Introduction
Oral cancer accounts for 2–4% of all cancer cases
worldwide (1). An estimated 263,900
new cases and 128,000 deaths from oral cancer occurred globally in
2008 (2). More than 90% of all oral
cancer cases are oral squamous cell carcinomas (OSCCs), which are a
result of multistep processes that develop from the combined
effects of a patient’s genetic predisposition and exposure to
environmental influences, including tobacco, alcohol, betel quid,
chronic inflammation and viral infection (3). Tongue squamous cell carcinoma (TSCC)
is a common OSCC. Imaging systems and treatment strategies in TSCC
are improving substantially, however, 5-year survival statistics
remain low. The molecular mechanisms underlying its clinical
characteristics including carcinogenesis, development, progression,
invasion, and metastasis have yet to be fully elucidated.
Epigenetics studies recently showed that aberrant DNA methylation
is involved in the progress of TSCC (4). DNA methylation is an important
regulator of gene transcription and its role in carcinogenesis and
development has been a topic of considerable interest in recent
years. DNA methylation, which represses transcription of the
promoter region of tumor suppressor genes leading to gene
silencing, has been studied extensively (5).
DNA methylation is a very stable epigenetic mark and
next generation sequencing studies have shown that several genes
are aberrantly methylated in various types of cancer (6,7).
Tissue specific DNA methylation patterns are stabilized during
embryonic development, and are maintained through cell divisions.
Aberrant DNA methylation patterns have been associated with a large
number of human malignancies and are found in two distinct forms:
hypermethylation and hypomethylation (8). The majority of DNA methylation studies
focus on the analysis of CpG islands located in the promoter areas
of candidate genes. However, differentially methylated areas may be
located within genes and at large distances from the nearest
neighboring genes (9). In the past
decade, researchers focused only on a few candidate genes of DNA
methylation in TSCC-related genes (4,5,10).
Nevertheless, systematic studies on the genome-wide distribution of
methylated loci in TSCC remain scarce. With the advent of next
generation sequencing, genome-wide screening has become an
attractive and useful tool for profiling TSCC.
In this study, methylated DNA immunoprecipitation
(MeDIP) in combination with microarray-based hybridization was
performed in adjacent normal control and tumor tissues from 9
patients with TSCC (11). Extensive
DNA methylation distribution information from human TSCC and its
adjacent normal tissues provided a preliminary genomic DNA
methylation profile. Selected differentially expressed genes were
further validated using methylation-specific PCR (MS-PCR) and
reverse transcription PCR (RT-PCR).
Materials and methods
Patients and samples
Samples were obtained from 20 patients diagnosed
with TSCC who underwent surgery at the Second Xiangya Hospital of
Central South University and the Hunan Provincial Tumor Hospital.
The study protocol was approved by the Ethics Committees of Central
South University and written informed consent was obtained from all
patients. None of the patients were given adjuvant chemotherapy or
radiation prior to the operation. For each TSCC case, fresh samples
were obtained from the primary tumor tissue and the adjacent normal
tongue mucosa with a clear surgical margin to tumor. Surgical
margins were considered as tumor-free or negative at least 5 mm of
histologically normal tissue according to the pathological
examination. The samples were snap frozen in liquid nitrogen and
stored at −80°C until subsequent analyses.
MeDIP and methylation microarray
hybridization
Genomic DNA from TSCC and adjacent normal control
tissue (n=9) were extracted and purified according to the DNeasy
Blood and Tissue kit (Qiagen, Germantown, MD, USA). Then, DNA
samples were pooled to one pair and sheared by sonication to obtain
fragments between 400 and 500 bp and were immunoprecipitated by
anti-5-methylcytidine antibody according to the MagMeDIP kit
(Diagenode, Liège, Belgium). Fully methylated DNA was obtained by
whole-genome amplification using the GenomePlex® Whole
Genome Amplification kit (Sigma-Aldrich, St. Louis, MO, USA). MeDIP
DNA and input DNA were labeled with Cy5 and Cy3, respectively, and
then analyzed using Human DNA Methylation 385K Promoter Plus CpG
Island Arrays (Roche NimbleGen, Madison, WI, USA), which were used
to detect 28,226 CpG sites. Gene chips were scanned by GenePix
4000B microarray scanner and hybridization signals were analyzed
using GenePix Pro 6.0 hybridization analysis software. Probe-rich
value setting was ≥2.0.
MS-PCR analysis
To determine the methylation status of the three
selected genes (FBLN1, ITIH5 and RUNX3), we used
MS-PCR in 20 pairs of samples from adjacent normal control and TSCC
tissues. Genomic DNA was isolated by using Wizard®
Genomic DNA Purification kit (Promega, Madison, WI, USA). Complete
bisulfite conversion and purification of DNA were performed
according to the EpiTect® Bisulfite kit (Qiagen). The
fully methylated and fully unmethylated DNA samples were used as
controls, and a water blank reaction was used as control for
contamination. Following amplification by PCR, products were
resolved on 1% agarose gels containing 0.5 μg/ml ethidium bromide
and visualized under UV transillumination. Primers for MS-PCR
analysis are shown in Table I.
 | Table IList of primers of methylated (M) and
unmethylated (U) sequences for MS-PCR analysis. |
Table I
List of primers of methylated (M) and
unmethylated (U) sequences for MS-PCR analysis.
| Gene | Primer | Sequence |
|---|
| FBLN1 | M primer | F:
5′-ATTAGGAGATTCGCGGTTTC-3′ |
| | R:
5′-GCTCCATAAACGACGAACG-3′ |
| U primer | F:
5′-GATTAGGAGATTTGTGGTTTTG-3′ |
| | R:
5′-CACACTCCATAAACAACAAACA-3′ |
| ITIH5 | M primer | F:
5′-TTGGCGATAGAAATTAAGTAAGTTC-3′ |
| | R:
5′-AACCACCTATATTAACCCACG-3′ |
| U primer | F:
5′-TTGGTGATAGAAATTAAGTAAGTTTGT-3′ |
| | R:
5′-AAAACCACCTATATTAACCCCACA-3′ |
| RUNX3 | M primer | F:
5′-TTACGAGGGGCGGTCGTACGCGGG-3′ |
| | R:
5′-AAAACGACCGACGCGAACGCCTCC-3′ |
| U primer | F:
5′-TTATGAGGGGTGGTTGTATGTGGG-3′ |
| | R:
5′-AAAACAACCAACACAAACACCTCC-3′ |
RT-PCR analysis
To determine the mRNA expression level of the six
selected genes (FBLN1, ITIH5, RUNX3, BCL2L14, CDCP1 and
DIRAS3), we used RT-PCR in 20 pairs of samples from adjacent
normal control and TSCC tissues. Total RNA was isolated from the
sample tissues using TRIzol® reagent (Invitrogen, Grand
Island, NY, USA) according to the manufacturer’s protocol. RNA was
efficiently reverse transcribed into cDNA using the Reverse
Transcription System (Promega). Primers for RT-PCR analysis and the
PCR conditions of the six selected genes are shown in Table II. The expression level of each
gene was confirmed by comparison to the expression amounts of
β-actin which served as an internal control. Optical density of
positive band was subsequently measured using Quantity One 4.62
(Bio-Rad, Hercules, CA, USA).
| Table IIList of primers for RT-PCR analysis
and the condition of PCR. |
Table II
List of primers for RT-PCR analysis
and the condition of PCR.
| Gene | Primer | Size of product
(bp) | Annealing
temperature (°C) | Cycles |
|---|
| FBLN1 | F:
5′-TCAATACAGTTGCCTAGAGCA-3′ | 409 | 60 | 28 |
| R:
5′-GAGGAACAAGAGGACCCAT-3′ | | | |
| ITIH5 | F:
5′-CACAGTTCTCCAGCGACA-3′ | 386 | 60 | 32 |
| R:
5′-CAGGACCGTTTCAGTATCAT-3′ | | | |
| RUNX3 | F:
5′-CAGAAGCTGGAGGACCAGAC-3′ | 180 | 62 | 32 |
| R:
5′-TCGGAGAATGGGTTCAGTTC-3′ | | | |
| BCL2L14 | F:
5′-TCCAGGTGTCTTTCTAACG-3′ | 407 | 58 | 34 |
| R:
5′-TTGACCTCTGATTCTCCC-3′ | | | |
| CDCP1 | F:
5′-CATAAGAGCATCGGTTTAGAG-3′ | 329 | 58 | 28 |
| R:
5′-GGGTAGTTGGCAGACATCA-3′ | | | |
| DIRAS3 | F:
5′-TCTCTCCGAGCAGCGCA-3′ | 410 | 62 | 34 |
| R:
5′-CGTCGCCACTCTTGCTGTCG-3′ | | | |
| β-actin | F:
5′-TAAGGAGAAGCTGTGCTACG-3′ | 459 | 58 | 28 |
| R:
5′-GACTCGTCATACTCCTGCTT-3′ | | | |
Statistical analysis
Data are expressed as the means ± standard deviation
(SD). Data were analyzed using SPSS 16.0 (SPSS, Chicago, IL, USA).
The results of MS-PCR analysis were analyzed using a two-tailed
Fisher exact test. The data of RT-PCR analysis were analyzed using
a Student’s t test. P<0.05 was considered to indicate
statistically significant differences.
Results
Global DNA methylation changes in TSCC
tissues
In order to investigate the aberrant genomic DNA
methylation in TSCC, we performed methylated DNA amplification
coupled with CpG island microarray analysis using a set of TSCC
tissue specimens (adjacent normal control tissue vs. tumor tissue)
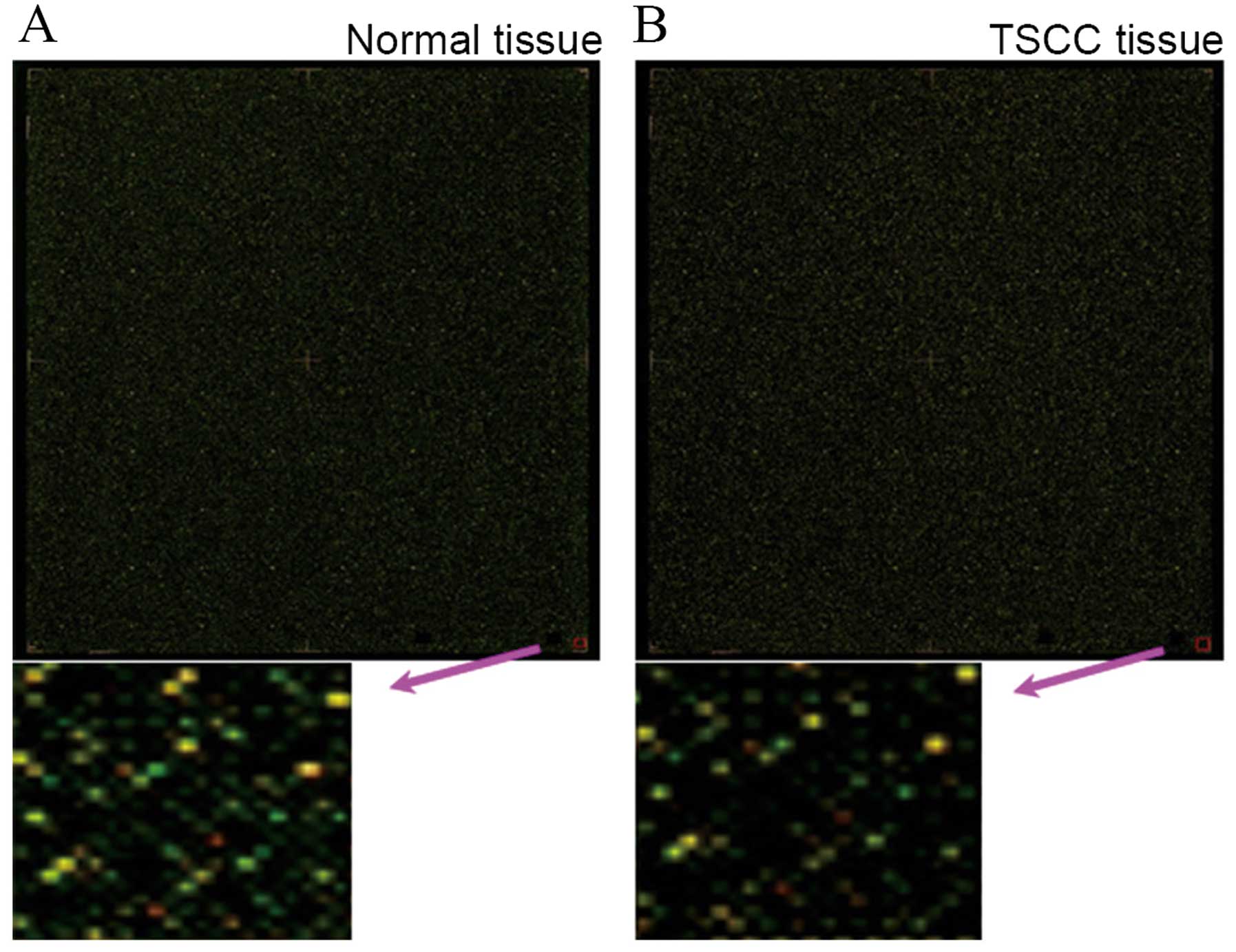
(Fig. 1). Methylation status of the
individual 28,226 CpG sites was compared between the 9 pairs of
samples. There were 2,654 DNA hypermethylated sites that
significantly differed in methylation level between adjacent normal
control and TSCC tissue. A total of 1,269 CpG sites covering 330
genes were found to be significantly hypermethylated in TSCC
tissues. In our analysis, these genes were located in different
chromosomes. Among these hypermethylated genes, 28 genes (8.48%)
and 27 genes (8.18%) were located on chromosome 1 and 19,
respectively. Furthermore, chromosome 17, 16, 7 and 2 had 26
(7.88%), 24 (7.27%), 23 (6.97%) and 22 (6.67%) hypermethylated
genes, respectively. Chromosome 18 had only 4 hypermethylated
genes, accounting for 1.21%. In addition, chromosome 15 or Y had 5
hypermethylated genes (1.52%). Of the 330 methylated genes in TSCC,
the hypermethylated sites of 218 genes (66.1%) were located in the
promoter region, 77 (23.3%) in the coding region, and 45 (13.6%) in
the downstream of gene, respectively.
In adjacent normal tissue, there were 1,385
hypermethylated CpG sites, covering 321 genes. Among these genes,
up to 40 genes were located on chromosome 19, accounting for
12.46%. Chromosome X and 1 had 32 and 23 genes, accounting for 9.97
and 7.17%, respectively. Moreover, chromosome 14 had only 2 genes
(0.62%). Among the 321 genes, 210 genes (65.4%) were located in the
promoter region. Familial aggregation was found in these genes of
differential methylation. For example, four genes of keratin
(KRT) family and three keratin-related proteins
(KRTAP) were shown to be hypermethylated in tumor tissue,
including KRT 14 and 31 on chromosome 17, KRT 72 and
75 on chromosome 12, KRTAP10-3 and 10-7 on chomosome
21 and KRTAP2-4 on chromosome 17. Seven members of
MAGE (melanoma antigen-encoding gene) family, MAGEA2,
MAGEA3, MAGEA4, MAGEA9, MAGEA9B, MAGEA10 and MAGEA11,
were found to be hypermethylated in adjacent normal tissue and
located on chromosome X.
Validation of aberrantly methylated genes
by MS-PCR
To verify the results obtained from our previous DNA
methylation microarray study, we selected three candidate genes
(FBLN1, ITIH5 and RUNX3), which were based on reports
of tumor-specific methylation genes, for further analysis by
MS-PCR. Twenty cases of TSCC and paired adjacent tissues were
detected by MS-PCR for these three genes. Of these three genes,
FBLN1 and ITIH5 were hypermethylated in TSCC tissue,
and RUNX3 was hypermethylated in adjacent normal tissue.
FBLN1 and ITIH5 showed a significantly higher
incidence of DNA methylation in TSCC, while RUNX3 had
markedly lower incidence of DNA methylation in TSCC than in
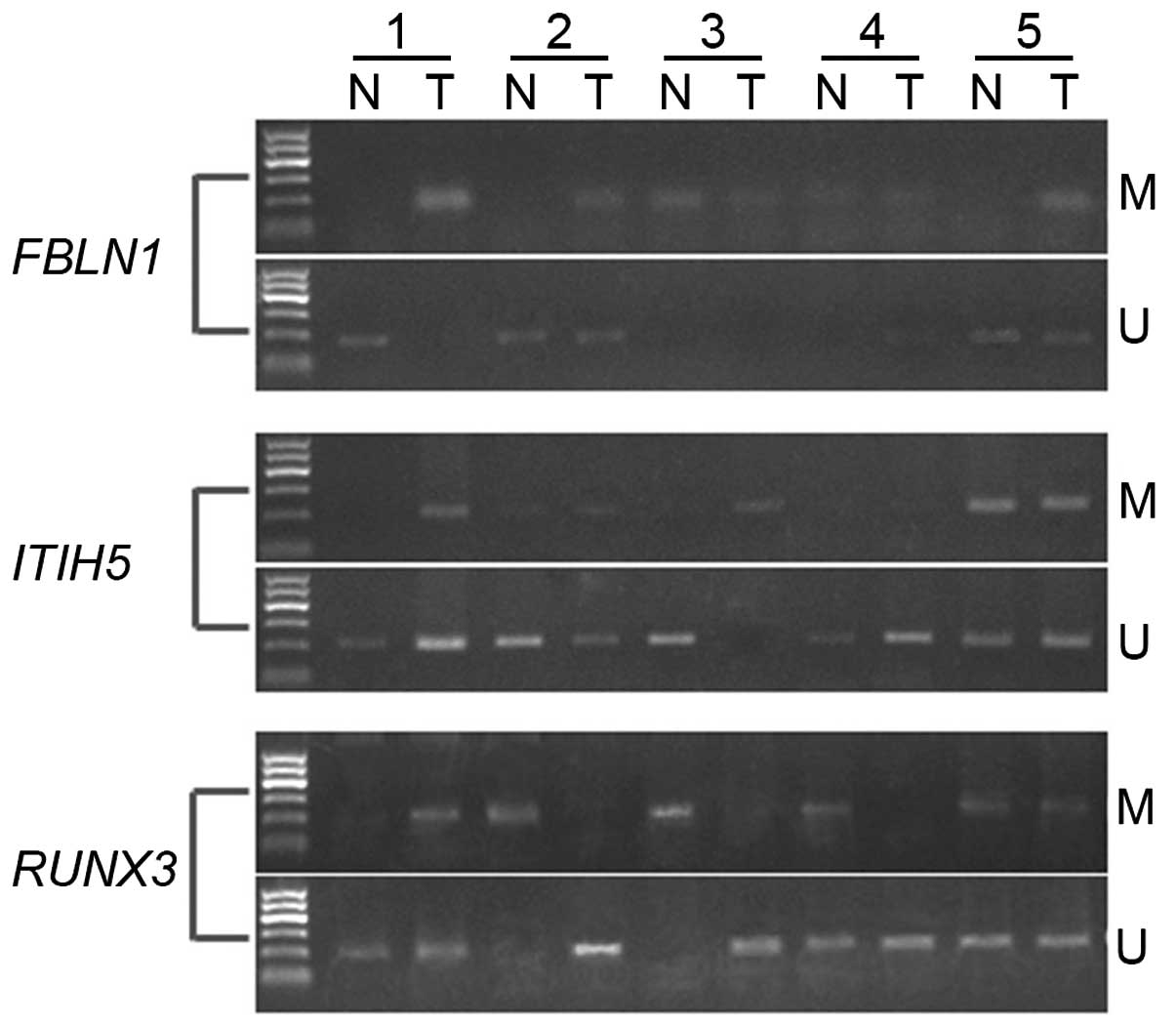
adjacent normal tissue. As shown in Fig. 2 and Table III, FBLN1 showed DNA
methylation in 11/20 cases (55%) in TSCC and in 4/20 cases (20%) in
adjacent normal tissue (P=0.048 <0.05). Moreover, methylation of
ITIH5 was detected in 14/20 cases in TSCC and in 5/20 cases
in adjacent control tissues, of which the incidence was 70 and 25%,
respectively (P=0.004 <0.01). In addition, we observed that
11/20 cases (55%) in adjacent normal tissues and only 3/20 cases
(15%) in TSCC showed DNA methylation of RUNX3 (P=0.019
<0.05). Differences in DNA methylation status of these three
genes were statistically significant between adjacent normal and
TSCC tissue. These findings were consistent with the microarray
study.
| Table IIIFrequency of DNA methylation status
of three candidate genes in paired adjacent normal and TSCC
tissues. |
Table III
Frequency of DNA methylation status
of three candidate genes in paired adjacent normal and TSCC
tissues.
| | FBLN1 | ITIH5 | RUNX3 |
|---|
| |
|
|
|
|---|
| Group | Count | U | M | U | M | U | M |
|---|
| | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) |
|---|
| N | 20 | 16 (80%) | 4 (20%) | 15 (75%) | 5 (25%) | 9 (45%) | 11 (55%) |
| T | 20 | 9 (45%) | 11 (55%) | 6 (30%) | 14 (70%) | 17 (85%) | 3 (15%) |
| | P=0.048a | P=0.004b | P=0.019a |
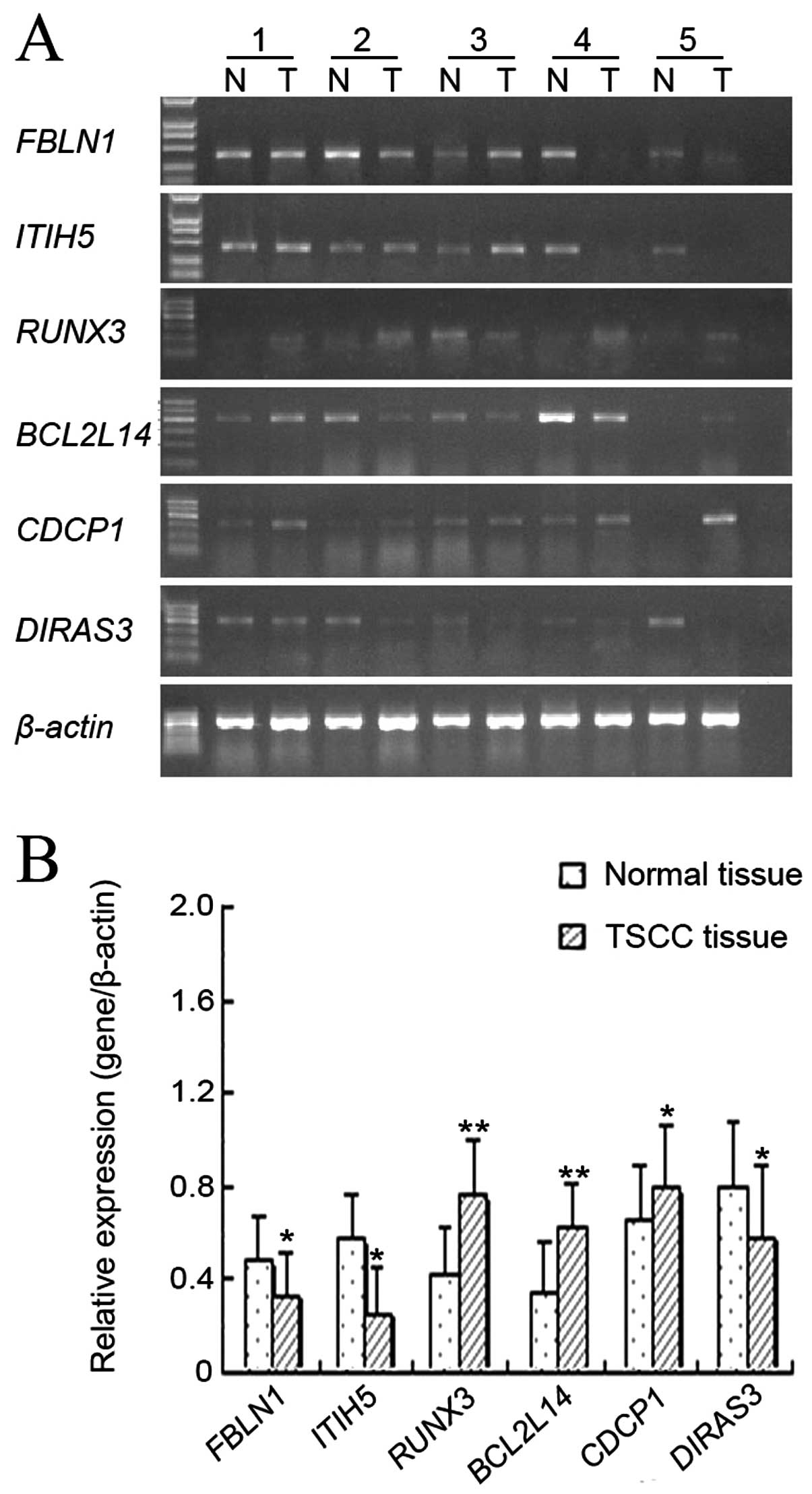
Analysis of mRNA expression level of
candidate genes by RT-PCR
According to our microarray analysis data, we
selected six genes, which were highly related to tumorigenesis and
development of TSCC, to detect their mRNA expression in the 20
paired samples. These genes included FBLN1, ITIH5, RUNX3,
BCL2L14, CDCP1 and DIRAS3. As compared with adjacent
normal tissue, TSCC tissue showed markedly upregulated expression
levels of RUNX3 (P=0.006 <0.01), BCL2L14 (P=0.001
<0.01) and CDCP1 (P=0.032 <0.05). By contrast,
expression levels of FBLN1 (P=0.042 <0.05), ITIH5
(P=0.021 <0.05) and DIRAS3 (P=0.037 <0.05) were
significantly downregulated in TSCC tissue as compared with
adjacent normal tissue (Fig. 3).
The gene expression levels of FBLN1, ITIH5 and RUNX3
in TSCC tissue were inversely correlated with the DNA methylation
status.
Discussion
In the present study, a total of 9 paired samples
from adjacent normal control and TSCC tissues were screened at the
individual 28,226 CpG sites. The results demonstrated that 2,654
CpG sites were significantly different in methylation level: 1,269
sites, covering 330 genes, were hypermethylated in TSCC tissue and
1,385 sites, covering 321 genes, were hypermethylated in adjacent
normal tissue. The microarray data could provide evidence and
insights to further clarify the molecular mechanism in TSCC.
DNA methylation is the most common gene modification
in eukaryotic cells. This epigenetic event has been studied
extensively in the field of cancer research. In the past decade,
several studies focused on the aberrant DNA methylation in various
types of cancer, suggesting that the CpG island hypermethylation in
gene promoter was closely related to inactivation of tumor
suppressor gene or candidate tumor suppressor gene expression
(12). Sardi et al(13) demonstrated that promoter
hypomethylation resulted in the activation of oncogene expression
in human bladder cancer and local hypermethylation may be
considered a potential mechanism for increasing genetic alterations
in bladder cancer formation. Furthermore, aberrant DNA methylation
can be detected before tumors are clinically evident. Kresty et
al(14) showed that
hypermethylation of the cell cycle-regulatory genes
p16INK4a and
p14ARF were detected in 57.7 and 3.8% of
patients, respectively, and the highest rates of
p16INK4a hypermethylation occurred in
lesions of the tongue and the floor of the mouth. The alterations
of p16INK4a and
p14ARF locus are frequent events preceding
the development of oral cancer. As described by Palmisano et
al(15), aberrant methylation
of the p16 and/or O6-methylguanine-DNA
methyltransferase (MGMT) promoters can be detected in DNA from
sputum in 100% of patients with lung squamous cell carcinoma up to
three years before clinical diagnosis. In addition, Rosas et
al(10) reported that aberrant
methylation of at least one of these genes,
p16CDKN2A, MGMT and death-associated
protein kinase (DAP-K), were detected in 17 (56%) of 30 head and
neck primary tumors; 14 (47%) of 30 at p16, 10 (33%) of 30
at Dap-K and 7 (23%) of 30 at MGMT. In 11 (65%) of 17 methylated
primary tumors, abnormally methylated DNA was detected in the
matched saliva samples.
In order to verify our microarray data, we selected
three genes (ITIH5, FBLN1 and RUNX3), which have
previously been reported in studies on aberrant DNA methylation in
various types of cancer, and tested their levels of DNA methylation
and mRNA expression in adjacent normal and TSCC tissues.
FBLN1, fibulin 1, was identified as a tumor
suppressor gene whose inactivation may contribute to
carcinogenesis. FBLN1 plays a tumor suppressive role in gastric
cancer (16) as well as in other
types of cancer such as breast (17) and prostate cancer (18). Cheng et al(16) demonstrated that aberrant promoter
hypermethylation resulted in downregulated expression of
FBLN1 in all gastric cancer cell lines and the primary
gastric carcinoma tissues and was significantly restored after
pharmacological demethylation. Kanda et al(19) reported that promoter
hypermethylation of FBLN1 was significantly associated with
advanced stage hepatocellular carcinoma, multiple tumors and
increased tumor size. The present study showed that FBLN1
was hypermethylated and its mRNA level was significantly decreased
in TSCC tissue compared with those in adjacent normal tissue. The
exact molecular mechanism of how FBLN1 suppresses
tumorigenesis remains unclear. Its expression could suppress cell
motility, inhibit the phosphorylation of extracellular
signal-regulated kinase and myosin light chain, and reduce the
intracellular calcium level (20).
The inter-α-trypsin inhibitors (ITIs) family
constitutes a group of plasma proteins consisting of one light
(bikunin) and two heavy chains (ITIHs). ITIH5 is a member of
the ITIH gene family (21)
and is a tumor suppressor gene. ITI plays a role in extracellular
matrix stabilization and in the prevention of tumor metastasis
(22). The studies of Veeck et
al(21) and Himmelfarb et
al(22) revealed that promoter
methylation-mediated downregulation of ITIH5 expression was
associated with unfavorable outcome in breast cancer patients. Hamm
et al(23) reported that
ITIH2, ITIH3, ITIH4 and ITIH5 were strongly
downregulated in a variety of human solid tumors (breast,
endometrium, ovary, cervix, stomach, small intestine, colon,
rectum, lung, thyroid, prostate, kidney and pancreas). ITIH5
could be involved in the progression, invasion and metastasis of
breast cancer, as its absence is associated with increased
proliferation rates and a prognostic value indicating poor clinical
outcome (21,22). Data from our study indicated that
aberrant ITIH5 promoter hypermethylation occurred in 14 out
of 20 cases (70%) in TSCC tissue. There were statistically
significant differences in ITIH5 expression between TSCC and
adjacent normal tissues.
RUNX3, runt-related transcription factor 3,
part of the runt-related (RUNX) family, appears to be an important
component of the transforming growth factor-β (TGF-β)-induced tumor
suppression pathway. RUNX3 has a critical role in the
regulation of cell proliferation and cell death by apoptosis, as
well as in angiogenesis, cell adhesion and invasion (24). RUNX3 acts as a tumor
suppressor gene in gastric cancer (25,26).
Aside from gastric cancer, it has been reported that downregulated
expression of RUNX3 is observed in bladder (27), gastric (28), liver (29–31),
colorectal (32,33) and lung (34) cancer. In these tumors, reduced
expression of RUNX3 was frequently caused by CpG island
hypermethylation. It was reported that RUNX3 expression
levels were upregulated in head and neck squamous cell carcinoma
(35), basal cell carcinoma of skin
(36) and ovarian cancer (37). RUNX3 may have an oncogenic
role in HNSCC as well as in basal cell carcinoma of skin (38,39).
In addition, RUNX3 expression was observed in the tongue and
palate epithelium of mouse embryos and disappears in newborn and
adult mice (24,40). The results of this study are in
accordance with these previous findings. RUNX3 was
hypomethylated and its expression was upregulated in TSCC tissue.
DNA hypermethylation of RUNX3 was observed in normal
epithelial cells of the tongue, suggesting that RUNX3 may be
silenced by methylation in normal oral mucosa. The exact molecular
mechanism of RUNX3 regulation in normal adult oral
epithelium and TSCC remains unclear and requires further
investigation.
Of the other three candidate genes, BCL2L14
and CDCP1 were hypermethylated in adjacent normal tissue and
DIRAS3 was hypermethylated in TSCC tissue. In the RT-PCR
analysis, the expressions of BCL2L14 and CDCP1 were
significantly upregulated in TSCC tissue compared to adjacent
normal tissue, whereas the expression of DIRAS3 was markedly
downregulated. Therefore, these findings indicate that the
expressions of the three genes in TSCC tissues may be caused by the
aberrant DNA methylation during the development of TSCC.
Apoptosis facilitator Bcl-2-like protein 14
(BCL2L14, also known as Bcl-G), is a protein encoded
by the BCL2L14 gene, and belongs to the Bcl-2 family. Bcl-2
family members act as anti- or pro-apoptotic regulators that are
involved in a wide variety of cellular activities (41). The human Bcl-G gene encodes
two proteins through alternative mRNA splicing, Bcl-G(L)
(long) and Bcl-G(S) (short). Bcl-G(L) mRNA is widely
expressed in adult human tissues, whereas Bcl-G(S) mRNA is
found only in testis. Overexpression of this gene has been shown to
induce apoptosis in cells. Bcl-G was downregulated in
prostate (42) and breast cancer
(43). However, in this study,
BCL2L14 was observed to be hypomethylated with upregulated
mRNA expression in TSCC tissue. CUB domain containing protein
(CDCP1), a transmembrane protein with intracellular tyrosine
residues which are phosphorylated upon activation, is assumed to be
involved in proliferative activities and resistance to apoptosis of
cancer cells (44). CDCP1 is
a novel stem cell marker that is expressed in several types of
cancer. CDCP1 mRNA highly overexpressed in human colon
cancer (45) and lung
adenocarcinoma (44). In the breast
cancer samples of Ikeda et al(46), tumors with high-level CDCP1
expression showed higher levels of proliferation. CDCP1 may
be involved in regulating the adhesion and motility of cancer cells
(47).
In our study, CDCP1 was shown to be
hypermethylated in CpG site using microarray analysis but its
expression was found to be upregulated using RT-PCR in TSCC tissue.
DIRAS3 (also known as ARHI and NOEY2),
GTP-binding protein Di-Ras3, is a novel imprinted tumor suppressor
gene that encodes a small GTPase with 60% homology to Ras and Rap
(48). Only the paternal allele of
DIRAS3 is expressed due to maternal imprinting.
DIRAS3 was absent or expressed at lower levels with allelic
loss and promoter hypermethylation in ovarian, breast, and liver
cancer as well as in other tumor tissues (49–51).
DIRAS3 may play an important role physiologically in
regulating cell growth through regulating expression of the cyclins
and cyclin dependent kinase inhibitors (49). Our results showed that DIRAS3
gene expression was downregulated in TSCC tissue with
hypomethylation status. The increased expression of BCL2L14
may be regulated by hypomethylation of its promoter region in TSCC
tissue. However, the mechanism by which CDCP1 expression was
upregulated with DNA hypermethylation and DIRAS3 expression
was downregulated with DNA hypomethylation in TSCC tissue compared
with that in adjacent normal tissue, remains unclear.
In summary, genome-wide DNA methylation microarray
was used to identify the methylated genes in TSCC. Distinctly
different DNA methylation profiles were obtained from adjacent
normal and TSCC tissues. Furthermore, we validated the methylation
status of three candidate genes by MS-PCR and the mRNA expression
level of six candidate genes by RT-PCR. These results are
consistent with the microarray data. Our study may provide useful
data for further investigation of TSCC biomarkers for diagnosis,
treatment and prognosis. Future gene-specific investigations are
required to explore the molecular mechanism of the tumorigenesis,
development and progression of TSCC.
References
|
1
|
Markopoulos AK: Current aspects on oral
squamous cell carcinoma. Open Dent J. 6:126–130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
3
|
Choi S and Myers JN: Molecular
pathogenesis of oral squamous cell carcinoma: implications for
therapy. J Dent Res. 87:14–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Demokan S and Dalay N: Role of DNA
methylation in head and neck cancer. Clin Epigenetics. 2:123–150.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
González-Ramírez I, García-Cuellar C,
Sánchez-Pérez Y and Granados-García M: DNA methylation in oral
squamous cell carcinoma: molecular mechanisms and clinical
implications. Oral Dis. 17:771–778. 2011.PubMed/NCBI
|
|
6
|
Lister R, Pelizzola M, Dowen RH, Hawkins
RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall
L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson
JA, Ren B and Ecker JR: Human DNA methylomes at base resolution
show widespread epigenomic differences. Nature. 462:315–322. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Weber M, Davies JJ, Wittig D, Oakeley EJ,
Haase M, Lam WL and Schubeler D: Chromosome-wide and
promoter-specific analyses identify sites of differential DNA
methylation in normal and transformed human cells. Nat Genet.
37:853–862. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar
|
|
9
|
Carvalho RH, Haberle V, Hou J, van Gent T,
Thongjuea S, van Ijcken W, Kockx C, Brouwer R, Rijkers E, Sieuwerts
A, Foekens J, van Vroonhoven M, Aerts J, Grosveld F, Lenhard B and
Philipsen S: Genome-wide DNA methylation profiling of non-small
cell lung carcinomas. Epigenetics Chromatin. 5:92012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosas SL, Koch W, da Costa Carvalho MG, Wu
L, Califano J, Westra W, Jen J and Sidransky D: Promoter
hypermethylation patterns of p16,
O6-methylguanine-DNAmethyltransferase, and death-associated protein
kinase in tumors and saliva of head and neck cancer patients.
Cancer Res. 61:939–942. 2001.
|
|
11
|
Jacinto FV, Ballestar E and Esteller M:
Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA
methylome. Biotechniques. 44:35–43. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sardi I, Dal Canto M, Bartoletti R and
Montali E: Abnormal c-myc oncogene DNA methylation in human bladder
cancer: possible role in tumor progression. Eur Urol. 31:224–230.
1997.PubMed/NCBI
|
|
14
|
Kresty LA, Mallery SR, Knobloch TJ, Song
H, Lloyd M, Casto BC and Weghorst CM: Alterations of p16(INK4a) and
p14(ARF) in patients with severe oral epithelial dysplasia. Cancer
Res. 62:5295–5300. 2002.PubMed/NCBI
|
|
15
|
Palmisano WA, Divine KK, Saccomanno G,
Gilliland FD, Baylin SB, Herman JG and Belinsky SA: Predicting lung
cancer by detecting aberrant promoter methylation in sputum. Cancer
Res. 60:5954–5958. 2000.PubMed/NCBI
|
|
16
|
Cheng YY, Jin H, Liu X, Siu JM, Wong YP,
Ng EK, Yu J, Leung WK, Sung JJ and Chan FK: Fibulin 1 is
downregulated through promoter hypermethylation in gastric cancer.
Br J Cancer. 99:2083–2087. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pupa SM, Argraves WS, Forti S, Casalini P,
Berno V, Agresti R, Aiello P, Invernizzi A, Baldassari P, Twal WO,
Mortarini R, Anichini A and Menard S: Immunological and
pathobiological roles of fibulin-1 in breast cancer. Oncogene.
23:2153–2160. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wlazlinski A, Engers R, Hoffmann MJ, Hader
C, Jung V, Muller M and Schulz WA: Downregulation of several
fibulin genes in prostate cancer. Prostate. 67:1770–1780. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kanda M, Nomoto S, Okamura Y, Hayashi M,
Hishida M, Fujii T, Nishikawa Y, Sugimoto H, Takeda S and Nakao A:
Promoter hypermethylation of fibulin 1 gene is associated with
tumor progression in hepatocellular carcinoma. Mol Carcinog.
50:571–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Twal WO, Czirok A, Hegedus B, Knaak C,
Chintalapudi MR, Okagawa H, Sugi Y and Argraves WS: Fibulin-1
suppression of fibronectin-regulated cell adhesion and motility. J
Cell Sci. 114:4587–4598. 2001.PubMed/NCBI
|
|
21
|
Veeck J, Chorovicer M, Naami A, Breuer E,
Zafrakas M, Bektas N, Dürst M, Kristiansen G, Wild PJ, Hartmann A,
Knuechel R and Dahl E: The extracellular matrix protein ITIH5 is a
novel prognostic marker in invasive node-negative breast cancer and
its aberrant expression is caused by promoter hypermethylation.
Oncogene. 27:865–876. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Himmelfarb M, Klopocki E, Grube S, Staub
E, Klaman I, Hinzmann B, Kristiansen G, Rosenthal A, Dürst M and
Dahl E: ITIH5, a novel member of the inter-alpha-trypsin inhibitor
heavy chain family is downregulated in breast cancer. Cancer Lett.
204:69–77. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hamm A, Veeck J, Bektas N, Wild PJ,
Hartmann A, Heindrichs U, Kristiansen G, Werbowetski-Ogilvie T, Del
Maestro R, Knuechel R and Dahl E: Frequent expression loss of
Inter-alpha-trypsin inhibitor heavy chain (ITIH) genes in multiple
human solid tumors: a systematic expression analysis. BMC Cancer.
8:252008. View Article : Google Scholar
|
|
24
|
Tsunematsu T, Kudo Y, Iizuka S, Ogawa I,
Fujita T, Kurihara H, Abiko Y and Takata T: RUNX3 has an oncogenic
role in head and neck cancer. PLoS One. 4:e58922009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li QL, Ito K, Sakakura C, Fukamachi H,
Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ,
Yamamoto H, Yamashita N, Yano T, Ikeda T, Itohara S, Inazawa J, Abe
T, Hagiwara A, Yamagishi H, Ooe A, Kaneda A, Sugimura T, Ushijima
T, Bae SC and Ito Y: Causal relationship between the loss of RUNX3
expression and gastric cancer. Cell. 109:113–124. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ito K, Liu Q, Salto-Tellez M, Yano T, Tada
K, Ida H, Huang C, Shah N, Inoue M, Rajnakova A, Hiong KC, Peh BK,
Han HC, Ito T, Teh M, Yeoh KG and Ito Y: RUNX3, a novel tumor
suppressor, is frequently inactivated in gastric cancer by protein
mislocalization. Cancer Res. 65:7743–7750. 2005.PubMed/NCBI
|
|
27
|
Kim WJ, Kim EJ, Jeong P, Quan C, Kim J, Li
QL, Yang JO, Ito Y and Bae SC: RUNX3 inactivation by point
mutations and aberrant DNA methylation in bladder tumors. Cancer
Res. 65:9347–9354. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen W, Gao N, Shen Y and Cen JN:
Hypermethylation downregulates Runx3 gene expression and its
restoration suppresses gastric epithelial cell growth by inducing
p27 and caspase3 in human gastric cancer. J Gastroenterol Hepatol.
25:823–831. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mori T, Nomoto S, Koshikawa K, Fujii T,
Sakai M, Nishikawa Y, Inoue S, Takeda S, Kaneko T and Nakao A:
Decreased expression and frequent allelic inactivation of the RUNX3
gene at 1p36 in human hepatocellular carcinoma. Liver Int.
5:380–388. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shiraha H, Nishina S and Yamamoto K: Loss
of runt-related transcription factor 3 causes development and
progression of hepatocellular carcinoma. J Cell Biochem.
112:745–749. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nishina S, Shiraha H, Nakanishi Y, Tanaka
S, Matsubara M, Takaoka N, Uemura M, Horiguchi S, Kataoka J,
Iwamuro M, Yagi T and Yamamoto K: Restored expression of the tumor
suppressor gene RUNX3 reduces cancer stem cells in hepatocellular
carcinoma by suppressing Jagged1-Notch signaling. Oncol Rep.
26:523–531. 2011.PubMed/NCBI
|
|
32
|
Ku JL, Kang SB, Shin YK, Kang HC, Hong SH,
Kim IJ, Shin JH, Han IO and Park JG: Promoter hypermethylation
downregulates RUNX3 gene expression in colorectal cancer cell
lines. Oncogene. 23:6736–6742. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee CW, Ito K and Ito Y: Role of RUNX3 in
bone morphogenetic protein signaling in colorectal cancer. Cancer
Res. 70:4243–4252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yanada M, Yaoi T, Shimada J, Sakakura C,
Nishimura M, Ito K, Terauchi K, Nishiyama K, Itoh K and Fushiki S:
Frequent hemizygous deletion at 1p36 and hypermethylation
downregulate RUNX3 expression in human lung cancer cell lines.
Oncol Rep. 14:817–822. 2005.PubMed/NCBI
|
|
35
|
Ginos MA, Page GP, Michalowicz BS, Patel
KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL and Gaffney PM:
Identification of a gene expression signature associated with
recurrent disease in squamous cell carcinoma of the head and neck.
Cancer Res. 64:55–63. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Salto-Tellez M, Peh BK, Ito K, Tan SH,
Chong PY, Han HC, Tada K, Ong WY, Soong R, Voon DC and Ito Y: RUNX3
protein is overexpressed in human basal cell carcinomas. Oncogene.
25:7646–7649. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee CW, Chuang LS, Kimura S, Lai SK, Ong
CW, Yan B, Salto-Tellez M, Choolani M and Ito Y: RUNX3 functions as
an oncogene in ovarian cancer. Gynecol Oncol. 122:410–417. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kudo Y, Tsunematsu T and Takata T:
Oncogenic role of RUNX3 in head and neck cancer. J Cell Biochem.
112:387–393. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ito K: RUNX3 in oncogenic and
anti-oncogenic signaling in gastrointestinal cancers. J Cell
Biochem. 112:1243–1249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lau QC, Raja E, Salto-Tellez M, Liu Q, Ito
K, Inoue M, Putti TC, Loh M, Ko TK, Huang C, Bhalla KN, Zhu T, Ito
Y and Sukumar S: RUNX3 is frequently inactivated by dual mechanisms
of protein mislocalization and promoter hypermethylation in breast
cancer. Cancer Res. 66:6512–6520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guo B, Godzik A and Reed JC: Bcl-G, a
novel pro-apoptotic member of the Bcl-2 family. J Biol Chem.
276:2780–2785. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pickard MR, Edwards SE, Cooper CS and
Williams GT: Apoptosis regulators Fau and Bcl-G are down-regulated
in prostate cancer. Prostate. 70:1513–1523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pickard MR, Green AR, Ellis IO, Caldas C,
Hedge VL, Mourtada-Maarabouni M and Williams GT: Dysregulated
expression of Fau and MELK is associated with poor prognosis in
breast cancer. Breast Cancer Res. 11:R602009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ikeda J, Oda T, Inoue M, Uekita T, Sakai
R, Okumura M, Aozasa K and Morii E: Expression of CUB domain
containing protein (CDCP1) is correlated with prognosis and
survival of patients with adenocarcinoma of lung. Cancer Sci.
100:429–433. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Perry SE, Robinson P, Melcher A, Quirke P,
Bühring HJ, Cook GP and Blair GE: Expression of the CUB domain
containing protein 1 (CDCP1) gene in colorectal tumour cells. FEBS
Lett. 581:1137–1142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ikeda JI, Morii E, Kimura H, Tomita Y,
Takakuwa T, Hasegawa JI, Kim YK, Miyoshi Y, Noguchi S, Nishida T
and Aozasa K: Epigenetic regulation of the expression of the novel
stem cell marker CDCP1 in cancer cells. J Pathol. 210:75–84. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Seidel J, Kunc K, Possinger K, Jehn C and
Lüftner D: Effect of the tyrosine kinase inhibitor lapatinib on
CUB-domain containing protein (CDCP1)-mediated breast cancer cell
survival and migration. Biochem Biophys Res Commun. 414:226–232.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yuan J, Luo RZ, Fujii S, Wang L, Hu W,
Andreeff M, Pan Y, Kadota M, Oshimura M, Sahin AA, Issa JP, Bast RC
Jr and Yu Y: Aberrant methylation and silencing of ARHI, an
imprinted tumor suppressor gene in which the function is lost in
breast cancers. Cancer Res. 63:4174–4180. 2003.PubMed/NCBI
|
|
49
|
Yu Y, Xu F, Peng H, Fang X, Zhao S, Li Y,
Cuevas B, Kuo WL, Gray JW, Siciliano M, Mills GB and Bast RC Jr:
NOEY2 (ARHI), an imprinted putative tumor suppressor gene in
ovarian and breast carcinomas. Proc Natl Acad Sci USA. 96:214–219.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Luo RZ, Fang X, Marquez R, Liu SY, Mills
GB, Liao WS, Yu Y and Bast RC: ARHI is a Ras-related small
G-protein with a novel N-terminal extension that inhibits growth of
ovarian and breast cancers. Oncogene. 22:2897–2909. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Huang J, Lin Y, Li L, Qing D, Teng XM,
Zhang YL, Hu X, Hu Y, Yang P and Han ZG: ARHI, as a novel
suppressor of cell growth and downregulated in human hepatocellular
carcinoma, could contribute to hepatocarcinogenesis. Mol Carcinog.
48:130–140. 2009. View Article : Google Scholar : PubMed/NCBI
|