Introduction
DNA topoisomerase II inhibitors, such as
daunorubicin (DNR), are potent antineoplastic agents widely used in
the treatment of patients with leukemia, lymphoma and diverse solid
tumor malignancies. They exert their function by introducing the
formation of a stable complex between enzyme DNA topoisomerase II
and DNA as well as by generating reactive oxygen species (ROS)
(1). The introduction of
double-strand DNA breaks and alterations in the replication and
transcription trigger apoptosis and cell death (2). However, the development of drug
resistance in leukemia cells poses a major obstacle in the
successful treatment of acute leukemia. It has been reported that
overexpression of constitutively active Raf-1 and P-gp could form
drug resistance to DNR, and the expression of the P-gp extrusion
pump was regulated by the Ras/Raf/MEK/ERK pathway (3–5).
DNR can activate the Raf/MEK/ERK pathway via the
protein kinase Cζ (PKCζ) pathway and combine with MEK inhibitor to
enhance the antitumor activity (6).
The constitutive activation of certain cellular signaling pathways
contributes to tumor development and resistance to chemotherapy
(7,8). Kornblau et al confirmed that
simultaneous activation of multiple signal transduction pathways
conferred poor prognosis in acute myelogenous leukemia (9). Upon growth factor stimulation,
Ras/Raf/MEK/ERK triggers a series of cascade events that govern
cell transformation, differentiation, proliferation and survival
(10–12). Approximately 30% of tumors harbor
active mutants of the Ras oncoprotein, and a downstream effector of
Ras, Raf, is also frequently mutated in tumors (13,14).
The active Ras/Raf/MEK/ERK kinase pathway inevitably accounts for a
hallmark of malignant phenotypes: abnormal cell growth, invasion
and angiogenesis (7). Therefore,
interruption of the Raf/MEK/ERK cascade has become a primary target
in acute leukemia therapy.
Sorafenib, an oral small molecular multi-kinase
inhibitor, has been approved by the Food and Drug Administration
(FDA) for the treatment of patients with renal cell carcinoma and
hepatoma (15,16). Sorafenib was initially identified as
a RAF kinase inhibitor, and was found to be able to inhibit the
MAPK pathway in multiple cancer cell lines. In human xenograft
models, including ovarian, colon, lung, pancreatic and melanoma,
sorafenib inhibited tumor growth through inhibition of either the
MAPK signaling or angiogenesis (17,18).
In order to reduce the p-ERK1/2 level induced by DNR
and extend the anti-leukemia activity of DNR, we combined sorafenib
with DNR, and explored the potent cellular and molecular effects in
K562 and U937 cell lines. Our present study suggested that DNR in
combination with sorafenib may represent a new and immediately
available therapeutic approach to treat acute leukemia with
activated ERK1/2.
Materials and methods
Reagents and antibodies
Sorafenib (Bayer, Pittsburgh, PA, USA) was dissolved
in dimethyl sulfoxide (DMSO) and stored at −20°C. DNR was purchased
from Sigma-Aldrich (St. Louis, MO, USA). U0126 (Alexis, San Diego,
CA, USA), a MEK1/2 inhibitor, was dissolved in DMSO with a final
concentration of 20 μM. The antibodies of rabbit anti-ERK1/2,
anti-phospho-ERK1/2 (Tyr202/Tyr204) were
obtained from Cell Signaling Technologies (Beverly, MA, USA).
DMRIE-C was purchased from Invitrogen (Carlsbad, CA, USA).
Cell culture
The leukemia cell lines K562 and U937, of myeloid
origin, were obtained from the American Type Culture Collection
(ATCC) and grown in RPMI-1640 medium supplemented with 10% fetal
bovine serum, 100 μg/ml penicillin and 100 μg/ml streptomycin.
Cells were cultured at 37°C in a humidified atmosphere containing
5% CO2. Control cultures received an equivalent amount
of DMSO only. Cells in logarithmic growth phase were used for
further experiments.
Cell viability assay
Cell viability was assessed by MTT assay. Briefly,
cells at 1×105/ml were treated with various
concentrations of DNR in 96-well plates for 24 h at 37°C. Then, MTT
working solution (5 mg/ml in PBS) was added to each well and cells
were incubated for 4 h. The water-insoluble formazan was formed
during incubation and was solubilized by adding DMSO to each well.
The amount of formazan was determined by measuring the absorbance
at 490 nm using a multiwell plate reader (Microplate Reader;
Bio-Rad, Hercules, CA, USA). Percent cell viability was calculated
as cell viability of the experimental samples/cell viability of the
control samples ×100. At least three independent experiments were
performed.
Assessment of apoptosis
Apoptotic cells were evaluated by Annexin V/PI
staining, and, in some cases, they were verified by Hoechst 33258
and Wright Giemsa staining according to the protocol. For flow
cytometry analysis, cells were treated with various concentrations
of DNR, sorafenib alone or in combination for 48 h. The cells were
harvested for Annexin V-Alexa Fluor-488/PI staining. The stained
cells were analyzed by a Becton Dickinson FACScan flow
cytometer.
Generation of transfection cell
lines
MEK2DD, pBABE-puro MEK2DD plasmid were gifts from
the Liuq’s laboratory (Sun Yat-sen Institute of Hematology,
Guangzhou, China) and pEGFP-N3 vector was purchased from BD
Biosciences Clontech (Bedford, MA, USA). The full-length MEK2DD
cDNA was subcloned into vector pEGFP-N3.
For stable transfection, pEGFP-N3 vector was
introduced into K562 cells using DMRIE-C reagent, according to the
manufacturer’s instructions. Two days later, the cells were
screened by G418 at a concentration of 1,000 μg/ml, and five days
later using G418 at a concentration of 500 μg/ml for continuous
screening. For transient transfection, MEK2 constitutive active
form, MEK2DD plasmid was introduced into K562 cells directly by
DMRIE-C, and 72 h later, the cells were exposed to different
concentrations of DNR.
Western blot analysis
K562 cells were treated with various concentrations
of sorafenib or DMSO (control) for 6, 12 and 24 h. Thereafter, the
cells were collected and lysed in a lysis buffer (1% Triton X-100;
50 mM HEPES, pH 7.4; 150 mM NaCl; 1 mM EGTA; 100 mM NaF; 10 mM
sodium pyrophosphate; 1 mM Na3VO4; 10%
glycerol; 1 mM PMSF; 10 μg/ml leupeptin). The levels of
phosphorylated and total ERK1/2 in cell lysates were determined by
immunoblotting with the corresponding antibodies. Briefly, cell
lysates (50 μg protein per well) were resolved by eletrophoresis on
10–12% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE;
Bio-Rad) and transferred to PVDF membranes. The membranes were
first incubated in TBST (50 mM Tris-HCl; 150 mM NaCl; 0.05%
Tween-20) containing 5% non-fat dry milk for 1 h to block
nonspecific protein binding; they were then washed with TBST three
times, and incubated with primary antibody (1:1000 dilution)
overnight at 4°C. The next day, the PVDF membranes were washed with
TBST three times, and incubated with HRP-conjugated secondary
antibody (1:2500 dilution) for 1 h at room temperature. The
membranes were then washed with TBST, and antibody binding was
visualized with the use of a super chemiluminescence detection
system.
Statistical analysis
Data are presented as means ± SD. One-way ANOVA
followed by Bonferroni multiple comparison was performed to assess
the differences between two groups under multiple conditions. If
the data failed the normality test, the Kruskal-Wallis one-way
ANOVA on ranks was used. P<0.05 was considered to indicate
statistically significant differences. The interaction between DNR
and sorafenib or U0126 was analyzed according to Jin’s formula,
determining whether the combination was additive or synergistic.
Jin’s formula was performed based on the following equation:
q=D1+2/(D1+D2−D1xD2),
where D1+2 indicated the effect when cells were used in
combination with drug 1 and 2, and D1, D2
indicated the effect when used alone. The value of q indicated
synergism when >1.15, antagonism when <0.85, and additivity
when located between 0.85 and 1.15.
Results
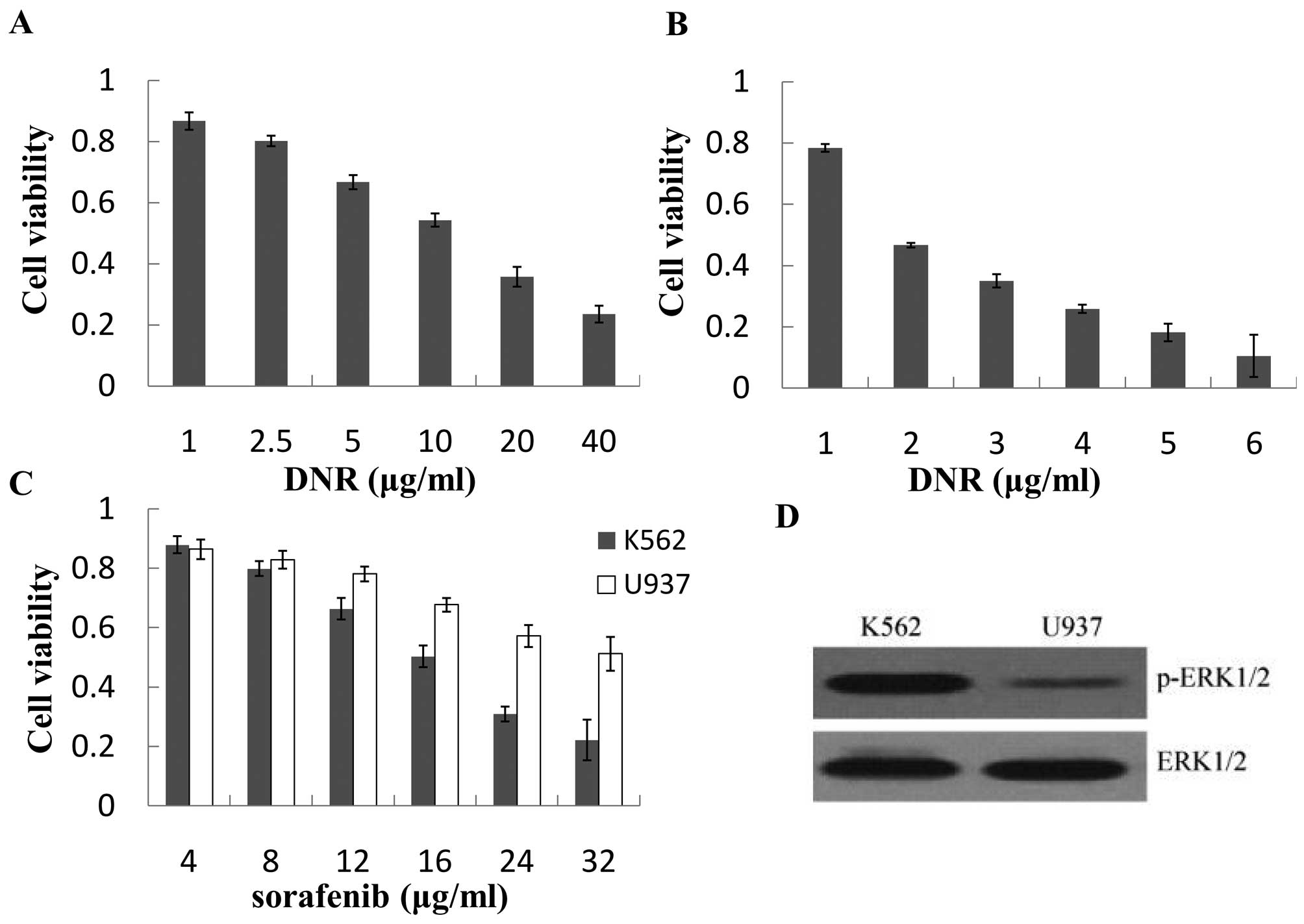
K562 cells are less sensitive to DNR than
U937 cells with high p-ERK1/2 levels
K562 and U937 cells were both exposed to DNR for 24
h. The cytotoxic effects of DNR were determined by MTT assay. As
shown in Fig. 1A and B, DNR
inhibited cell proliferation in a dose-dependent manner. The
IC10 values of DNR for K562 and U937 cells were
0.82±0.12 μg/ml and 0.055±0.011 μg/ml, respectively. The
antiproliferative effects of DNR on K562 were stronger than those
on U937 cells. However, K562 cells were more sensitive to sorafenib
than U937 cells (Fig. 1C). To
investigate the reason why K562 and U937 cells had different
reactions to DNR or sorafenib, the p-ERK1/2 levels were evaluated
by western blot assay. We found that the p-ERK1/2 level of K562
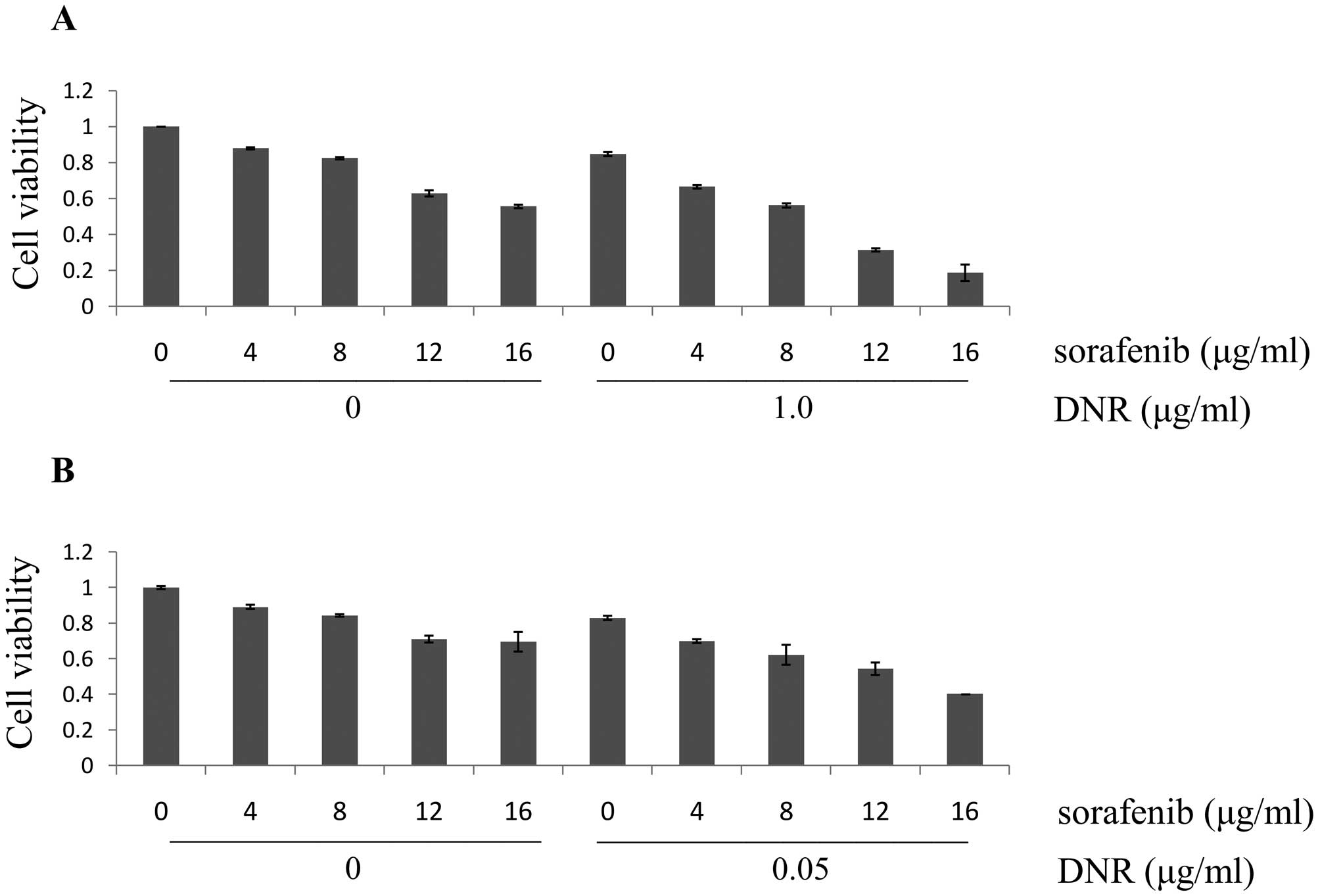
cells was higher than that of U937 cells (Fig. 1D). In addition, cell lines were
treated with combinations of these two agents at different doses
but in a constant ratio (sorafenib to DNR: 4.0–16.0 μg/ml to 1.0
μg/ml in K562 cells, 4.0–16.0 μg/ml to 0.05 μg/ml in U937 cells,
respectively). As shown in Fig. 2,
the combination of sorafenib with DNR inhibited the growth of K562
and U937 cells in a dose-dependent manner. Moreover, Jin’s formula
was used to determine synergy. At each dose level, the combined
effects resulted in synergism (q >1.15). This indicates that the
effects of the combination on cell viability are likely
synergistic.
Sorafenib synergistically enhances the
apoptotic effect of DNR on K562 cells
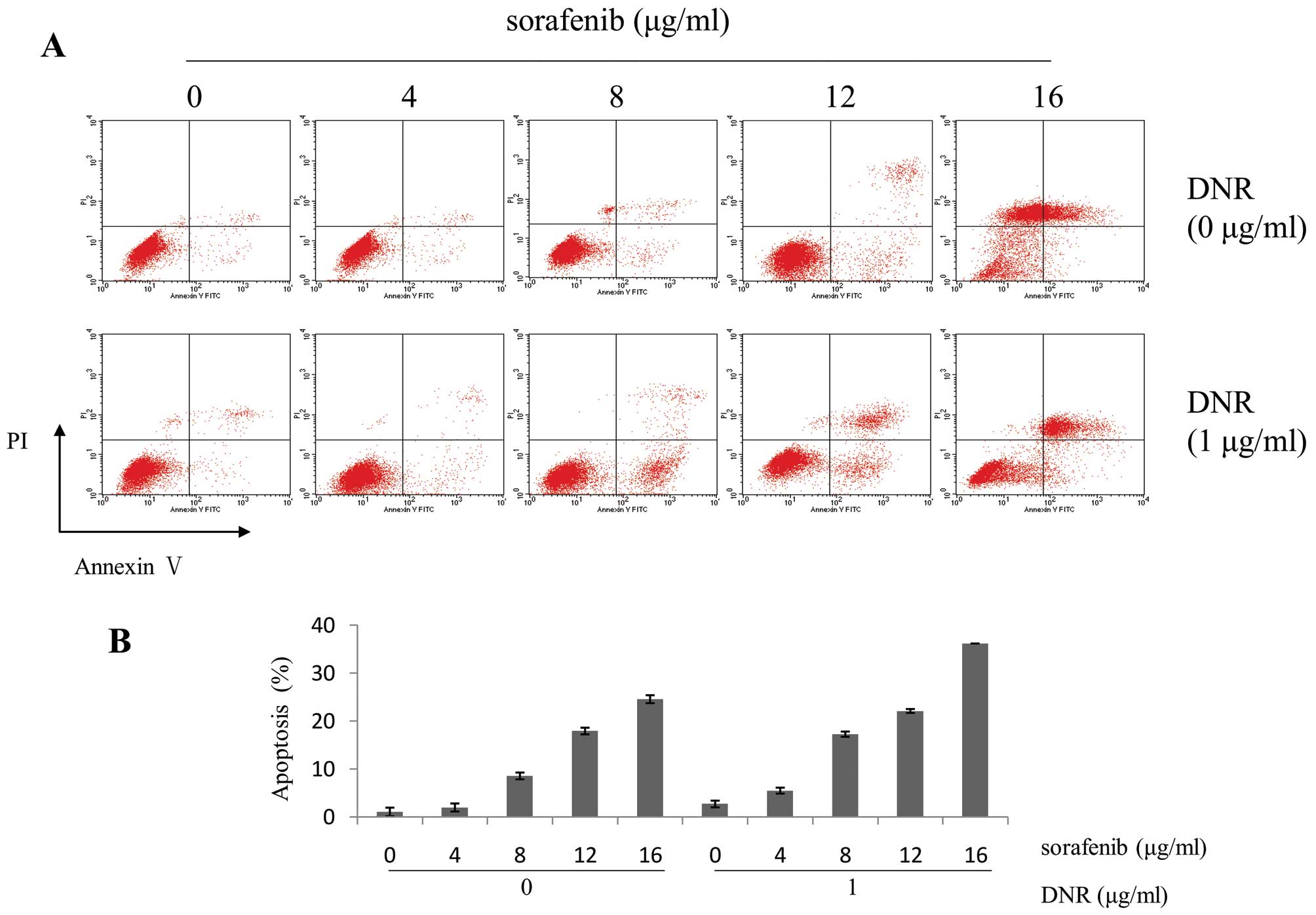
To assess whether combined treatment enhanced
apoptosis, apoptotic analysis was conducted by Annexin V/PI,
Wright-Giemsa and Hoechst 33258 staining on K562 cells treated with
sorafenib, either alone or together with DNR for 48 h. Flow
cytometry showed that the combination of 16 μg/ml sorafenib with 1
μg/ml DNR in K562 cells resulted in apoptosis rates of 36.2%,
compared to sorafenib (24.55%) or DNR (2.67%) alone (Fig. 3). According to Jin’s method,
sorafenib in combination with DNR had synergistic effects of
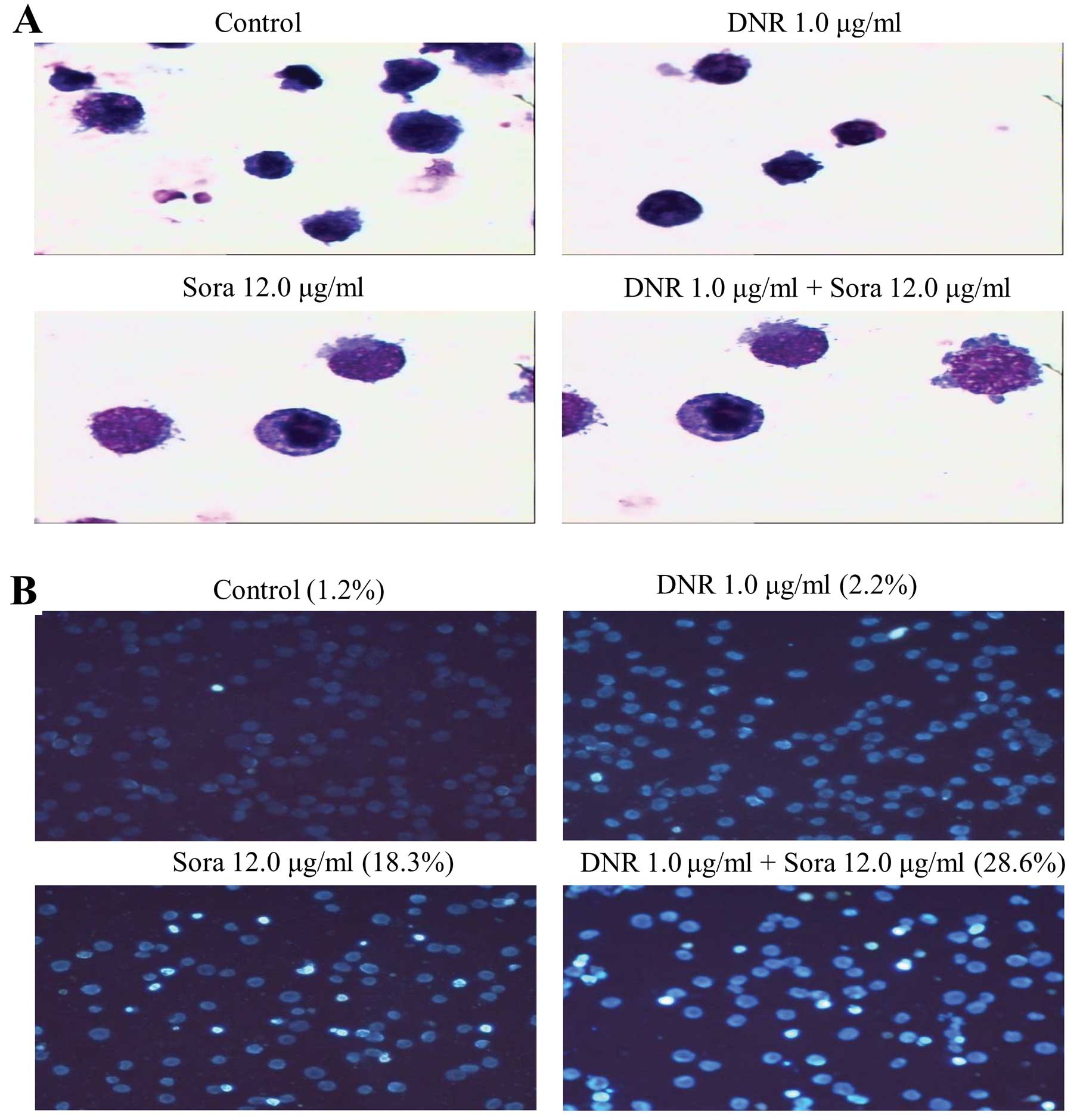
apoptosis on K562 cells (q>1.15, p<0.01). The morphological
changes demonstrated by Wright-Giemsa and Hoechst 33258 staining
also confirmed this conclusion. An apparent increase in the
percentage of cells with typical chromatin condensation and
fragmentation of nuclei was observed in cell cultures treated with
the combination of sorafenib and DNR compared to either agent alone
(Fig. 4A). As shown in Fig. 4B, marked morphological changes of
cell apoptosis, such as cell shrinkage and nuclear condensation,
were observed. These results suggest that sorafenib sensitizes K562
cells to DNR-induced apoptosis.
Effects of sorafenib and DNR on ERK1/2
phosphorylation and activity
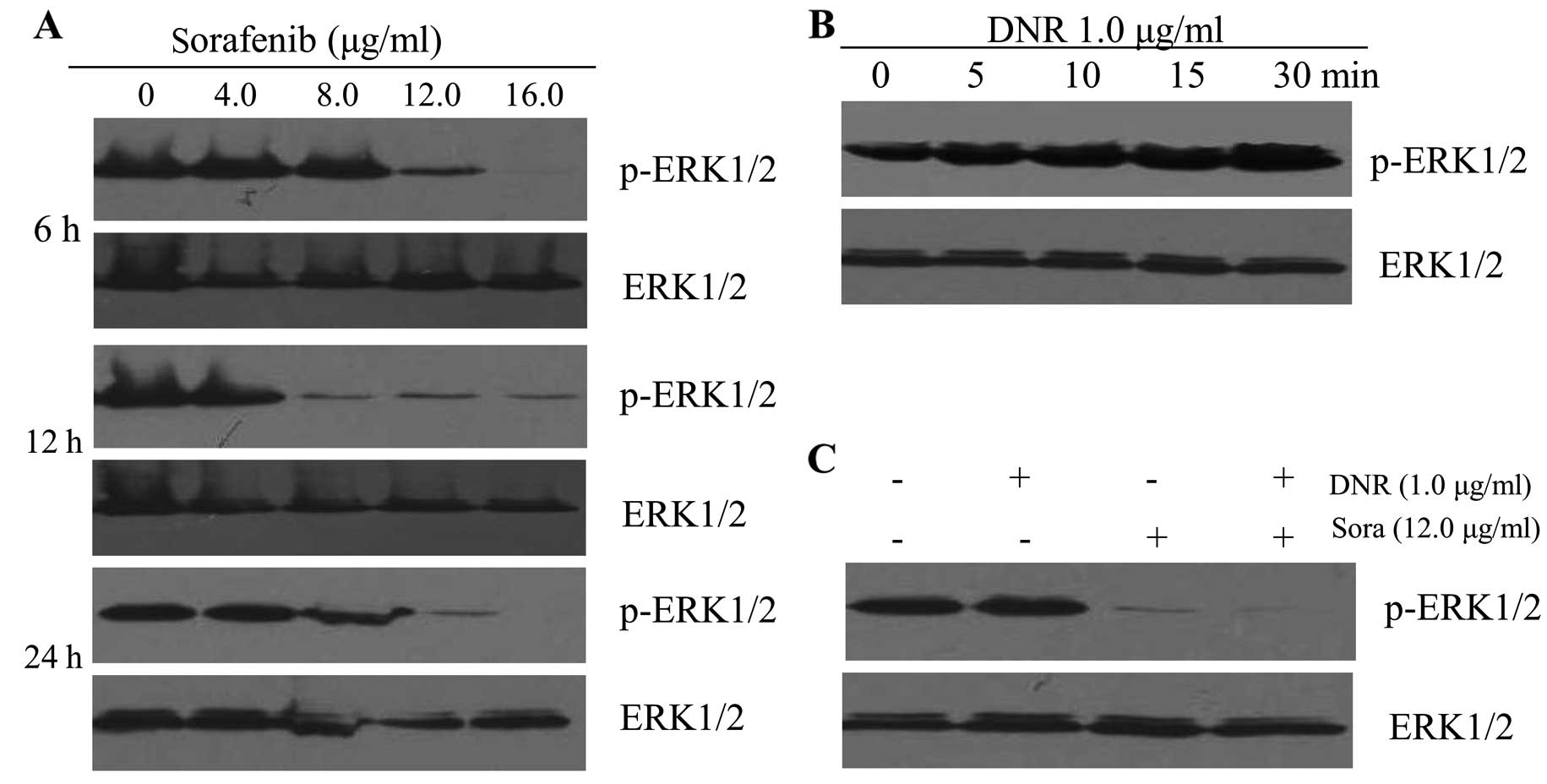
We found that sorafenib, a type of oral multi-kinase
inhibitor which was recently approved by the FDA, suppressed
p-ERK1/2 expression in a dose- and time-dependent manner on K562
cells. Sorafenib (12 μg/ml) downregulated the p-ERK1/2 level as
early as 6 h. After 24 h, p-ERK1/2 expression was completely
inhibited by sorafenib at 16 μg/ml (Fig. 5A). We then used antibody directed
against phosphorylated forms of both ERK1 and ERK2, and found that
incubation of K562 cells with 1.0 μg/ml DNR induced ERK1/2
activation, and ERK1/2 tyrosine phosphorylation was observed within
5 min and remained relatively stable for at least 30 min (Fig. 5B). The phosphorylation of ERK1/2
following treatment with DNR increased in a time-dependent manner.
As shown in Fig. 5C, co-treatment
with 12 μg/ml sorafenib and 1.0 μg/ml DNR caused the same
attenuation of p-ERK1/2 protein levels as treatment with sorafenib
alone.
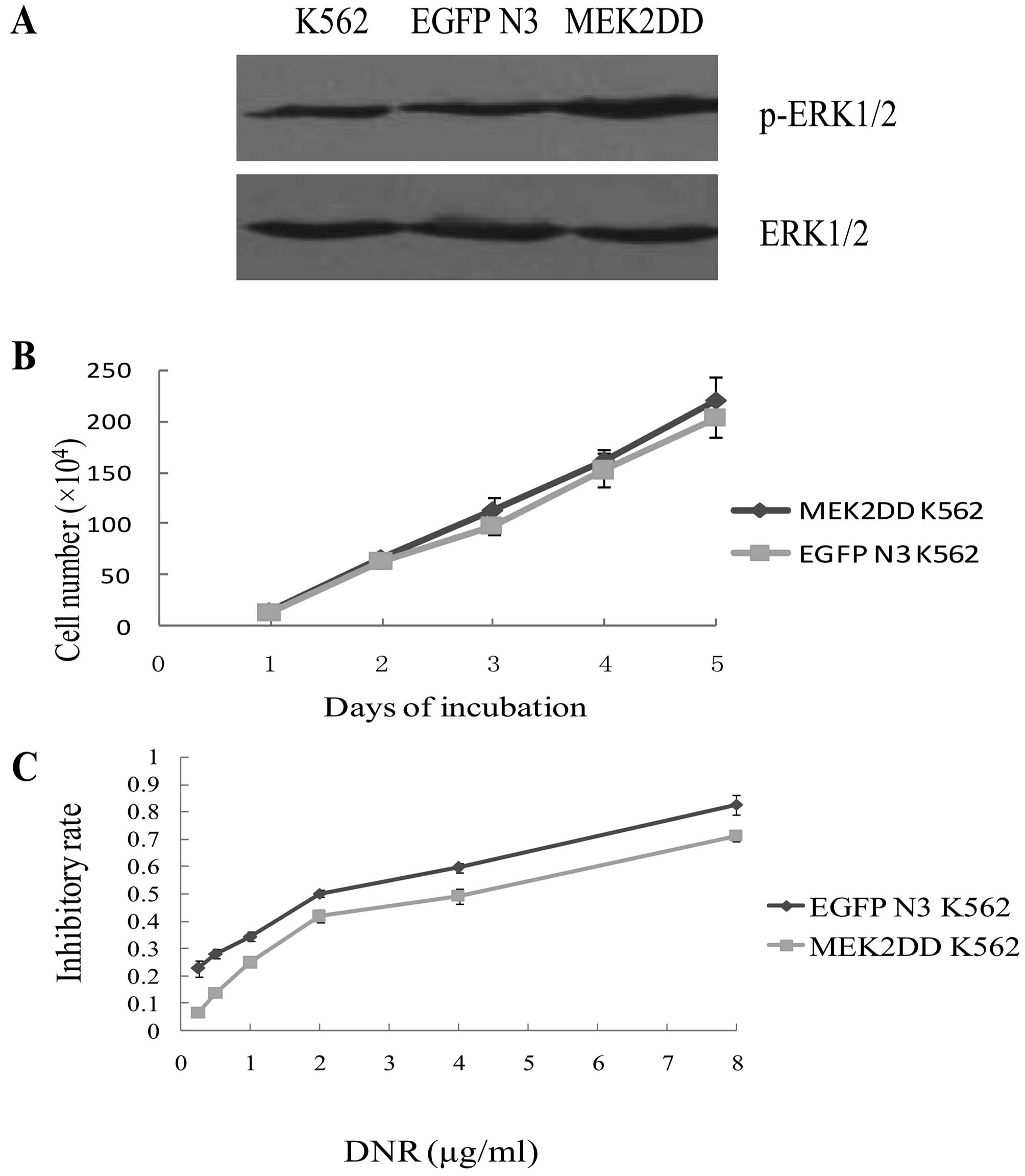
Upregulation of p-ERK1/2 levels in K562
cells attenuates the cytotoxic effect of DNR
To determine whether the observed upregulation of
p-ERK1/2 could reduce the cytotoxic effect of DNR on K562 cells, we
transfected K562 cells with empty EGFP-N3 control or an active
MEK2DD vector. The p-ERK1/2 level after K562 cells transfected with
MEK2DD vector was higher than control cells, as shown in Fig. 6A. However, there was no significant
difference in the growth rate between these two types of
transfected cells (p>0.05) (Fig.
6B). Subsequently, K562 cells expressing empty vector or a
MEK2DD vector encoding construct were exposed to increasing
concentrations of DNR for 48 h. MTT assay showed that
IC50 (3.33±0.2 μg/ml) of DNR on MEK2DD K562 was higher
than that on EGFP-K562 (1.71±0.14 μg/ml) (p<0.01).
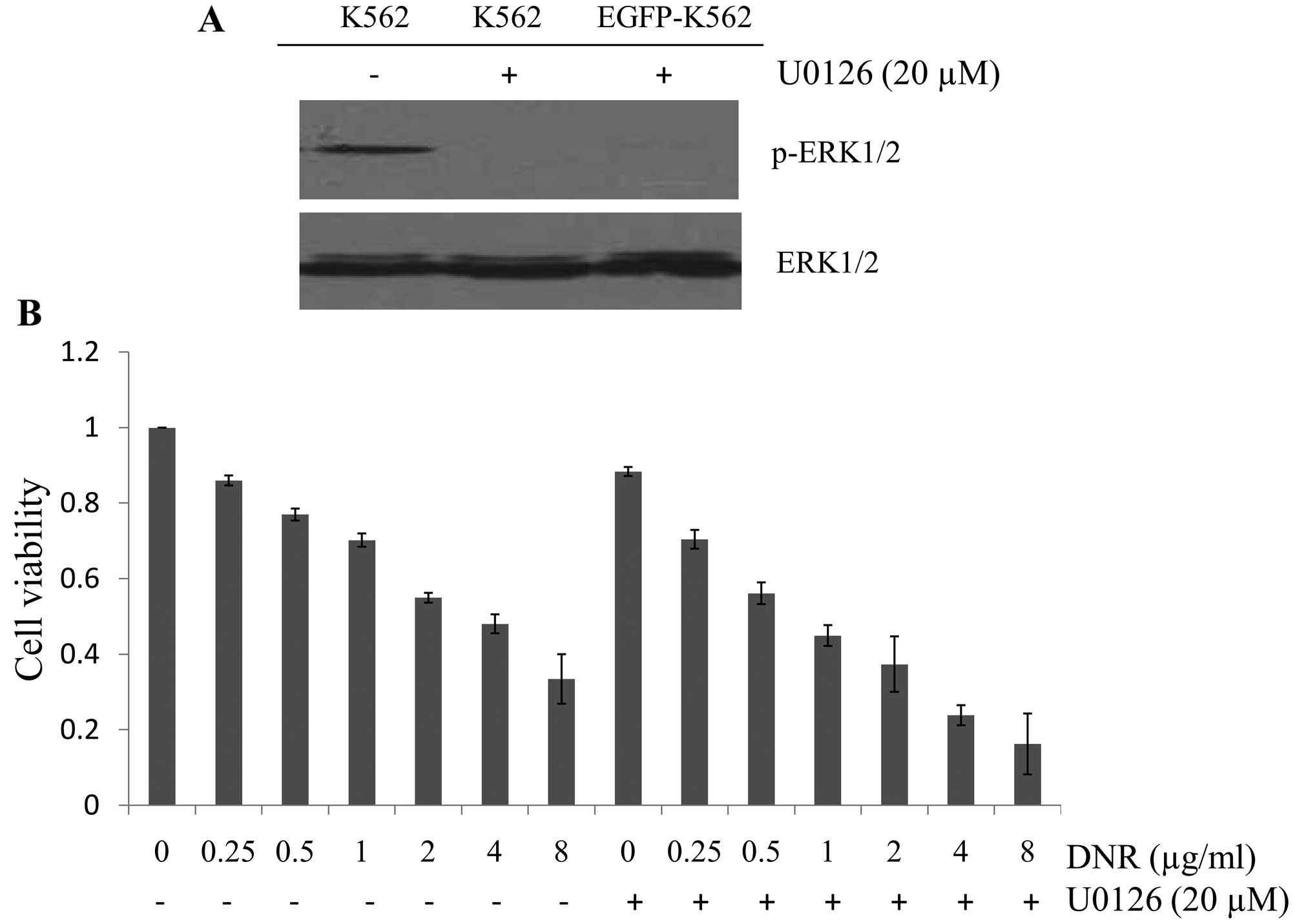
MEK1/2 inhibitor U0126 sensitizes K562
cells to DNR
To evaluate whether blocking of the RAF/MEK/ERK
pathway could increase the efficacy of DNR to inhibit leukemia
cells, U0126 (MEK1/2 inhibitor) in combination with DNR was used.
p-ERK1/2 expression was completely downregulated after incubation
with 20 μM U0126 for 24 h in K562 and EGFP-K562 cells (Fig. 7A). Subsequently, K562 cells were
exposed to DNR (0.25–8.0 μg/ml) in combination with 20 μM U0126
simultaneously for 48 h. As expected, the inhibitory rate of the
combination was higher than any concentration of DNR alone. Jin’s
formula was used to determine synergy, and the results were
consistent. The value q of each drug concentration was >1.15
(p<0.01), thereby suggesting that downregulation of the p-ERK1/2
level could enhance sensitivity to DNR in K562 cells.
Discussion
In the present study, we demonstrated that sorafenib
effectively downregulated p-ERK1/2 levels and that this activity
directly contributed to the sensitization of K562 cells to DNR. DNR
represents one of the major antitumor agents widely used in the
treatment of acute myeloid leukemia. Cytotoxicity induced by DNR is
related to DNA damage due to intercalations of the drug and its
interaction with nuclear topoisomerase II (19). DNR has been reported to induce
apoptosis in myeloid leukemia cell lines (20). However, whether apoptosis simply
reflects DNA lesions or represents an independent cytotoxic
mechanism triggered by a specific signaling pathway remains
controversial (21).
We found that incubation of K562 cells with 1.0
μg/ml DNR induced ERK1/2 activation, and p44/p42 ERK1/2 tyrosine
phosphorylation was observed within 5 min and remained relatively
stable for at least 30 min. Mas et al confirmed that the
activation of ERK1/2 by DNR was relevant to PKC (6). Han et al first reported that
DNR induced cytoprotective autophagy by activation of ERK in
myeloid leukemia cells (22).
ERK1/2 is a member of the mitogen-activated protein kinase (MAPK)
family. The classical MAPK module consists of a Raf-1 kinase,
mitogen-induced extracellular kinase (MEK), and extracellular
regulated kinase (ERK). The majority of the water-soluble growth
factors, which regulate the growth, proliferation and
differentiation of normal and transformed cells, exert at least
part of their effects by signaling through the highly conserved
Ras/Raf/MEK/ERK pathway. A cascade of kinase activation occurs
after the cognate receptor ligated. Therefore, the Raf/MEK/ERK
pathway can govern drug resistance, apoptosis and sensitivity to
target therapy (23,24). Some studies have suggested that this
signal transduction pathway may be involved in the regulation of
several aspects of drug resistance. For instance, expression of the
P-gp extrusion pump is regulated by the Ras/Raf/MEK/ERK pathway
(5).
Sorafenib is an orally active multi-kinase inhibitor
which was originally developed as an inhibitor of Raf-1. However,
it has been subsequently shown to inhibit multiple other kinases,
including platelet-derived growth factor β, vascular endothelial
growth factor receptor 2 and 3, and FMS-like tyrosine kinase 3
(FLT3) (17). It has been used in
combination with several anticancer drugs and has shown marked
antitumor activity (25,26). In order to improve the antitumor
effect and slow down the formation of drug resistance, we combined
sorafenib with DNR. Consequently, the combination of sorafenib with
DNR resulted in the inhibition of proliferation and induction of
apoptosis in a dose-dependent manner on K562 cells. The combination
effect could be synergistic according to Jin’s formula. The value q
of each drug concentration was more than 1.15 (p<0.01).
Apoptotic effects were measured by flow cytometry, Hoechst 33258
and Wright Giemsa staining, and all results were consistent. The
present study demonstrated that sorafenib suppressed p-ERK1/2
expression in a dose- and time-dependent manner in K562 cells.
Furthermore, sorafenib alone or in combination with DNR suppressed
the p-ERK1/2 expression with no difference.
To investigate whether upregulating the p-ERK1/2
level may reduce the cytotoxity of DNR, K562 cells which
transfected with empty vector or a MEK2DD vector encoding construct
were exposed to increasing concentrations of DNR for 48 h. We found
that K562 cells transfected with a constitutively active MEK2DD
plasmid showed increasing IC50 values following DNR
treatment compared with control cells. These results confirmed that
high p-ERK1/2 levels could reduce the antitumor effect of DNR on
K562 cells. Conversely, suppression of the p-ERK1/2 expression with
U0126 enhanced DNR-induced apoptosis in K562 cells.
Raf-1/ERK activation contributes to cell survival
when drug concentrations are reduced due to low-dose schedules,
pharmacokinetic alterations, or altered bio-distribution. In this
regard, we speculated that constitutive MAPK activation, as
evidenced in fresh leukemia cells, may significantly alter DNR
cytotoxicity in clinical settings (27,28).
For this reason, co-treatment of sorafenib and DNR in patients with
leukemia is very efficient and effective. Thus, the combination
mechanism between sorafenib and DNR should be further studied in
primary leukemia specimens and xenograft models.
In summary, this study indicated that the
combination of DNR and sorafenib contributed to a synergistic
anti-leukemia activity in vitro and ERK1/2 could be a
potential target for pharmacologic manipulation to improve
DNR-induced cytotoxity on leukemia cells.
References
|
1
|
Thornalley PJ and Dodd NJ: Free radical
production from normal and adriamycin-treated rat cardiac
sarcosomes. Biochem Pharmacol. 34:669–674. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pommier Y: DNA topoisomerase I and II in
cancer chemotherapy: update and perspectives. Cancer Chemother
Pharmacol. 32:103–108. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weinstein-Oppenheimer CR, Henríquez-Roldán
CF, Davis JM, et al: Role of the Raf signal transduction cascade in
the in vitro resistance to the anticancer drug doxorubicin. Clin
Cancer Res. 7:2898–2907. 2001.PubMed/NCBI
|
|
4
|
Kim SH, Lee SH, Kwak NH, Kang CD and Chung
BS: Effects of the activated Raf protein kinase on the human
multidrug resistance 1 (MDR1) gene promoter. Cancer Lett.
98:199–205. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cornwell MM and Smith DE: A signal
transduction pathway for activation of the mdr1 promoter involves
the proto-oncogene c-raf kinase. J Biol Chem. 268:15347–15350.
1993.PubMed/NCBI
|
|
6
|
Mas VM, Hernandez H, Plo I, et al: Protein
kinase Czeta mediated Raf-1/extracellular-regulated kinase
activation by daunorubicin. Blood. 101:1543–1550. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
8
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kornblau SM, Womble M, Qiu YH, et al:
Simultaneous activation of multiple signal transduction pathways
confers poor prognosis in acute myelogenous leukemia. Blood.
108:2358–2365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kolch W: Coordinating ERK/MAPK signalling
through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 6:827–837.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoon S and Seger R: The extracellular
signal-regulated kinase: multiple substrates regulate diverse
cellular functions. Growth Factors. 24:21–44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giehl K: Oncogenic Ras in tumour
progression and metastasis. Biol Chem. 386:193–205. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thompson N and Lyons J: Recent progress in
targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug
discovery. Curr Opin Pharmacol. 5:350–356. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Escudier B, Eisen T, Stadler WM, et al:
Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J
Med. 356:125–134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Strumberg D, Clark JW, Awada A, et al:
Safety, pharmaco-kinetics, and preliminary antitumor activity of
sorafenib: a review of four phase I trials in patients with
advanced refractory solid tumors. Oncologist. 12:426–437. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wilhelm SM, Carter C, Tang L, et al: BAY
43-9006 exhibits broad spectrum oral antitumor activity and targets
the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in
tumor progression and angiogenesis. Cancer Res. 64:7099–7109. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wilhelm S, Carter C, Lynch M, et al:
Discovery and development of sorafenib: a multikinase inhibitor for
treating cancer. Nat Rev Drug Discov. 5:835–844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cummings J, Anderson L, Willmott N and
Smyth JF: The molecular pharmacology of doxorubicin in vivo. Eur J
Cancer. 27:532–535. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Quillet-Mary A, Mansat V, Duchayne E, et
al: Daunorubicin-induced internucleosomal DNA fragmentation in
acute myeloid cell lines. Leukemia. 10:417–425. 1996.PubMed/NCBI
|
|
21
|
Hannun YA: Functions of ceramide in
coordinating cellular responses to stress. Science. 274:1855–1861.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han W, Sun J, Feng L, et al: Autophagy
inhibition enhances daunorubicin-induced apoptosis in K562 cells.
PLoS One. 6:e284912011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weinstein-Oppenheimer CR, Blalock BL,
Steelman LS, et al: The Raf signal transduction cascade as a target
for chemotherapeutic intervention in growth factor responsive
tumors. Pharmacol Ther. 88:229–279. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abrams SL, Steelman LS, Shelton JG, et al:
The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and
sensitivity to targeted therapy. Cell Cycle. 9:1781–1791. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dasmahapatra G, Yerram N, Dai Y, Dent P
and Grant S: Synergistic interactions between vorinostat and
sorafenib in chronic myelogenous leukemia cells involve Mcl-1 and
p21CIP1 down-regulation. Clin Cancer Res. 13:4280–4290. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu C, Friday BB, Lai JP, et al: Cytotoxic
synergy between the multikinase inhibitor sorafenib and the
proteasome inhibitor bortezomib in vitro: induction of apoptosis
through Akt and c-Jun NH2-terminal kinase pathways. Mol
Cancer Ther. 5:2378–2387. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Towatari M, Iida H, Tanimoto M, et al:
Constitutive activation of mitogen-activated protein kinase pathway
in acute leukemia cells. Leukemia. 11:479–484. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Laurent G and Jaffrézou JP: Signaling
pathways activated by daunorubicin. Blood. 98:913–924. 2001.
View Article : Google Scholar : PubMed/NCBI
|