Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of cancer worldwide, with nearly one million new cases
and over 600,000 deaths annually (1–5).
Although surgical resection offers the best prognosis for long-term
survival, the majority of HCC patients are not eligible for surgery
as at the time of diagnosis the tumor may be too large, or it may
have expanded into nearby major blood vessels or metastasized
(6). Therefore, chemotherapy
remains a major therapeutic approach for patients with advanced
HCC. Regimens including doxorubicin, cisplatin or fluorouracil, as
single agents or in combination, are the standard treatments for
these patients. However, due to drug resistance, systemic
chemotherapy using the above regimens produces a disappointing low
response rate (7). Moreover,
several currently used anticancer agents have potent cytotoxic
effects in normal cells (8). These
problems limit the effectiveness of current HCC chemotherapy,
increasing the necessity for the development of novel anticancer
agents. Natural products have received attention as they have
relatively few side-effects and have long been used as alternative
remedies for a variety of diseases including cancer (9,10).
Thus, identifying naturally occurring agents is a promising
approach for anticancer treatment.
The biological role of apoptosis is to eliminate
redundant or damaged cells and, hence, is crucial for maintaining
tissue homeostasis. Disturbed regulation of this vital
physiological process underlies numerous diseases including cancer
(11–13). Apoptosis can be triggered by either
intrinsic stimuli, such as cytokine deprivation and DNA damage, or
by extrinsic stimuli, such as death ligand-receptor engagement.
Both intrinsic and extrinsic signals eventually lead to the
activation of cysteine-dependent aspartate-directed proteases
(caspases) and nucleases, resulting in destruction of the cell
(13,14). The best understood intrinsic
apoptotic pathway is centered at the mitochondria, which is
therefore referred to as mitochondrial-dependent apoptosis. Bcl-2
family proteins are key regulators of mitochondrial-dependent
apoptosis (12,13), functioning as either suppressors,
such as Bcl-2, or promoters, such as Bax. One mechanism by which
Bcl-2 family proteins regulate apoptosis is through their effect on
the permeability of the mitochondrial outer membrane (MOM) via
homo- or hetero-association (15).
Activation of either of the pro-apoptotic proteins Bax or Bak is
sufficient to induce mitochondrial outer membrane permeabilization
(MOMP) (16–19). This event leads to the release of
pro-apoptotic proteins such as cytochrome c and Diablo/Smac
that, in turn, trigger the activation of the caspase cascade
(19–23). The anti-apoptotic Bcl-2 protein
protects cells from apoptosis by interacting with Bax and
inhibiting Bax-mediated MOMP (16,23,24).
The ratio of active anti- and pro-apoptotic Bcl-2 family proteins
determines the fate of cells. Alteration of this ratio by aberrant
expression of these proteins impairs the normal cellular apoptotic
program and may contribute to various apoptosis-related diseases
including several types of cancer (25,26).
Therefore, promoting cell apoptosis via regulation of the Bcl-2
family proteins has been a major focus in the development of
anticancer therapies.
Livistona chinensis, belonging to the
monocotyledonous Palmaceae family, is a medicinal herb
widely distributed in Eastern Asia. The seed of Livistona
chinensis has long been used in China to clinically treat
various types of cancer (27).
Extracts of the Livistona chinensis seed have been shown to
inhibit the growth of several cancer cells (28–31).
However, the precise mechanisms of its tumoricidal activity remain
largely unknown. Using an HCC mouse xenograft model and a human HCC
cell line, in the present study we evaluated the efficacy of the
ethanol extract of Livistona chinensis seed (EELC) against
tumor growth in vivo and in vitro, and investigated
the underlying molecular mechanisms.
Materials and methods
Materials and reagents
Dulbecco’s modified Eagle’s medium (DMEM), fetal
bovine serum (FBS), penicillin-streptomycin, Trypsin-EDTA, TRIzol
reagent, JC-1, caspase-3 and -9 colorimetric protease assay kits
were purchased from Invitrogen (Carlsbad, CA, USA). SuperScript II
reverse transcriptase was obtained from Promega (Madison, WI, USA).
Bcl-2 and Bax antibodies, horseradish peroxidase (HRP)-conjugated
secondary antibodies were obtained from Cell Signaling Technology
(Beverly, MA, USA). The TUNEL assay kit was purchased from R&D
Systems (Minneapolis, MN, USA). A fluorescein isothiocyanate
(FITC)-conjugated Annexin V apoptosis detection kit was obtained
from Becton-Dickinson (San Jose, CA, USA). Tricin was provided by
Professor Xinhua Lin from the Department of Pharmacology, Fujian
Medical University. All other chemicals, unless otherwise stated,
were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of EELC. Livistona chinensis
seeds (500 g) were extracted with 5,000 ml of 85% ethanol using a
refluxing method and were filtered. The resultant solution was
concentrated to a relative density of 1.05, and the dried powder of
EELC was obtained by spraying desiccation method using a spray
dryer (Buchi, Model B-290, Flawil, Switzerland). For animal
experiments, the powder of EELC was dissolved in saline to a
working concentration of 300 mg/ml. The stock solution of EELC in
cell-based experiments was prepared by dissolving EELC powder in
50% DMSO to a stock concentration of 500 mg/ml and the working
concentrations were made by diluting the stock solution in the cell
culture medium. The final concentration of DMSO in the medium for
all cell experiments was <0.5%.
HPLC-TOF/MS analysis
The samples were analyzed by HPLC-TOF/MS using a
micrOTOF-Q spectrometer from Bruker Daltonics (Bremen, Germany)
with an electrospray ionization (ESI) interface coupled with an
HPLC Dionex UltiMate 3000 (Fig. 1).
A Wonda Sil Herbal Medicine Column (150×4.6 mm; 5 μm, GL Sciences)
was used for gradient separation. A linear gradient system
consisted of mobile phase A (0.1% formic acid aqueous solution) and
mobile phase B (acetonitrile containing 0.1% formic acid). The
gradient elution profile was as follows: 0–5 min, 10% B; 5–45 min,
10–95% B; 45–50 min, 95% B. The column was recycled with 10%
solvent B for 10 min, and equilibrated for another 2 min before
using again. The column was maintained at 25°C with a flow rate of
0.4 ml/min during the gradient separation and column equilibration.
The injection volume was 10 μl. Fig. 1A
and B shows HPLC profiles of EELC and a control sample tricin.
The MS operating conditions were optimized as follows: the dry gas
temperature was set at 180°C, the flow rate was 3.0 l/min, the
nebulizer pressure was set at 2.0 bar, and the capillary voltage
was at −3.5 kV. Data were analyzed using Bruker Daltonics
DataAnalysis 3.0 software.
Cell culture
Human HCC HepG2 cells were obtained from the
American Type Culture Collection (ATCC, Manassas, VA, USA). The
cells were grown in DMEM containing 10% (v/v) FBS, and 100 U/ml
penicillin and 100 μg/ml streptomycin in a 37°C humidified
incubator with 5% CO2. The cells were subcultured at
80–90% confluency.
Animals
Male BALB/c athymic (nude) mice (with an initial
body weight of 20–22 g) were obtained from Shanghai SLAC Laboratory
Animal Co., Ltd. (Shanghai, China) and housed under pathogen-free
conditions with controlled temperature (22°C), humidity, and a 12-h
light/dark cycle. Food and water were provided ad libitum
throughout the experiment. All animal treatments were performed
strictly in accordance with the international ethical guidelines
and the National Institutes of Health Guide concerning the Care and
Use of Laboratory Animals. The experiments were approved by the
Institutional Animal Care and Use Committee of Fujian University of
Traditional Chinese Medicine.
In vivo nude mice xenograft study
HCC xenograft mice were produced with HepG2 cells.
The cells were grown in culture and then detached by
trypsinization, washed, and resuspended in serum-free DMEM.
Resuspended cells (4×106) mixed with Matrigel (1:1) were
subcutaneously injected into the right flank of mice to initiate
tumor growth. After 7 days of xenograft implantation when tumor
size reached ~3 mm in diameter, mice were randomized into two
groups (n=10) and intragastrically administered 3 g/kg of EELC or
saline daily, 5 days a week for 21 days. Body weight and tumor size
were measured. Tumor size was determined by measuring the major (L)
and minor (W) diameter with a caliper. The tumor volume was
calculated according to the following formula: Tumor volume = π/6 ×
L × W2. At the end of the experiment, the animals were
anesthetized with pelltobarbitalum natricum, and the tumor issue
was removed and weighed. A portion of each tumor was fixed in 10%
buffered formalin and the remaining tissue was snap-frozen in
liquid nitrogen and stored at −80°C.
In situ apoptosis detection by TUNEL
The TUNEL reaction was carried out following
treatment with EELC as previously described (32). The 4-μm sections of tumor samples
were analyzed by TUNEL staining using TumorTACS In situ Apoptosis
kit (R&D Systems). Apoptotic cells were counted as DAB-positive
cells (brown stained) at five arbitrarily selected microscopic
fields at a magnification of ×400. TUNEL-positive cells were
counted as a percentage of the total cells.
Evaluation of cell viability by MTT
assay
Cell viability was assessed by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
colorimetric assay. HepG2 cells were seeded into 96-well plates at
a density of 1×104 cells/well in 0.1 ml medium. The
cells were treated with various concentrations of EELC for 6, 12 or
24 h. Treatment with 0.5% DMSO was included as vehicle control. At
the end of the treatment, 10 μl MTT (5 mg/ml in phosphate-buffered
saline; PBS) were added to each well, and the samples were
incubated for an additional 4 h at 37°C. The purple-blue MTT
formazan precipitate was dissolved in 100 μl DMSO. The absorbance
was measured at 570 nm using an ELISA reader (BioTek, Model ELx800,
Winooski, Vermont, USA).
Detection of apoptosis by flow cytometry
analysis with Annexin V/PI staining
Following incubation with various concentrations of
EELC, apoptosis of HepG2 cells was determined by flow cytometry
analysis using a fluorescence-activated cell sorting (FACS) caliber
(Becton-Dickinson) and Annexin V-fluorescein isothiocyanate
(FITC)/Propidium iodide (PI) kit. Staining was performed according
to the manufacturer’s instructions. The percentage of cells in
early apoptosis was calculated by Annexin V-positivity and
PI-negativity, while the percentage of cells in late apoptosis was
calculated by Annexin V-positivity and PI-positivity.
Measurement of mitochondrial membrane
potential (ΔΨm) by flow cytometric analysis with JC-1 staining
JC-1 is a cationic dye that exhibits
potential-dependent accumulation in mitochondria, indicated by a
fluorescence emission shift from green to red, which can thus be
used as an indicator of mitochondrial potential. In this
experiment, 1×106 treated HepG2 cells were resuspended
after trypsinization in 1 ml of medium and incubated with 10 μg/ml
of JC-1 at 37°C, 5% CO2, for 30 min. Both red and green
fluorescence emissions were analyzed by flow cytometry following
JC-1 staining.
Analysis of caspase activation
The activities of caspase-3 and -9 were determined
by a colorimetric assay using the caspase-3 and -9 activation kits,
following the manufacturer’s instructions. Briefly, after treatment
with various concentrations of EELC for 24 h, HepG2 cells were
lysed with the lysis buffer provided by the manufacturer for 30 min
on ice. The lysed cells were centrifuged at 16,000 × g for 10 min.
The protein concentration of the clarified supernate was determined
and 100 μg of the protein were incubated with 50 μl of the
colorimetric tetrapeptides, Asp-Glu-Val-Asp (DEVD)-p-nitroaniline
(pNA) (specific substrate of caspase-3) or Leu-Glu-His-Asp
(LEHD)-pNA (specific substrate of caspase-9) at 37°C in the dark
for 2 h. Samples were read at 405 nm in an ELISA plate reader
(BioTek, Model ELx800). The data were normalized to the activity of
the caspases in control cells (treated with 0.5% DMSO vehicle) and
represented as ‘fold of control’.
RNA extraction and RT-PCR analysis
The expression of Bax and Bcl-2 genes were detected
by RT-PCR as previously described (32). Briefly, total RNA from tumor tissues
or HepG2 cells was isolated with TRIzol reagent. Oligo (dT)-primed
RNA (1 μg) was reverse-transcribed with SuperScript II reverse
transcriptase (Promega) according to the manufacturer’s
instructions. The obtained cDNA was used to determine the mRNA
amount of Bcl-2 or Bax by PCR. GAPDH was used as an internal
control. The sequences of the primers used for amplification of
Bcl-2, Bax, and GAPDH transcripts were: Bcl-2 forward, 5′-CAG CTG
CAC CTG ACG CCC TT-3′ and reverse, 5′-GCC TCC GTT ATC CTG GAT
CC-3′; Bax forward, 5′-TGC TTC AGG GTT TCA TCC AGG-3′ and reverse,
5′-TGG CAA AGT AGA AAA GGG CGA-3′; GAPDH forward, 5′-GT CAT CCA TGA
CAA CTT TGG-3′ and reverse, 5′-GA GCT TGA CAA AGT GGT CGT-3′.
Immunohistochemistry
Immunohistochemical staining (IHS) for Bcl-2 and Bax
was performed as previously described (33). Briefly, after fixing with 10%
formaldehyde for 12 h, tumor samples were processed conventionally
for paraffin-embedded tumor slides. The slides were subjected to
antigen retrieval and the endogenous peroxidase activity was
quenched with hydrogen peroxide. After blocking non-specific
proteins with normal serum in PBS (0.1% Tween-20), slides were
incubated with rabbit polyclonal antibodies against Bcl-2 and Bax
(all in 1:200 dilution). After washing with PBS, slides were
incubated with biotinylated secondary antibody followed by
conjugated horseradish peroxidase (HRP)-labelled streptavidin
(Dako), and then washed with PBS. The slides were then incubated
with diamino-benzidine (DAB, Sigma) as the chromogen, followed by
counterstaining with diluted Harris hematoxylin (Sigma). After
staining, five high-power fields (x400) were randomly selected in
each slide, and the average proportion of positive cells in each
field was counted using the true color multi-functional cell image
analysis management system (Image-Pro Plus, Media Cybernetics,
USA). To rule out any non-specific staining, PBS was used to
replace the primary antibody as a negative control.
Western blot analysis
HepG2 cells (2×105) were seeded into
6-well plates in 2 ml medium and treated with various
concentrations of EELC for 24 h. Treated cells were lysed with
mammalian cell lysis buffer containing protease and phosphatase
inhibitor cocktails. The lysates were resolved in 12% SDS-PAGE gels
and electroblotted. The PVDF membranes were blocked with 5% skimmed
milk and probed with primary antibodies against Bcl-2, Bax or
β-actin (1:1,000) overnight at 4°C and then with the appropriate
HRP-conjugated secondary antibody followed by enhanced
chemiluminescence detection.
Statistical analysis
All data are the means of three determinations. The
data were analyzed using the SPSS package for Windows (Version
11.5). Statistical analysis of the data was performed with the
Student’s t-test and ANOVA. P<0.05 was considered to indicate
statistically significant differences.
Results and Discussion
EELC inhibits the growth of HCC in vivo
and in vitro
The in vivo therapeutic efficacy of EELC
against tumor growth was determined through comparison of tumor
weight and volume in treated and control HCC xenograft mice, while
its adverse effects were evaluated by measuring changes of body
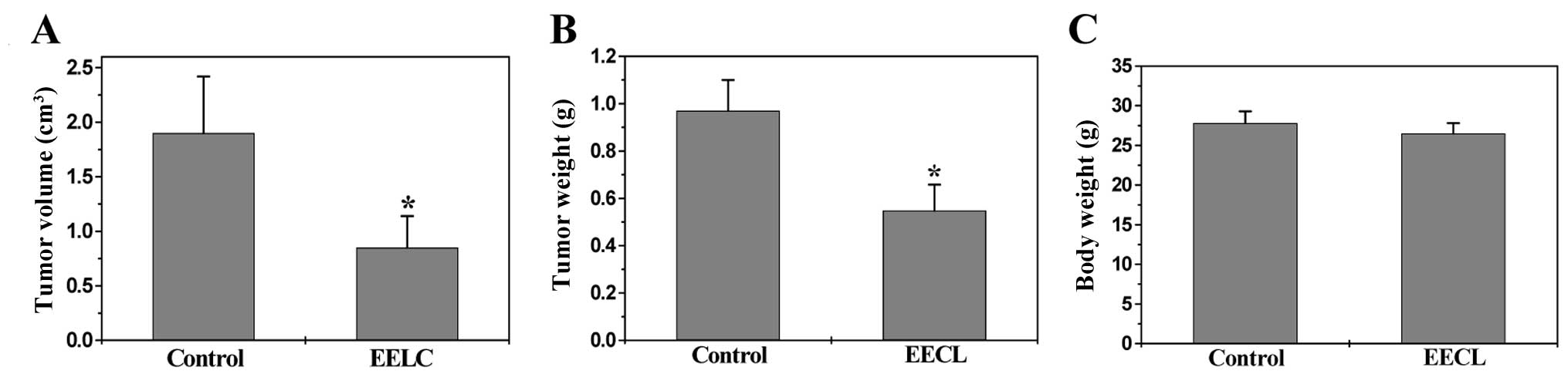
weight. As shown in Fig. 2A, EELC
treatment resulted in a 50% decrease of tumor volume as compared to
control (control, 1.90±0.52 cm3; EELC-treatment,
0.85±0.29 cm3; P<0.01). Accordingly, the tumor weight
per mouse in the EELC-treatment group was 43% less than that in the
control group (control, 0.97±0.13 g; EELC-treatment, 0.55±0.11 g;
P<0.01) (Fig. 2B). However, EELC
treatment had no effect on the changes of body weight (Fig. 2C). Taken together, it is suggested
that EELC is potent in suppressing HCC growth in vivo,
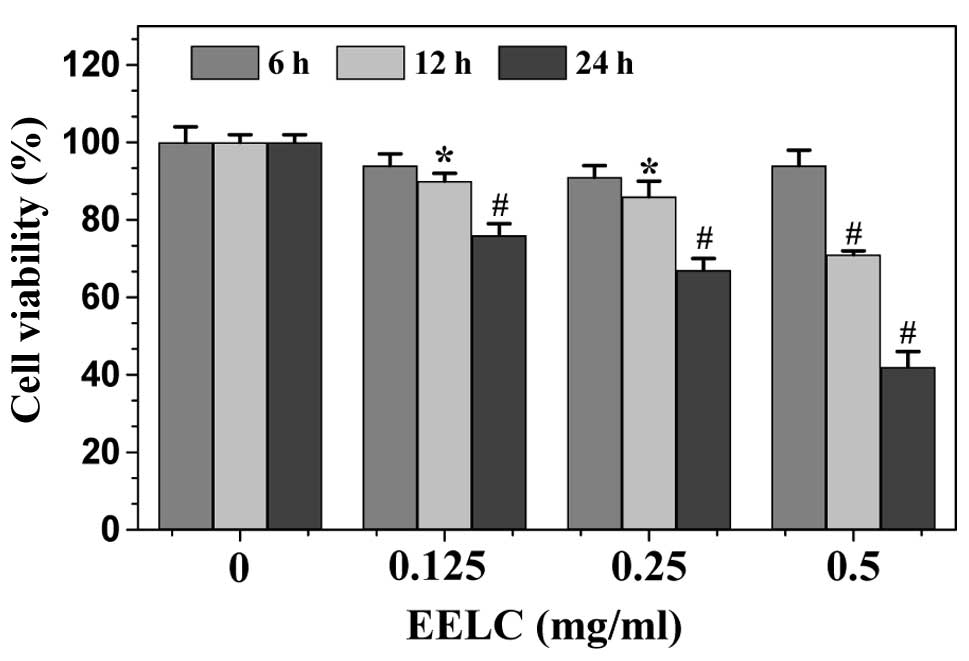
without apparent signs of toxicity. To evaluate the in vitro
antitumor activity of EELC, we performed MTT assay to examine its
effect on the viability of human HCC HepG2 cells. As shown in
Fig. 3, treatment with 0, 0.125,
0.25 and 0.5 mg/ml of EELC for 6, 12 or 24 h, respectively, reduced
cell viability by 6–24, 9–33 or 6–58%, compared to untreated
control cells (P<0.01), suggesting that EELC inhibits HCC cell
growth in vitro in a dose- and a time-dependent manner.
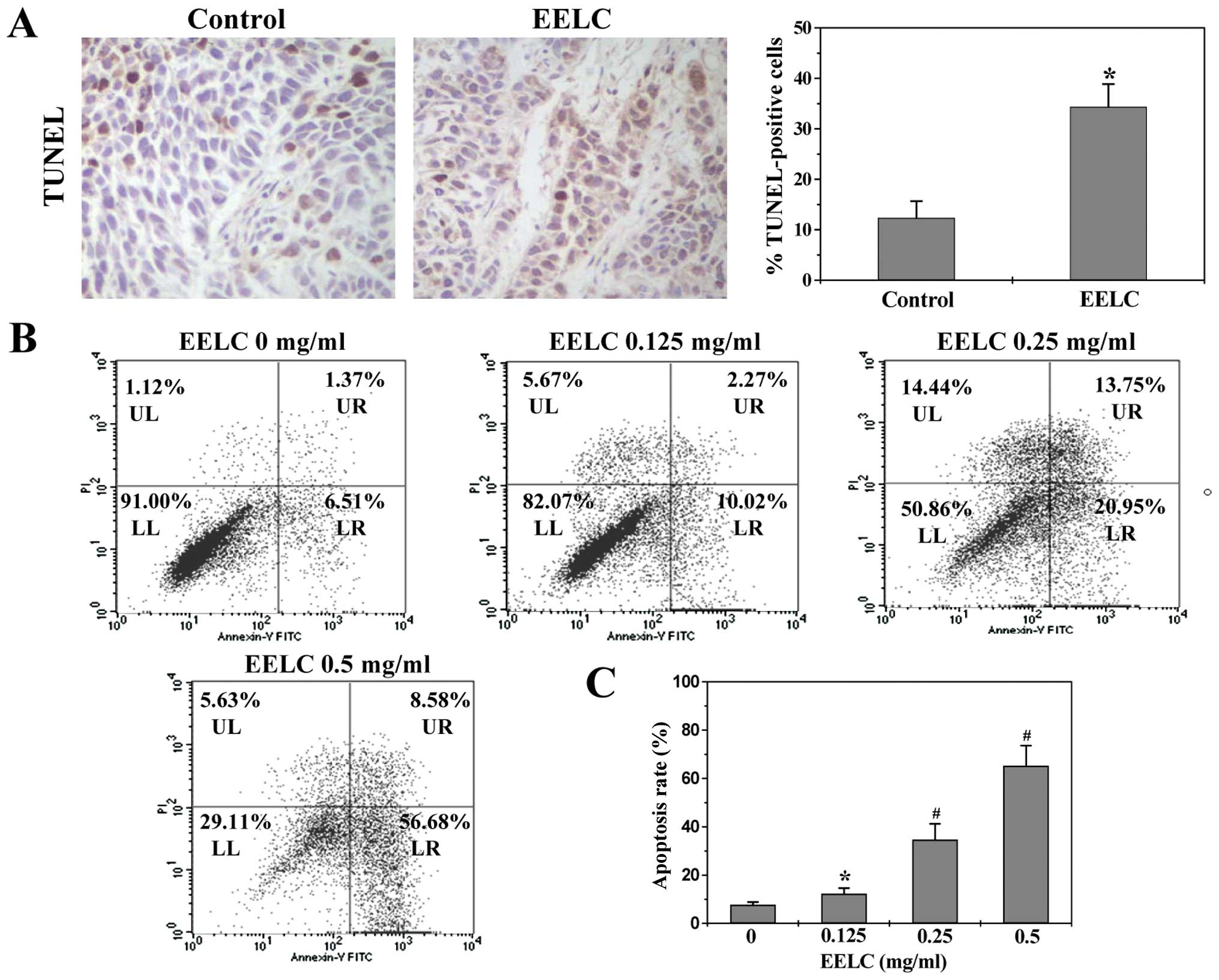
EELC induces apoptosis in HCC xenograft
tumor tissues and HepG2 cells
Cell apoptosis in tumors from HCC xenograft mice was
evaluated via IHS for TUNEL. As shown in Fig. 4A, the percentage of TUNEL-positive
cells was greater in tumors from EELC-treated mice as compared to
controls (EELC-treatment, 34.33±4.52%; control, 12.33±3.32%;
P<0.01). Apoptosis of HepG2 cells was examined using Annexin
V/PI staining followed by FACS analysis. In this assay, Annexin
V/PI double-negative population (labeled as LL in the FACS diagram)
indicates viable cells, whereas Annexin V-positive/PI-negative or
Annexin V/PI double-positive population (labeled as LR or UR in the
FACS diagram) represents cells undergoing early or late apoptosis,
respectively. As shown in Fig. 4B and
C, following treatment with 0, 0.125, 0.25 and 0.5 mg/ml of
EELC, the percentage of cells undergoing apoptosis (including the
early and late apoptotic cells) was 7.8±1.05, 12.3±2.34, 34.7±6.53
and 65.3±8.35%, respectively (P<0.01 or 0.05). These data
demonstrate that EELC promotes HCC cell apoptosis both in
vivo and in vitro.
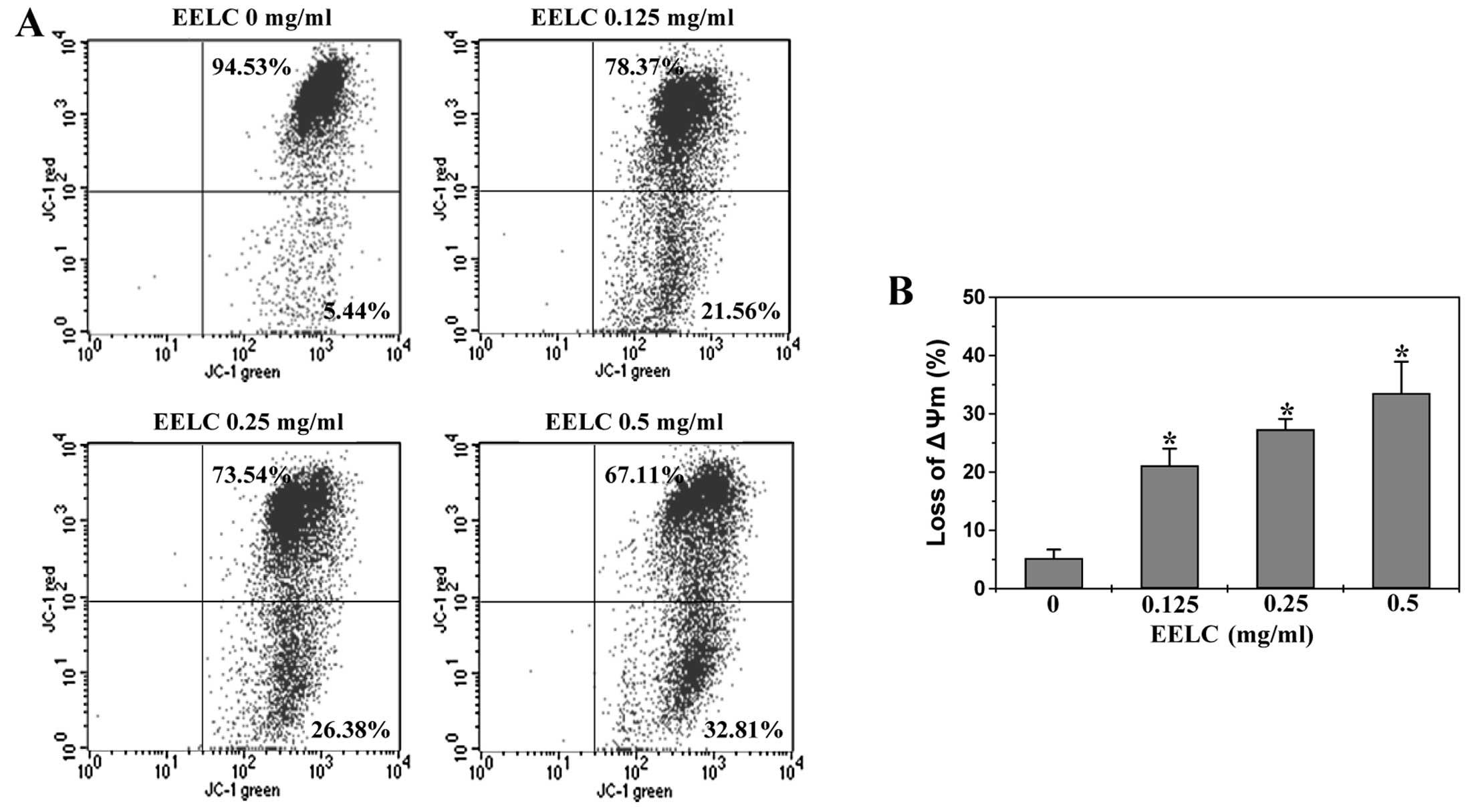
EELC induces the loss of ΔΨm in HepG2
cells
The mitochondrial-dependent pathway is the most
common apoptotic pathway in vertebrate animal cells. Mitochondrial
outer membrane permeabilization (MOMP), accompanied by the collapse
of electrochemical gradient across the mitochondrial membrane, is a
key commitment step in the induction of mitochondrial-dependent
apoptosis, as it is the point of convergence for a large variety of
intracellular apoptotic signaling pathways leading to the release
of several apoptogenic proteins from the mitochondrial
intermembrane space (34,35). To investigate the mechanism of
EELC’s pro-apoptotic activity, we used FACS analysis with JC-1
staining to examine the change in ΔΨm following EELC treatment. The
membrane-permeant JC-1 dye displays potential-dependent
accumulation in mitochondria, indicated by a fluorescence emission
shift from green (~525 nm) to red (~590 nm). Therefore, collapse of
mitochondrial potential during apoptosis could be represented by a
decrease in red fluorescence intensity. As shown in Fig. 5, upon treatment with EELC, the JC-1
fluorescence profile in HepG2 cells shifted from
red-bright/green-bright signal to red-dim/green-bright pattern. The
percentage of cells with reduced JC-1 red fluorescence following
treatment with 0, 0.125, 0.25 and 0.5 mg/ml of EELC was 5.23±1.47,
21.12±2.88, 27.33±1.78 and 33.57±5.73%, respectively (P<0.01),
suggesting that EELC dose-dependently induces the loss of ΔΨm in
HCC cells.
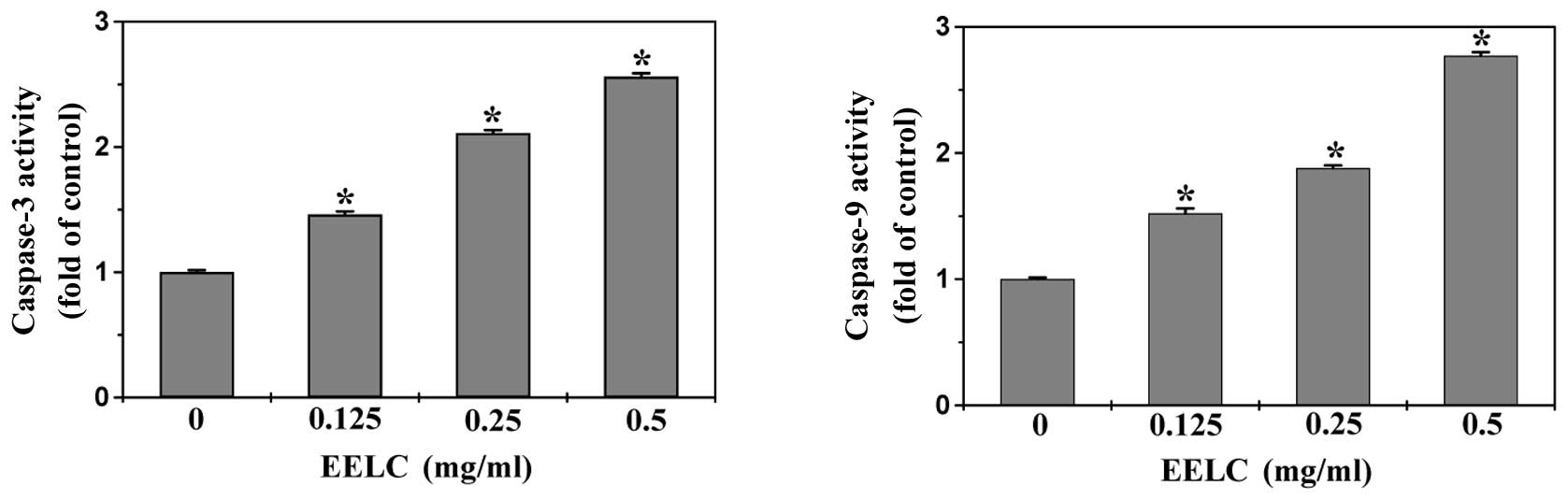
EELC induces the activation of caspase-9
and caspase-3 in HepG2 cells
Caspases, represented by a family of cysteine
proteases, are the key proteins that modulate the apoptotic
response. Caspase-3, a key executioner of apoptosis, is activated
by an initiator caspase such as caspase-9 during
mitochondrial-mediated apoptosis. To identify the downstream
effectors in the apoptotic signaling pathway, the activation of
caspase-9 and caspase-3 was examined by a colorimetric assay using
specific chromophores, DEVD-pNA (specific substrate of caspase-3)
and LEHD-pNA (specific substrate of caspase-9). As shown in
Fig. 6, EELC treatment
significantly and dose-dependently induced activation of both
caspase-9 and caspase-3 in HepG2 cells (P<0.05, vs. untreated
control cells).
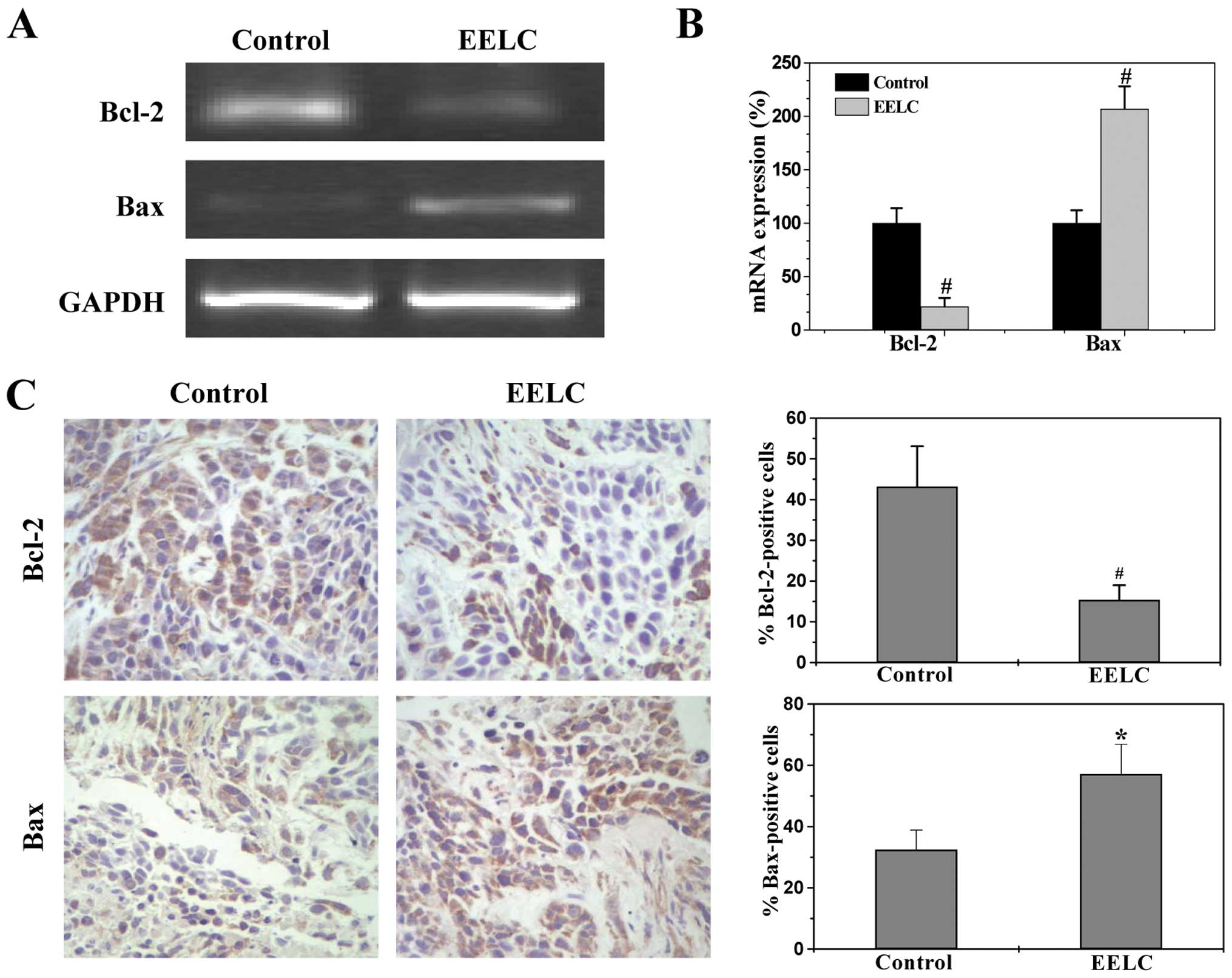
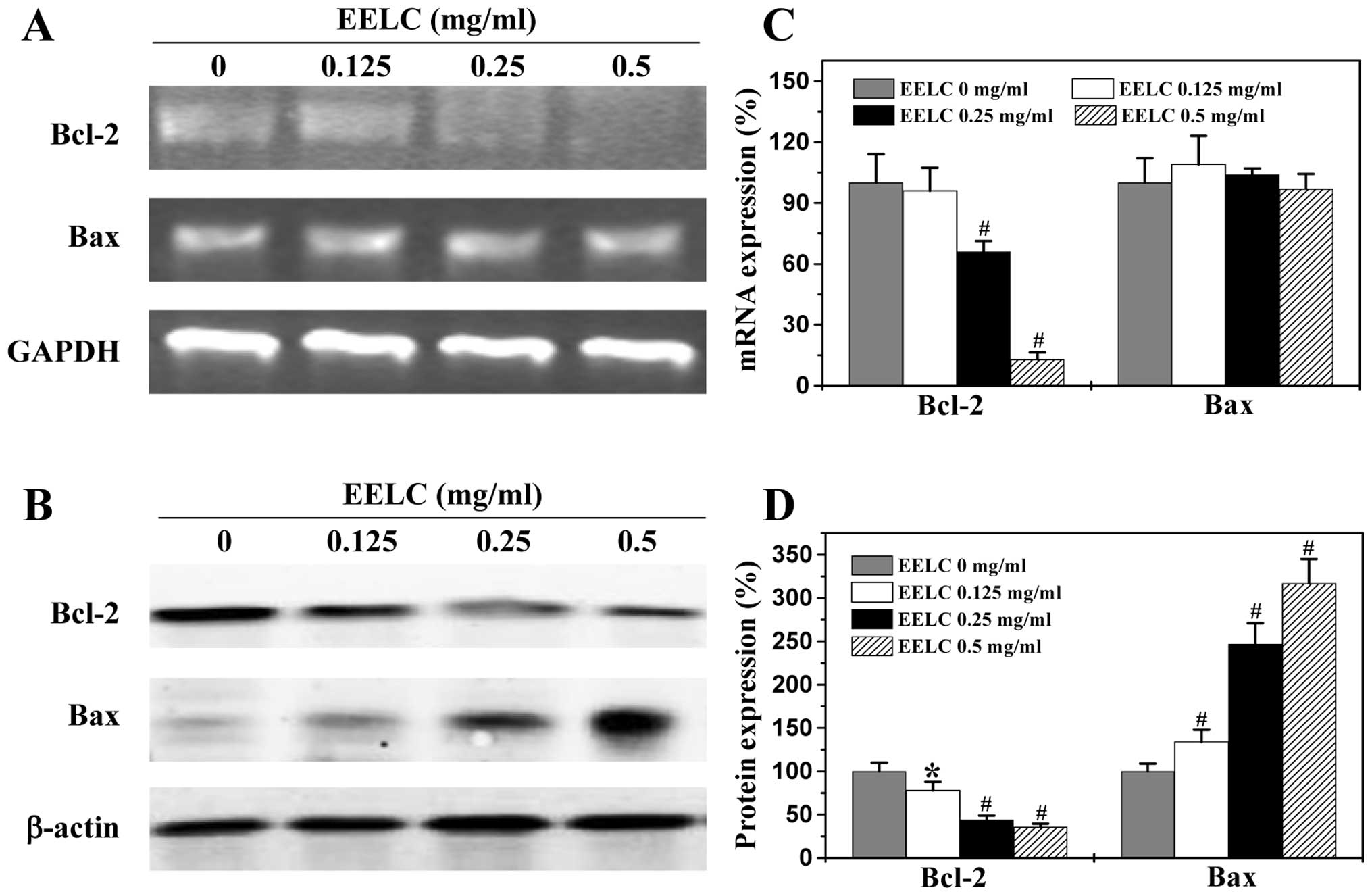
EELC enhances the pro-apoptotic Bax/Bcl-2
ratio in HCC xenograft tumor tissues and HepG2 cells
Bcl-2 family proteins are key regulators of
mitochondrial-mediated apoptosis, including anti-apoptotic members
such as Bcl-2 and pro-apoptotic members such as Bax. MOMP is
considered to occur through the formation of pores in the
mitochondria by pro-apoptotic Bax-like proteins that can be
inhibited by anti-apoptotic Bcl-2-like members. Therefore, the
ratio of active anti- and pro-apoptotic Bcl-2 family members is
critical for determining the fate of cells. Higher Bcl-2 to Bax
ratios are often found in various types of cancer (36), which not only confer a survival
advantage to the cancer cells, but also cause resistance to chemo-
and radio-therapies. To further study the mechanism of EELC’s
pro-apoptotic activity, we performed RT-PCR and IHS or western
blotting to examine the mRNA and protein expression of Bcl-2 and
Bax in the HCC xenograft tumor tissues and the HepG2 cells. As
shown in Figs. 7 and 8, EELC significantly reduced
anti-apoptotic Bcl-2 mRNA levels both in the tumors of HCC mice and
in the HepG2 cells, whereas the level of pro-apoptotic Bax mRNA was
significantly increased following EELC treatment. The protein
expression patterns of Bcl-2 and Bax were similar to the patterns
observed for the respective mRNA. Collectively, these data
demonstrate that EELC promotes mitochondrial-dependent apoptosis of
HCC cells through upregulation in the pro-apoptotic Bax/Bcl-2
ratio.
In conclusion, herein we demonstrated for the first
time that EELC inhibits HCC growth both in vivo and in
vitro by promoting the mitochondrial-dependent apoptosis of
cancer cells. Our findings suggest that the Livistona
chinensis seed may be a potential novel therapeutic agent for
the treatment of HCC and other types of cancer.
Acknowledgements
This study was sponsored by the National Natural
Science Foundation of China (81073097 and 81202790).
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
EELC
|
ethanol extract of the seeds of
Livistona chinensis
|
|
DMSO
|
dimethyl sulfoxide
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
References
|
1
|
Gomaa A, Khan S, Toledano M, Waked I and
Taylor-Robinson S: Hepatocellular carcinoma: epidemiology, risk
factors and pathogenesis. World J Gastroenterol. 14:4300–4308.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Montalto G, Cervello M, Giannitrapani L,
Dantona F, Terranova A and Castagnetta LA: Epidemiology, risk
factors, and natural history of hepatocellular carcinoma. Ann NY
Acad Sci. 963:13–20. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sherman M: Hepatocellular carcinoma:
epidemiology, risk factors, and screening. Semin Liver Dis.
25:143–154. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Parkin D, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
5
|
Yeh C, Chen T, Chang M, Hsu C and Yeh T:
Identification of NV-F virus DNA in hepatocellular carcinoma. J Med
Virol. 79:92–96. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levin B and Amos C: Therapy of
unresectable hepatocellular carcinoma. N Engl J Med. 332:1294–1296.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abou-Alfa G, Huitzil-Melendez F, O’Reilly
E and Saltz L: Current management of advanced hepatocellular
carcinoma. Gastrointest Cancer Res. 2:64–70. 2008.PubMed/NCBI
|
|
8
|
Boose G and Stopper H: Genotoxicity of
several clinically used topoisomerase II inhibitors. Toxicol Lett.
116:7–16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gordaliza M: Natural products as leads to
anticancer drugs. Clin Transl Oncol. 9:767–776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Newman D, Cragg G and Snader K: The
influence of natural products upon drug discovery. Nat Prod Rep.
17:215–234. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adams J and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cory S and Adams J: The Bcl-2 family:
regulators of the cellular life-of-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reed J: Mechanisms of apoptosis. Am J
Pathol. 157:1415–1430. 2000. View Article : Google Scholar
|
|
14
|
Borner C: Bcl-2 family members:
integrators of survival and death. Biochim Biophys Acta.
1644:71–72. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vaux D and Korsmeyer S: Cell death in
development. Cell. 96:245–254. 1999. View Article : Google Scholar
|
|
16
|
Gross A, McDonnell J and Korsmeyer S:
Bcl-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hsu Y, Wolter K and Youle R: Cytosol to
membrane redistribution of members of the Bcl-2 family during
apoptosis. Proc Natl Acad Sci USA. 94:3668–3672. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wolter K, Hsu YT, Smith CL, Nechushtan A,
Xi XG and Youle RJ: Movement of Bax from the cytosol to
mitochondria. J Cell Biol. 139:1281–1292. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan C, Dlugosz PJ, Peng J, Zhang Z,
Lapolla SM, Johnson AE, Andrews DW and Lin J: Auto-activation of
the apoptosis protein Bax increases mitochondrial membrane
permeability and is inhibited by Bcl-2. J Biol Chem.
281:14764–14775. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Antonsson B, Montessuit S, Lauper S, Eskes
R and Martinou JC: Bax oligomerization is required for
channel-forming activity in liposomes and to trigger cytochrome c
release from mitochondria. Biochem J. 345:271–278. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jürgensmeier JM, Xie Z, Deveraux Q,
Ellerby L, Bredesen D and Reed JC: Bax directly induces release of
cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA.
95:4997–5002. 1998.PubMed/NCBI
|
|
22
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: a
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI and Jones DP: Prevention of apoptosis by Bcl-2:
release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thomenius MJ, Wang NS, Reineks EZ, Wang Z
and Distelhorst CW: Bcl-2 on the endoplasmic reticulum regulates
Bax activity by binding to BH3-only proteins. J Biol Chem.
278:6243–6250. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao G, Dai S and Chen R: Dictionary of
traditional Chinese medicine. Shanghai Scientific and Technical
Publishers. 2:2459–2460. 2006.
|
|
28
|
Cheueng S and Tai J: In vitro
studies of the dry fruit of Chinese fan palm Livistona
chinensis. Oncol Rep. 5:1331–1336. 2005.
|
|
29
|
Sartippour MR, Liu C and Shao ZM:
Livistona extract inhibits angiogenesis and cancer growth.
Oncol Rep. 6:1355–1357. 2001.
|
|
30
|
Wen CH, Rae MH and Lang MC: Selective
downregulation of EGF receptor and downstream MAPK pathway in human
cancer cell lines by active components partially purified from the
seeds of Livistona chinensis R. Brown. Cancer Lett.
248:137–146. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang H, Li A, Dong XP and Xu XY: Screening
of anti-tumor parts from the seeds of Livistona chinensisand
its anti-angiogenesis effect. Zhong Yao Cai. 315:718–722. 2008.(In
Chinese).
|
|
32
|
Cai Q, Lin J, Wei L, Zhang L, Wang L, Zhan
Y, Zeng J, Xu W, Shen A, Hong Z and Peng J: Hedyotis diffusa
willd inhibits colorectal cancer growth in vivo via inhibition of
STAT3 signaling pathway. Int J Mol Sci. 13:6117–6128. 2012.
View Article : Google Scholar
|
|
33
|
Wei L, Lin J, Xu W, Cai Q, Shen A, Hong Z
and Peng J: Scutellaria barbata D. don inhibits tumor
angiogenesis via suppression of hedgehog pathway in a mouse model
of colorectal cancer. Int J Mol Sci. 13:9419–9430. 2012. View Article : Google Scholar
|
|
34
|
Mantymaa P, Siitonen T, Guttorm T, Saily
M, Kinnula V, Savolainen ER and Koistinen P: Induction of
mitochondrial manganese superoxide dismutase confers resistance to
apoptosis in acute myeloblastic leukaemia cells exposed to
etoposide. Br J Haematol. 108:574–581. 2000. View Article : Google Scholar
|
|
35
|
Korper S, Nolte F, Rojewski MT, Thiel E
and Schrezenmeier H: The K+ channel openers diazoxide
and NS1619 induce depolarization of mitochondria and have
differential effects on cell Ca2+ in CD34+
cell line KG-1a. Exp Hematol. 31:815–823. 2003.
|
|
36
|
Kitada S, Pedersen IM, Schimmer AD and
Reed JC: Dysregulation of apoptosis genes in hematopoietic
malignancies. Oncogene. 21:3459–3474. 2002. View Article : Google Scholar : PubMed/NCBI
|