AA (50 μM) (Fig. 4B). In comparison with control cells,
U937 cells exposed to AA showed an 18 to 22-fold increase in
fluorescence, which was due to COX-1 as could be largely prevented
by ASA and not by celecoxib (Fig.
4B). Following monensin treatment, the AA exposure caused an
increase in fluorescence 45 to 55-fold that of control cells
(untreated and not exposed to AA). Such an increase was largely due
to COX-2, as celecoxib prevented it by 55–65% and was also, in
part, due to COX-1, as ASA inhibited it by 35–40% (Fig. 4B). These results indicate that COX-2
upregulated in monensin-stressed cells is highly active and suggest
that, by transforming or avoiding the accumulation of AA, keeps
stressed cells alive.
Discussion
In the present investigation we showed that U937
cells treated with subcytotoxic concentrations of monensin were
growth-arrested in the G1 cell cycle phase and escaped from death
by activation of a survival pathway, in which p38 MAPK and COX-2
were involved.
We investigated the role of cyclooxygenases as it is
known that these can be activated by phosphorylated p38 MAPK and
can mediate survival signals (31,32).
COX-2, an enzyme known to be expressed by several tumor cells, was
not apparently expressed in untreated U937 cells, while it was
upregulated within 6 h of monensin treatment. Such expression
depended on p38, as it could be prevented by SB203580. This result
is in agreement with a previously reported finding that p38 causes
stabilization of COX-2 transcript (31). The viability assessment after
inhibition of COX-2 or COX-1 revealed that the former played a
pro-survival role in monensin-treated cells.
It is known that monensin causes an increased
release of arachidonic acid (AA) (33) and, therefore, it is plausible that
the upregulated COX-2 is needed to decrease free AA. This
conclusion is supported by the detection that COX-1 was less active
than COX-2 to transform AA. We assayed the activity of COX
isoenzymes in alive cells; the specific peroxidative function of
COX-1 or COX-2 was explored by loading the cells with the probe
CDCF-DA. In this assay, the analysis of cells pretreated with
celecoxib or NS398, inhibiting COX-2, or with ASA, inhibiting
COX-1, revealed that in monensin-treated cells, COX-2 was more
active than the constitutively expressed COX-1. This conclusion was
reached considering the activity of the isoenzymes towards
endogenous and exogenous AA. It is noteworthy that COX-1, although
present, is much less active than COX-2. However, this aspect has
already been investigated (34) and
some possible explanations are that the Km value of COX-1 for AA is
known to be higher than that of COX-2 (10 and 2 μM, respectively);
mice deficient in COX-1, the constitutive isoform believed to be
responsible for homeostasis, are healthy and live a normal life
span, whereas mice lacking COX-2 expression, the inducible isoform,
thought to be expressed mainly in disease states, display various
developmental problems, female reproductive disorders, and a
shortened life span; COX-1 and COX-2 utilize different pools of AA
for synthesizing prostanoids: in murine fibroblasts and
macrophages, ligand-induced prostaglandin production occurs via
expression of COX-2, while COX-1 present in these cells cannot
metabolize ligand-released AA.
The COX isoforms have both overlapping as well as
distinct physiological and pathological functions; while COX-1 is
involved in the homeostasis of various physiological functions,
COX-2 is responsible for a number of pathological processes such as
inflammation and cancer. COX-2 mRNA levels, as well as COX-2
protein levels, are markedly increased in human colorectal
adenocarcinomas relative to normal colonic mucosa and
overexpression of COX-2 has been identified as an early central
event in colon carcinogenesis (35,36). A
previous study showed that constitutive expression of COX-2 in
human colon cancer promotes tumor invasion and the metastatic
potential of these cells (37). As
well as in colon cancer, increased COX-2 expression is also found
in other types of cancer, including breast, lung and the mucous
membrane of the head and neck (38–40).
However, the functions of this enzyme in tumor cells are not
completely understood. For example, it is not clear whether COX-2
in tumor cells plays an antiapoptotic role. Furthermore, COX-2 is
not expressed in some tumor cells. Thus, in these cases the use of
COX-2 inhibitors would be of no benefit. However, in the present
study, we detected that the stress caused by monensin in U937
leukemia cells induced COX-2 increased expression upon p38 MAPK
activation. This pathway may also be activated by other
antineoplastic drugs and should be considered in combination
therapy with COX-2 or p38 MAPK inhibitors.
Acknowledgements
This study was supported by grants to L.D.R. from
Sapienza Ateneo 2010 and 2011 (8.1.1.1.32.5 and 8.1.1.1.34.1).
References
|
1
|
Kennedy SG, Wagner AJ, Conzen SD, Jordan
J, Bellacosa A, Tsichlis PN and Hay N: The PI 3-kinase/Akt
signaling pathway delivers an anti-apoptotic signal. Genes Dev.
11:701–713. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: a play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Finkel T and Holbrook NJ: Oxidants,
oxidative stress and the biology of ageing. Nature. 408:239–247.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Widmann C, Gibson S, Jarpe MB and Johnson
GL: Mitogen-activated protein kinase: conservation of a
three-kinase module from yeast to human. Physiol Rev. 79:143–180.
1999.PubMed/NCBI
|
|
7
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction patyhways activated by stress and inflammation:
a 10-year update. Physiol Rev. 92:689–737. 2012.PubMed/NCBI
|
|
8
|
Lewis TS, Shapiro PS and Ahn NG: Signal
transduction through MAP kinase cascades. Adv Cancer Res.
74:49–139. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu J, Suzuki H, Akhand AA, Zhou YW,
Hossain K and Nakashima I: Modes of activation of mitogen-activated
protein kinases and their roles in cepharanthine-induced apoptosis
in human leukemia cells. Cell Signal. 14:509–515. 2002. View Article : Google Scholar
|
|
10
|
Platanias LC: MAP kinase signaling
pathways and hematologic malignancies. Blood. 101:4667–4679. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shrode LD, Rubie EA, Woodgett JR and
Grinstein S: Cytosolic alkalinization increases stress-activated
protein kinase/c-Jun NH2-terminal kinase (SAPK/JNK)
activity and p38 mitogen-activated protein kinase activity by a
calcium-independent mechanism. J Biol Chem. 272:13653–13659.
1997.PubMed/NCBI
|
|
12
|
Raciti M, Lotti LV, Valia S, Pulcinelli FM
and Di Renzo L: JNK2 is activated during ER stress and promotes
cell survival. Cell Death Dis. 3:e4292012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okamoto S, Krainc D, Sherman K and Lipton
SA: Antiapoptotic role of the p38 mitogen-activated protein
kinase-myocyte enhancer factor 2 transcription factor pathway
during neuronal differentiation. Proc Natl Acad Sci USA.
97:7561–7566. 2000. View Article : Google Scholar
|
|
14
|
Nakazato K and Hatano Y: Monensin-mediated
antiport of Na+ and H+ across liposome
membrane. Biochim Biophys Acta. 1064:103–110. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu WH and Loh TT: Effects of
Na+/H+ antiport and intracellular pH in the
regulation of HL-60 cell apoptosis. Biochim Biophys Acta.
1269:122–128. 1995.
|
|
16
|
Park WH, Lee MS, Park K, Kim ES, Kim BK
and Lee YY: Monensin-mediated growth inhibition in acute
myelogenous leukemia cells via cell cycle arrest and apoptosis. Int
J Cancer. 101:235–242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park WH, Seol JG, Kim ES, et al:
Monensin-mediated growth inhibition in human lymphoma cells through
cell cycle arrest and apoptosis. Br J Haematol. 119:400–407. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iljin K, Ketola K, Vainio P, et al:
High-throughput cell-based screening of 4910 known drugs and
drug-like small molecules identifies disulfiram as an inhibitor of
prostate cancer cell growth. Clin Cancer Res. 15:6070–6078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ketola K, Hilvo M, Hyotylainen T, et al:
Salinomycin inhibits prostate cancer growth and migration via
induction of oxidative stress. Br J Cancer. 106:99–106. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sundstrom C and Nilsson K: Establishment
and characterization of a human histiocytic lymphoma cell line
(U-937). Int J Cancer. 17:565–577. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nicoletti I, Migliorati G, Pagliacci MC,
Grignani F and Riccardi C: A rapid and simple method for measuring
thymocyte apoptosis by propidium iodide staining and flow
cytometry. J Immunol Methods. 139:271–279. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cirone M, Di Renzo L, Lotti LV, et al:
Primary effusion lymphoma cell death induced by bortezomib and AG
490 activates dendritic cells through CD91. PLoS One. 7:e317322012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matusali G, Arena G, De Leo A, Di Renzo L
and Mattia E: Inhibition of p38 MAP kinase pathway induces
apoptosis and prevents Epstein Barr virus reactivation in Raji
cells exposed to lytic cycle inducing compounds. Mol Cancer.
8:182009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marfè G, Morgante E, Di Stefano C, Di
Renzo L, et al: Sorbitol-induced apoptosis of human leukemia is
mediated by caspase activation and cytochrome c release. Arch
Toxicol. 82:371–377. 2008.PubMed/NCBI
|
|
25
|
Morita I, Schindler M, Regier MK, Otto JC,
Hori T, DeWitt DL and Smith WL: Different intracellular locations
for prostaglandin endoperoxide H synthase-1 and -2. J Biol Chem.
270:10902–10908. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davies SP, Reddy H, Caivano M and Cohen P:
Specificity and mechanism of action of some commonly used protein
kinase inhibitors. Biochem J. 351:95–105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Smith WL, DeWitt DL and Garavito RM:
Cyclooxygenases: structural, cellular, and molecular biology. Annu
Rev Biochem. 69:145–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen W, Tang Q, Gonzales MS and Bowden GT:
Role of p38 MAPK and ERK in mediating ultraviolet-B induced
cyclooxygenase-2 gene expression. Oncogene. 20:3921–3926. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hung JH, Su IJ, Lei HY, et al: Endoplasmic
reticulum stress stimulates the expression of cyclooxygenase-2
through activation of NF-kappaB and pp38 mitogen-activated protein
kinase. J Biol Chem. 279:46384–46392. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rizzo MT, Pudlo N, Farrell L and Leaver A:
Specificity of arachidonic acid-induced inhibition of growth and
activation of c-jun kinases and p38 mitogen-activated protein
kinase in hematopoietic cells. Prostaglandins Leukot Essent Fatty
Acids. 66:31–40. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gauthier ML, Pickering CR, Miller CJ,
Fordyce CA, Chew KL, Berman HK and Tlsty TD: p38 regulates
cyclooxygenase-2 in human mammary epithelial cells and is activated
in premalignant tissue. Cancer Res. 65:1792–1799. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hendrickx N, Volanti C, Moens U, et al:
Up-regulation of cyclooxygenase-2 and apoptosis resistance by p38
MAPK in hypericin-mediated photodynamic therapy of human cancer
cells. J Biol Chem. 278:52231–52239. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang XD, Kiang JG, Scheibel LW and
Smallridge RC: Phospholipase C activation by
Na+/Ca2+ exchange is essential for
monensin-induced Ca2+ influx and arachidonic acid
release in FRTL-5 thyroid cells. J Investig Med. 47:388–396.
1999.PubMed/NCBI
|
|
34
|
Smith WL and Langenbach R: Why there are
two cyclooxygenase isozymes. J Clin Invest. 107:1491–1495. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Reddy BS, Rao CV and Seibert K: Evaluation
of cyclooxygenase-2 inhibitor for potential chemopreventive
properties in colon carcinogenesis. Cancer Res. 56:4566–4569.
1996.PubMed/NCBI
|
|
36
|
Tsujii M, Kawano S and DuBois R:
Cyclooxygenase-2 expression in human colon cancer cells increases
metastatic potential. Proc Natl Acad Sci USA. 94:3336–3340. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oshima M, Dinchuk JE, Kargman SL, et al:
Suppression of intestinal polyposis in Apc delta716 knockout mice
by inhibition of cyclooxygenase 2 (COX-2). Cell. 87:803–809. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wolff H, Saukkonen K, Anttila S,
Karjalainen A, Vainio H and Ristimaki A: Expression of
cyclooxygenase-2 in human lung carcinoma. Cancer Res. 58:4997–5001.
1998.PubMed/NCBI
|
|
39
|
Chan G, Boyle JO, Yang EK, Zhang F, et al:
Cyclooxygenase-2 expression is up-regulated in squamous cell
carcinoma of the head and neck. Cancer Res. 59:991–994.
1999.PubMed/NCBI
|
|
40
|
Higashi Y, Kanekura T and Kanzaki T:
Enhanced expression of cyclooxygenase (COX)-2 in human skin
epidermal cancer cells: evidence for growth suppression by
inhibiting COX-2 expression. Int J Cancer. 86:667–671. 2000.
View Article : Google Scholar : PubMed/NCBI
|