Introduction
Glioblastoma multiforme (GBM) is one of the most
prevalent and aggressive malignant primary brain tumor in adult
patients, usually with poor prognosis and a low 5-year survival
rate (1,2). During the past two decades, many
advances had been made to GBM treatment, including new
neurosurgerical techniques, radiation therapy and novel
chemotherapeutics, yet the patient survival time from initial
diagnosis has not significantly increased; the vast majority of
patients succumb to the disease within 2 years (3–6). Thus,
the development of new therapeutic strategies specifically
targeting the apoptotic or senescence pathway which may improve
treatment responses has become the focus of antitumor research.
Unlike normal brain cells, glioblastoma cells
exhibit certain stem cell-like features, such as self-renewal
ability, unlimited proliferation, differentiation capacity, and
repressed premature senescence and apoptosis. Consequently, the
induction of glioblastoma cells into a senescent state may be
effective as a novel therapeutic strategy. Several critical
regulatory genes associated with apoptotic and senescence have been
identified in human glioblastoma. These genes mainly promote tumor
growth, invasion and resistance to conventional chemotherapy and
radiotherapy (1,7–9).
Cellular senescence is a normal physiological
process characterized by a decrease in the number of dividing
cells, unresponsiveness to mitogenic stimuli that finally triggers
self-apoptosis. Compared with proliferating cells, cells entering
into a senescence state undergo dramatic structural alterations,
such as a decrease in mass, and a change in the function of
subcellular organelles, and in particular exhibit enlarged flat
morphology, reactive oxygen production, lipofuscin accumulation,
and enhanced activity of senescence-associated β-galactosidase (SA
β-gal), and alter the expression of several senescence-associated
genes (10,11) such as p16, p21, transforming growth
factor β-1 (TGF-β1), insulin growth factor binding protein 3
(IGFBP3) and apolipoprotein J (ApoJ). Particularly in stem cells,
Bmi-1 is a critical gene that controls self-renewal.
Copper functions as a co-factor of different enzymes
in cellular metabolism, such as cytochrome c oxidase and
Cu/Zn superoxide dismutase (12).
In contrast, cellular ionic copper in excess damages different
types of biomolecules through cytotoxicity and by mediating the
generation of the highly reactive hydroxyl radical (13,14).
Thus, copper is an essential trace element for the process of
cellular senescence due to its dual role in the biological system.
In many age-associated disorders such as Alzheimer’s and
Parkinson’s disease, excess accumulation of copper is a crucial
factor in pathogenesis (15,16).
In the present study, we investigated the
alterations in cell morphology and molecular genesis of the
glioblastoma cell line U87-MG following exposure to subcytotoxic
doses of copper sulfate. We demonstrated that copper sulfate
induces glioblastoma cell lines into a replicative senescent state,
and we further verified a change in the expression of
senescence-associated genes and confirmed copper exposure-induced
senescence through the Bmi-1 pathway.
Materials and methods
Cell culture and proliferation
U87-MG glioma cell lines were obtained from the
American Type Culture Collection and were routinely cultured in a
10-cm culture dish containing 10 ml Minimum Essential Medium (MEM),
α-modified MEM, supplemented with 10% fetal bovine serum (FBS), 1%
non-essential amino acid (NEAA), 1% glutamine and 1% antibiotics
(penicillin-streptomycin) at 37°C in an atmosphere containing 5%
CO2. Cells were subcultured into 3 new dishes when
confluent.
Copper cytotoxicity
The cell survival rate was measured after cells were
exposed to 5 different doses of copper sulfate (0, 100, 250, 500
and 750 μM) to optimize copper cytotoxicity based on a previous
report (17), and was compared with
the controls (750 μM Na2SO4). We replaced the
new whole medium without copper sulfate after a 24-h exposure, and
then added neutral red staining dye (40 μg/ml), and continued the
incubation for 3 h at 37°C. Cells were washed with DPBS after
staining, lysised with acetic acid (1% v/v) and dissolved in 50%
(v/v) ethanol. The optical absorbance was recorded by a microplate
reader (GeneAmp 5700 Sequence Detector) at 540 nm. The cells
treated with sodium sulfate were set as the control.
Cell proliferation assay
The effect of copper exposure on the proliferation
of the U87-MG cells was assessed by MTT assay. MTT assay is a
classical method used for cell density determination, by testing
activity of dehydrogenase enzymes. Cells (2×104/well)
were plated onto 96-well plates. After cells attached, different
doses of copper sulfate were added into the supernatants, and then
fixed with MTT at different time-points (1, 2, 3 and 4 days) After
MTT fixation for 4 h at 37°C, the supernatant was discarded and the
sediment was dissolved with 150 μl DMSO. Then, the absorbance at
570 nm for each well was recorded using a microplate reader
(GeneAmp 5700 Sequence Detector). Growth curves were constructed
according to the collected data.
Senescence-associated
β-galactosidase
After a 24-h copper exposure in 6-well plates, the
supernatant was discarded including the ionic copper, and the cells
were washed with DPBS twice. Whole medium was added and culture was
continued for another 48 h. After culture, the activity of
senescence-associated β-galactosidase (SA β-gal) was determined as
described by Dimri et al(18). The proportion of SA β-gal-positive
cells was determined by counting 400 cells/well under a
microscope.
Real-time PCR analysis
RNA was extracted by TRIzol reagent (Sigma) from
cells after 3 days of exposure to copper. Total RNA (2 μg) was
converted to cDNA by reverse transcription reaction. Real-time
polymerase chain reaction (PCR) was employed using 1X Express
SYBR-Green ER (Invitrogen), primers (10 pM) and an optimal
concentration of cDNA (4 ng) as follows: hot start at 95°C for 10
min, followed by 95°C for 15 sec, 60°C for 1 min for 40 cycles, and
9°C for 15 sec, 60°C for 15 sec. We designed the primers as
follows: apolipoprotein J-F, GGA TGA AGG ACC AGT GTG ACA AG and
apolipoprotein J-R, CAG CGA CCT GGA GGG ATT C; TGF-β1-F, AGG GCT
ACC ATG CCA ACT TCT and TGF-β1-R, CCG GGT TAT GCT GGT TGTACA;
p21-F, CTG GAG ACT CTC AGG GTC GAA and p21-R, CCA GGA CTG CAG GCT
TCC T; IGFBP3-F, CAG AGC ACA GAT ACC CAG AAC TTC and IGFBP3-R, CAC
ATT GAG GAA CTT CAG GTG ATT; p16-F, CTA CTG AGG AGC CAG CGT CTA GG
and p16-R, AGA GTG GCG GGG TCG GCG CAG TT; Bmi-1-F, GAG ATC GGG GCG
AGA CAA TG and Bmi-1-R, GAC CTC CAA CGC CTC CTC CT; actin-F, CAG
GTC ATC ACC ATT GGC AAT GAG C and actin-R, CGG ATG TCC ACG TCA CAC
TTC ATG A. The specificity of amplification was checked by
performing melting curves and electrophoresis of the amplification
products.
Western blot analysis
Treated U87-MG cells were washed with ice-cold DPBS
twice, and then replaced with 1 ml 0.25% trypsin, and incubated for
5 min at 37°C. The cells were collected into 2-ml tubes and spun
down for 15 min at 2,500 rpm at 4°C. The pellet was lysed with 100
μl RIPA buffer (10 mM NaPO4, pH 7.2, 0.3 M NaCl, 0.1%
SDS, 1% NP40, 1% deoxycholate, 2 mM EDTA) on ice for at least 30
min. The titer of the protein extracts was quantified by Bradford
assay (19). An equal amount of
protein (50 μg/lane) was loaded on SDS-PAGE gels for
electrophoresis. After appropriate separation, proteins were
transferred to a nitrocellulose membrane, and then blocking with 5%
non-fat dry milk diluted in Tris buffer saline 0.1 M added to 0.1%
Tween-20 (TBST). Proteins were coated with special monoclonal
primary antibodies against Bmi-1, p16 and p21 (Abcam, Cambridge,
MA, USA) and actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA)
overnight at 4°C. After washing with TBST for 3 times, a
peroxidase-conjugated secondary antibody was added to detect the
proteins at room temperature for 1 h. Immunoblots were detected by
ECL Western Blotting Kit (Pierce™-Thermo Scientific). Results were
recorded by exposure to X-ray film. Actin was detected as the
control.
Statistical analysis
The t-test was performed to compare normal and
senescent states. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of copper sulfate on cellular
viability
In order to determine the optimal appropriate
subcytotoxic concentration of copper sulfate, we used 5 different
concentrations of copper sulfate: 0, 100, 250, 500 and 750 μM for
24 h based on previous research (17), and we determined the mean cell
viability from three independent experiments, setting the control
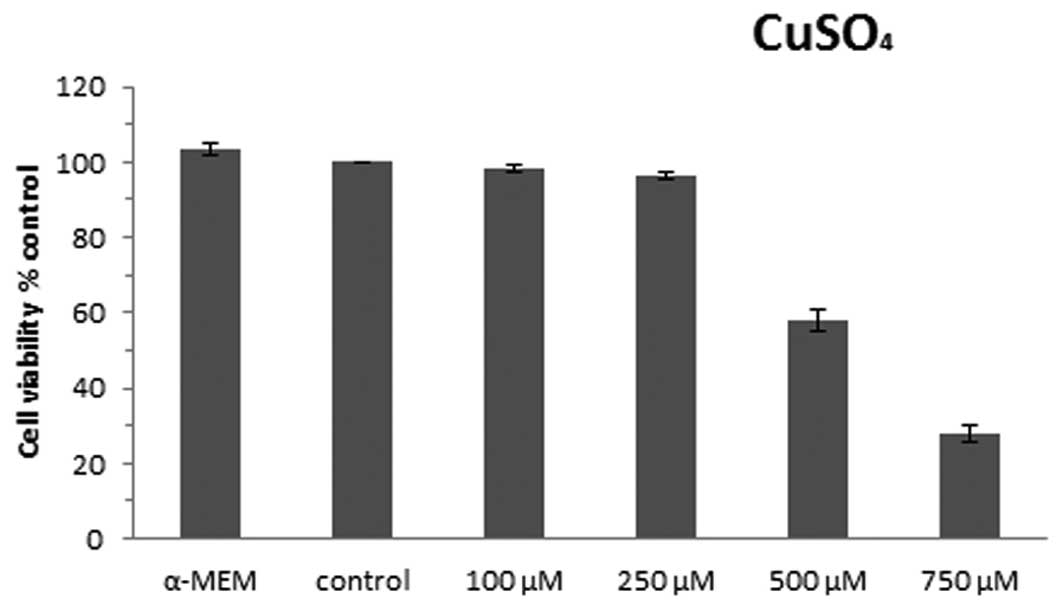
cells as 100%. Cell viability was assessed using neutral red assay
as shown in Fig. 1. The control
cells did not have significant differences in viability when
compared with cells cultured in α-MEM medium (103.3%). The doses of
copper sulfate used and the viability of the U87-MG cells were
found to have a dose-effect relationship; the viability of the
cells decreased according to the increasing doses of copper sulfate
used. When the cells were exposed to 100 and 250 μM copper sulfate,
the cell viability was found to be 98.2 and 96.4% compared with the
control cells, indicating that no significant differences in cell
viability were observed. The viability of the cells exposed to 500
and 750 μM copper sulfate significantly decreased to 58.0 and
28.1%, respectively, when compared with the control cells. Thus,
250 μM CuSO4 was determined to be the optimal
subcytotoxic concentration used in this study.
Proliferation of U87-MG cells exposed to
copper sulfate
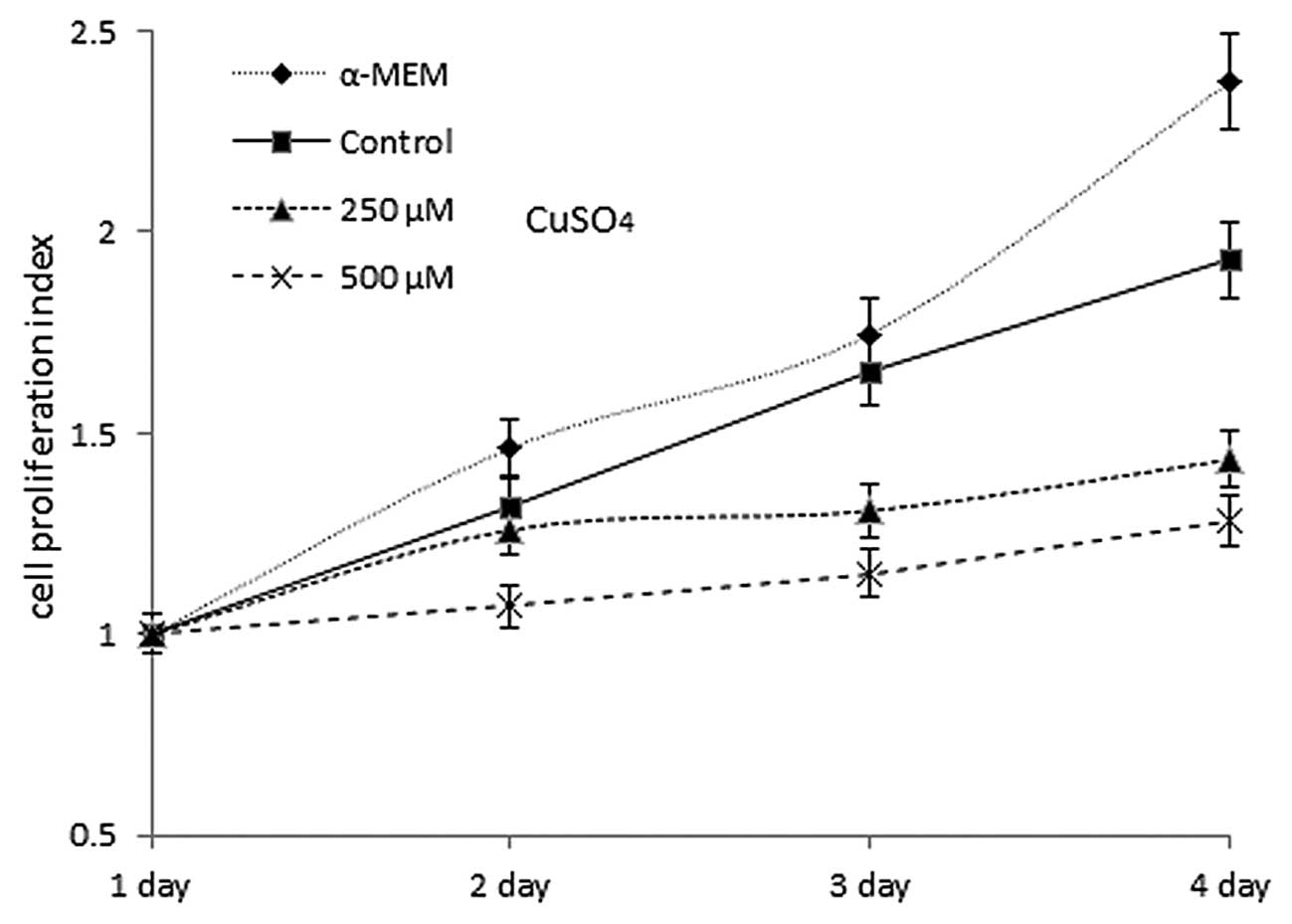
To assess the effect of copper on the proliferation
of U87-MG cells, we determined the cell activity of dehydrogenase
enzymes by MTT assay at 1, 2, 3 and 4 days after exposure. For each
condition, it was assumed that, on day 1, the proliferation index
was 1. Cell proliferation curves for the different conditions
during exposure are shown in Fig.
2. Cells cultured in α-MEM and control cells exhibited
approximately the same proliferation rate, showing an increase in
activity of dehydrogenase enzymes from 1- (day 1) to 2.37- and
1.93-fold (day 4), respectively, while cells exposed to 250 or 500
μM copper sulfate only had an increase in proliferation from 1-
(day 1) to 1.43- and 1.28-fold on day 4, respectively.
Alterations in cell morphology and
senescence-associated β-galactosidase activity in U87-MG cells
treated with copper sulfate
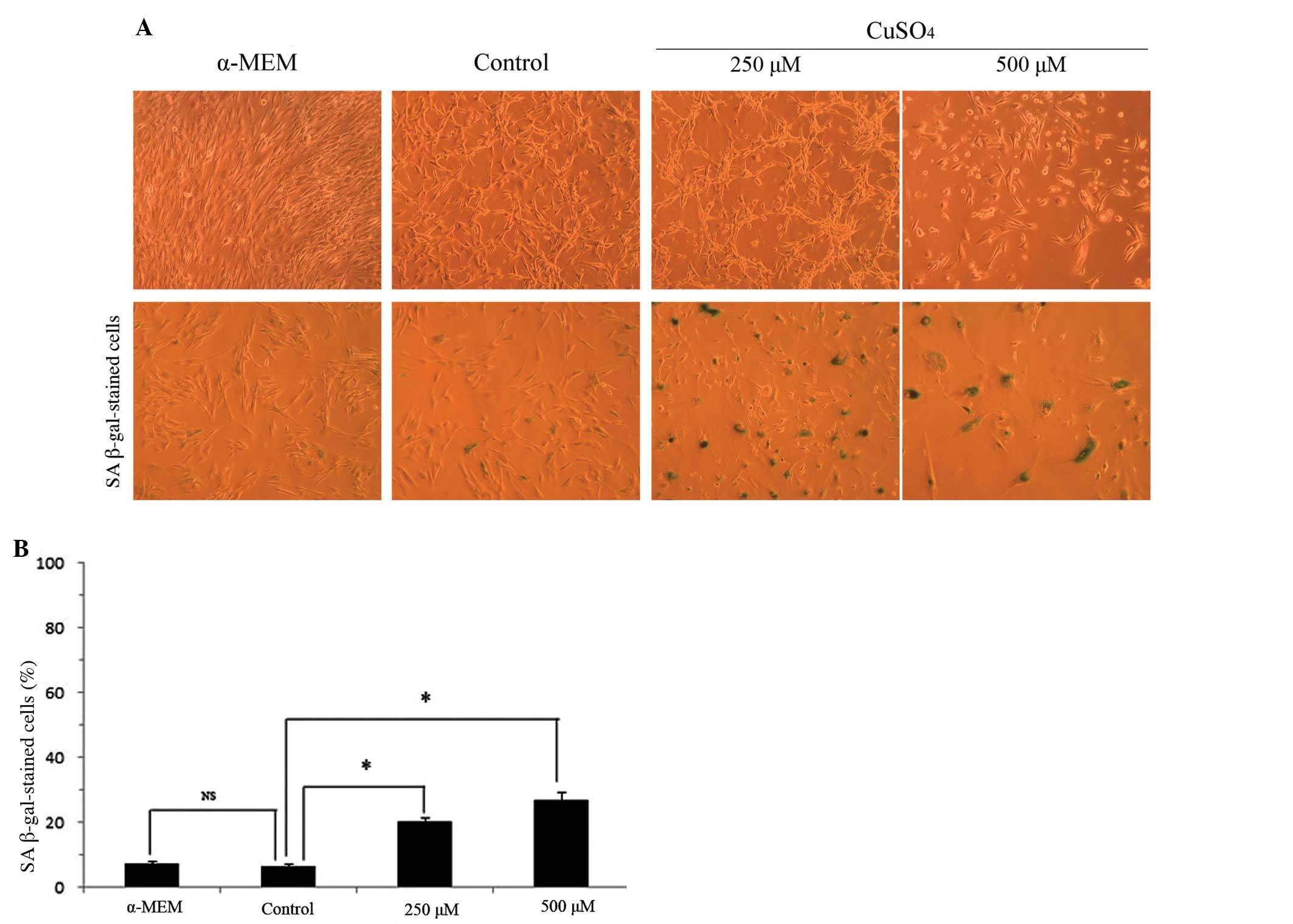
The main morphological cell alterations observed in
cellular senescence include an increase in the cell surface and a
change from a small spindle-fusiform into a large flat spread.
Alterations in the cell morphological features were noted in the
U87-MG cells exposed to 250 or 500 μM copper sulfate (Fig. 3A), including an enlarged cell
surface and stellate outline with thin extensions. Cells incubated
with 500 μM copper sulfate had a lower cell density when compared
with the cells cultured with α-MEM, 250 μM copper sulfate and the
control cells (Fig. 3A), similarly
to the results of the cell viability. SA β-gal staining is commonly
used to provide evidence of cell senescence and whose activity
increases with the process of aging. We compared the difference in
β-gal activity among the α-MEM-treated, the control, and the cells
exposed to 250 and 500 μM copper sulfate. No significant difference
was detected between the α-MEM and control group (P=0.11). In
contrast, the percentage of SA β-gal positive-stained cells
increased significantly to 20 (P<0.05) and 27% (P<0.05)
following exposure to 250 and 500 μM copper sulfate, respectively,
when compared with the control group.
Senescence-associated gene expression in
the U87-MG cell line exposed to copper sulfate
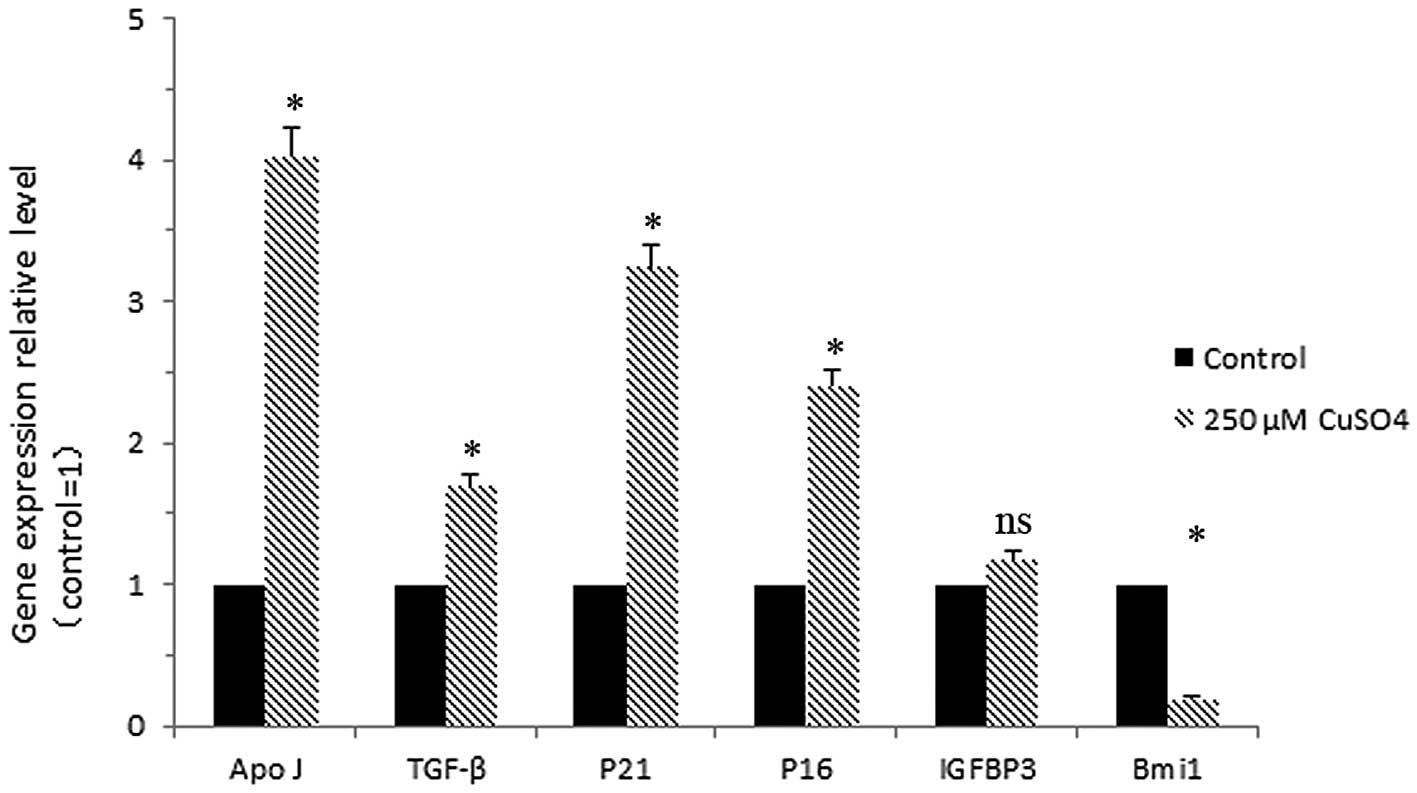
Alteration in the expression levels of several
senescence-associated genes is a typical feature of senescence. The
genes encoding apolipoprotein J (ApoJ), transforming growth factor
β1 (TGF-β1), cyclin-dependent kinase inhibitor 1A (p21),
cyclin-dependent kinase inhibitor 2A (p16), insulin growth factor
binding protein 3 (IGFBP3) and B lymphoma Mo-MLV insertion region-1
(Bmi-1) were detected by real-time PCR. Transcript levels of these
genes were quantified 72 h after cell exposure to copper sulfate.
We mainly focused on the difference between the control and the
cells exposed to 250 μM CuSO4. Cells treated with 250 μM
CuSO4 exhibited a statistically significant difference
in mRNA levels of the senescence-associated genes ApoJ, TGF-β1, p21
and p16 (4.0-, 1.7-, 3.2- and 2.4-fold, respectively) but a marked
decrease in Bmi-1 (0.2-fold) when compared with the control cells.
A non-significant increase in IGFBP3 mRNA expression (1.2-fold) was
also observed (Fig. 4).
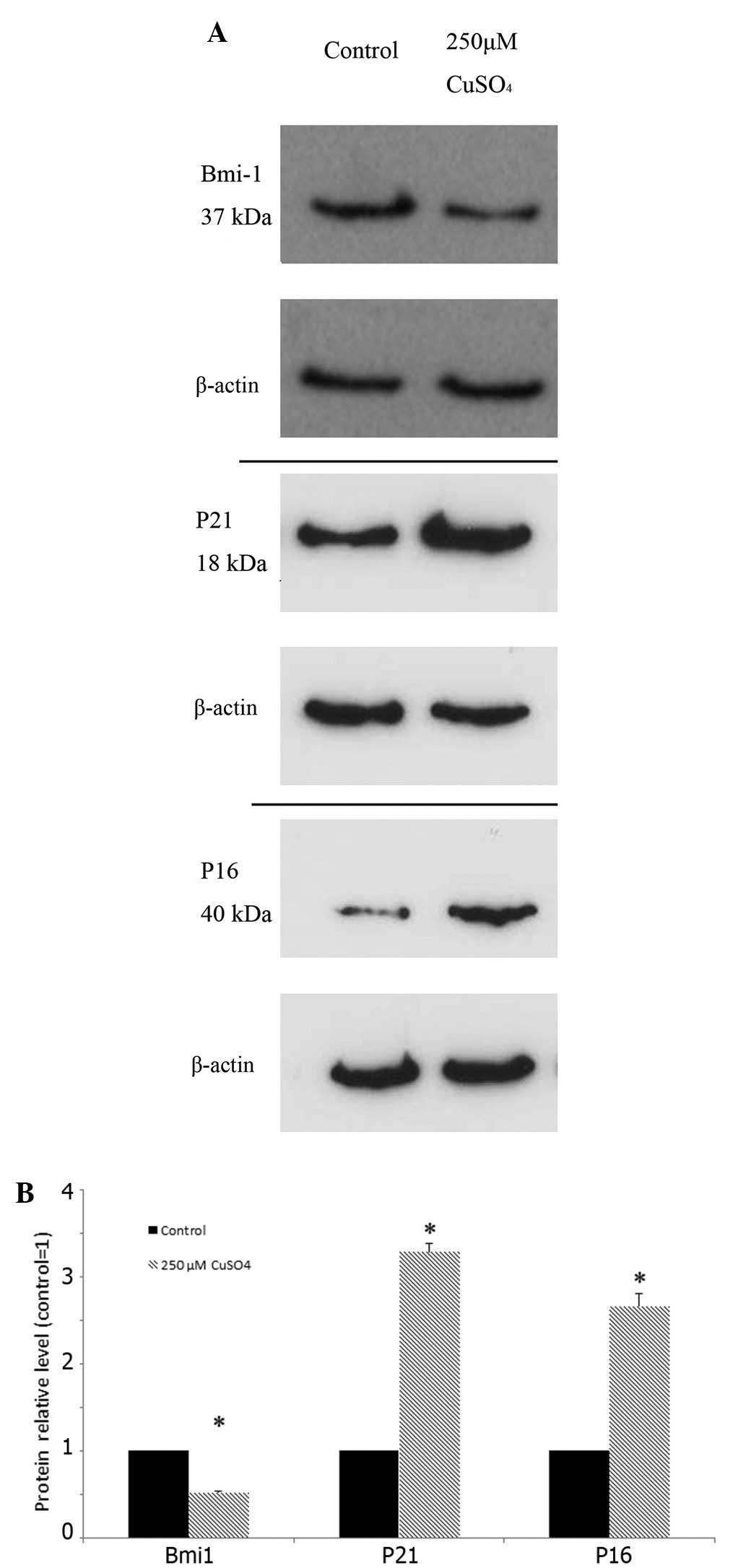
Western blot analysis of U87-MG cells
exposed to copper sulfate
In order to analyze the mechanism of U87-MG
senescence induced by copper sulfate, we assessed the protein
levels of several senescence-associated genes, including Bmi-1, p16
and p21, by western blot analysis. The variation in the protein
levels is shown in Fig. 5A, and the
relative densitometric data were plotted (Fig. 5B). The intracellular level of actin
did not have a statistically significant difference between the
cells exposed to 250 μM CuSO4 and the control cells.
However, the 250 μM CuSO4-treated cells showed increased
protein levels of p21 and p16, but a decreased level of Bmi-1, when
compared with the control cells.
Discussion
In the present study, we demonstrated that
appropriate doses of copper sulfate induced the U87-MG cell line
into a senescent state. High doses (≥500 μM) of copper sulfate may
be toxic to cells and cause a dramatic decrease in cell viability.
Like many other oxidative agents, a subcytotoxic amount (250 μM in
this study) of copper sulfate induced changes in cell structure and
gene expression pattern which are specifically noted in cell aging,
but do not significantly affect cell viability. In the present
investigation, cell proliferation was found substantially reduced
during the 4 days after treatment with 250 μM of CuSO4,
when compared with the control cells. Then, we choose 250 μM as the
most appropriate subcytotoxic dose employed thereafter to research
copper-induced glioblastoma senescence.
Copper is an essential metal that functions as a
transition element in many physiological activities, but it is also
toxic to cells when intracellularly overloaded. Since copper
contributes to the production of an excess of damaging oxidant
radicals, similar to other oxidant agents, copper also induces
normal cells into a premature senescent state (17). Data collected from in vitro
experiments and cell culture studies prove that copper has the
capacity to initiate oxidative damage and interfere with important
cellular events (14).
Glioblastoma is one of the most aggressive brain
tumors, driven by tumor stem cells as confirmed by stem cell
research (20–22). To some extent, many features of
these tumors are similar to normal stem cells, such as self-renewal
ability, unlimited proliferation, and capability for
differentiation. Therefore, we assumed that certain key genes and
pathways which regulate stem cell proliferation and maintenance may
also be fundamental to glioblastoma anti-senescence. Thus, we
focused on the alteration of the PcG gene Bmi-1 in ionic
copper-exposed glioblastoma cells. Since the main regulators of
cell cycle progression through G1 phase are heterodimers, a
cyclin-dependent kinases (10), the
overexpression of CDK inhibitor p16 and p21 may function as
biomarkers in replicative senescence. In order to confirm the
glioblastoma cell senescence state, we also assessed the expression
of other typical senescence-associated genes and proteins,
including Apo J, TGF-β, p16, p21 and IGFBP3.
B lymphoma Mo-MLV insertion region-1 (Bmi-1), is a
proto-oncogene, located on chromosome 10p11.23. It encodes a
324-amino acid nuclear protein which modifies the process of
self-renewal in many types of stem cells (23–27).
Previous reports have confirmed that the polycomb factor Bmi-1
inhibits cell cycle inhibitors p16, p19 and p21, promoting neuron
stem cell (NSC) self-renewal. With increasing age and NSC passage,
Bmi-1 enhancement of NSC self-renewal is significantly increased
(23,24,27).
Some studies have shown that the NSCs markedly expand and
demonstrate aggressive proliferation when Bmi-1 is overexpressed.
Bmi-1 was found to sustain human glioblastoma self-renewal, and
high expression in CD133+ tumor-initiating cells has
been confirmed (28).
Collectively, the results showed that Bmi-1 was
downregulated in copper-induced senescent U87-MG cells, at both the
gene expression and protein level. Since Bmi-1 is considered to be
a functional gene required for tumor cell growth (28–31),
we hypothesized that the copper-induced glioblastoma cell aging
occurs through the downregulation of Bmi-1. Yet, the detailed
mechanism of how ionic copper downregulates Bmi-1 gene expression
remains unclear.
We showed that a subcytotoxic dose of copper sulfate
is able to induce senescence in the U87-MG glioblastoma cell line.
The adaptative conditions of senescence were also observed, such as
limitation of proliferation, enhanced SA β-gal activity, and
alteration in expression of related genes. In the present study, we
investigated the primary molecular mechanisms of copper-induced
senescence in glioblastoma cells. By investigating the process of
senescence of glioblastoma cells induced by ionic copper, we offer
a new vision of how ionic metals affect tumor growth and
proliferation. We also found that downregulation of Bmi-1 is a key
mechanism of senescence in glioblastoma. Further research will be
undertaken to explore the detailed mechanism of how ionic copper
regulates senescence-associated genes in glioblastoma cells. Thus,
efficient novel therapeutic strategies may be developed.
Acknowledgements
The present study was supported by the Program for
New Century Excellent Talents in the University (no. NECT-06-0611),
the National Science Foundation of China (nos. 81071008 and
81171177), the 211 Project-phase III of Zhengzhou University, the
Basic and Clinical Research of Stem Cells, the Excellent Youth
Foundation of Henan Scientific Committee (no. 114100510005), the
Young Excellent Teachers in University Funded Projects of Henan
Province, the Bureau of Science and Technology Development Project
from Henan Province (no. 200902310250).
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, et al:
The 2007 WHO classification of tumours of the central nervous
system. Acta Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohgaki H, Dessen P, Jourde B, et al:
Genetic pathways to glioblastoma: a population-based study. Cancer
Res. 64:6892–6899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stupp R, Hegi ME, Mason WP, et al: Effects
of radiotherapy with concomitant and adjuvant temozolomide versus
radiotherapy alone on survival in glioblastoma in a randomised
phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet
Oncol. 10:459–466. 2009.
|
|
5
|
Stupp R, Hegi ME, Gilbert MR and
Chakravarti A: Chemoradiotherapy in malignant glioma: standard of
care and future directions. J Clin Oncol. 25:4127–4136. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ohgaki H and Kleihues P: Population-based
studies on incidence, survival rates, and genetic alterations in
astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol.
64:479–489. 2005.PubMed/NCBI
|
|
7
|
Bruggeman SW, Hulsman D, Tanger E, et al:
Bmi-1 controls tumor development in an Ink4a/Arf-independent manner
in a mouse model for glioma. Cancer Cell. 12:328–341. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cancer Genome Atlas Research Network.
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohgaki H and Kleihues P: Genetic
alterations and signaling pathways in the evolution of gliomas.
Cancer Sci. 100:2235–2241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hwang ES, Yoon G and Kang HT: A
comparative analysis of the cell biology of senescence and aging.
Cell Mol Life Sci. 66:2503–2524. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakahara Y, Shiraishi T, Okamoto H, et al:
Detrended fluctuation analysis of genome-wide copy number profiles
of glioblastomas using array-based comparative genomic
hybridization. Neuro Oncol. 6:281–289. 2004. View Article : Google Scholar
|
|
12
|
Gupta A and Lutsenko S: Human copper
transporters: mechanism, role in human diseases and therapeutic
potential. Future Med Chem. 1:1125–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Valko M, Morris H and Cronin MT: Metals,
toxicity and oxidative stress. Curr Med Chem. 12:1161–1208. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gaetke LM and Chow CK: Copper toxicity,
oxidative stress, and antioxidant nutrients. Toxicology.
189:147–163. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barnham KJ and Bush AI: Metals in
Alzheimer’s and Parkinson’s diseases. Curr Opin Chem Biol.
12:222–228. 2008.
|
|
16
|
Brewer GJ: Risks of copper and iron
toxicity during aging in humans. Chem Res Toxicol. 23:319–326.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matos L, Gouveia A and Almeida H: Copper
ability to induce premature senescence in human fibroblasts. Age.
34:783–794. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dimri GP, Lee X, Basile G, et al: A
biomarker that identifies senescent human cells in culture and in
aging skin in vivo. Proc Natl Acad Sci USA. 92:9363–9367. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Galli R, Binda E, Orfanelli U, et al:
Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hemmati HD, Nakano I, Lazareff JA, et al:
Cancerous stem cells can arise from pediatric brain tumors. Proc
Natl Acad Sci USA. 100:15178–15183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh SK, Hawkins C, Clarke ID, et al:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Simon JA and Tamkun JW: Programming off
and on states in chromatin: mechanisms of Polycomb and trithorax
group complexes. Curr Opin Genet Dev. 12:210–218. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Orlando V: Polycomb, epigenomes, and
control of cell identity. Cell. 112:599–606. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lessard J, Baban S and Sauvageau G:
Stage-specific expression of polycomb group genes in human bone
marrow cells. Blood. 91:1216–1224. 1998.PubMed/NCBI
|
|
26
|
Park IK, Qian D, Kiel M, et al: Bmi-1 is
required for maintenance of adult self-renewing haematopoietic stem
cells. Nature. 423:302–305. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Molofsky AV, Pardal R, Iwashita T, Park
IK, Clarke MF and Morrison SJ: Bmi-1 dependence distinguishes
neural stem cell self-renewal from progenitor proliferation.
Nature. 425:962–967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abdouh M, Facchino S, Chatoo W, Balasingam
V, Ferreira J and Bernier G: Bmi-1 sustains human glioblastoma
multiforme stem cell renewal. J Neurosci. 29:8884–8896. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vonlanthen S, Heighway J, Altermatt HJ, et
al: The bmi-1 oncoprotein is differentially expressed in non-small
cell lung cancer and correlates with INK4A-ARF locus expression. Br
J Cancer. 84:1372–1376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim JH, Yoon SY, Jeong SH, et al:
Overexpression of Bmi-1 oncoprotein correlates with axillary lymph
node metastases in invasive ductal breast cancer. Breast.
13:383–388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leung C, Lingbeek M, Shakhova O, et al:
Bmi-1 is essential for cerebellar development and is overexpressed
in human medulloblastomas. Nature. 428:337–341. 2004. View Article : Google Scholar : PubMed/NCBI
|