Introduction
Lung cancer is the leading cause of morbidity and
mortality in malignant tumors. Non-small cell lung cancer (NSCLC)
accounts for 80% of lung cancer cases. Epidemic studies reveal
gender differences in NSCLC patients, particularly in lung
adenocarcinoma. Women are more susceptible to smoke or other
environmental factors (1), in view
of the fact that estrogen and progesterone are well-known
prognostic factors for breast, endometrial and ovarian cancer,
suggesting a possible involvement of gender-dependent factors in
the pathogenesis and/or development of NSCLCs (2).
Sex hormones and their receptors have been the focus
of considerable cancer research. Among sex steroids, estrogens play
an important role in the development of breast and endometrial
carcinoma, whereas androgens significantly contribute to the
development of prostate cancer (3).
By contrast, progesterone generally promotes differentiation and
inhibits cellular proliferation through the nuclear progesterone
receptor (nPR) (4). Previous
studies showed that progesterone-mediated growth inhibition was
mainly preceded by decreased expression of cyclins and/or induction
of cyclin-dependent kinase inhibitors (5–7).
Administration of progestins, including medroxyprogesterone
acetate, is currently used as an endocrine therapy in breast and
endometrial carcinoma patients (8,9).
During embryogenesis, sex hormones influence the development of
lung tissue, but during adulthood, the lung is not a target organ
for sex hormones. Notably, female adenocarcinoma has a better
prognosis than male lung cancer or other female pathologic types of
lung cancer, indicating gender as an independent prognostic factor
(10). It is reported that
progesterone can mediate growth inhibition in PR-positive tumors in
mice through decreased expression of cyclins A, D1 and E and/or
induction of cyclin-dependent kinase inhibitors such as p21 and p27
(11). By contrast, PR antagonist
mifepristone can inhibit spontaneous growth of lung cancer in mice
(12). Combination of estrogen and
progestins in NSCLC cells may cooperate in promoting expression of
vascular endothelial growth factor (VEGF) which is essential for
the progression of lung carcinoma, mainly by increasing
proliferation of endothelial cells from neighboring blood vessels
(13). Differences of progesterone
actions may partly be explained by different types of progesterone
receptors in various tissues.
Progesterone receptors include the nPR family, mPR
family and progesterone membrane receptor components family
(PGMRCs). The nPR family has been studied extensively in lung
cancer, with a focus on the pathological characteristics, clinical
stage and lymph node metastasis (14), while studies on the mPR family in
lung cancer are few. Recently, mPRα was cloned from the ovarian
tissue of spotted seatrout oocytes and recognized as the earliest
and most thoroughly-studied progesterone membrane receptor by
binding P4 in the membrane and subsequently inducing a series of
alterations in the secondary messenger pathways through activation
of the pertussis toxin-sensitive inhibitory G-proteins (15–17).
Our preliminary study proved that mPRα acted as an
epithelial-mesenchymal transition (EMT) negative regulatory
protein, mediated progesterone’s effect to reverse the EMT process
and inhibited tumor development in breast cancer MDA-MB468 (MB468)
cells (18). To date, there are no
previous reports on whether mPRα-mediated progesterone signal plays
a role in tumor invasion and metastasis. Our study examined the
expression and location of mPRα in the lung adenocarcinoma cell
line A549. Further research is focused on whether mPRα can mediate
the effects of P4 on lung adenocarcinoma cell migration and
invasion as well as its molecular pathway mechanism.
Materials and methods
Antibodies and inhibitors
Mifepristone (MIF) and pyrazolo-pyrimidine compound
(PP1) were purchased from EMD Chemicals (Gibbstown, NJ, USA).
Anti-mPRα goat polyclonal IgG, anti-MMP-9 goat polyclonal IgG,
anti-GAPDH goat polyclonal IgG, anti-mPRα blocking peptide, donkey
anti-goat IgG-HRP, goat anti-rabbit IgG-HRP and anti-mouse IgG were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Anti-FAK rabbit polyclonal and anti-p-FAK rabbit polyclonal IgG
were from Cell Signaling (Danvers, MA, USA). P4-BSA-FITC conjugate
and anti-α-tubulin mouse monoclonal IgM were purchased from Sigma
(St. Louis, MO, USA).
Cell culture
The human lung adenocarcinoma cancer cell line A549
and the breast cancer cell line MDA-MB231 (MB231) were obtained
from the American Type Culture Collection (Rockville, MD, USA). The
MB231 cell line is negative for mPRα. MB231 w/mPRα cells are
derived from MB231 cells transfected with mPRα cDNA plasmid, with
mPRα mRNA and protein strong expression. These cancer cells were
cultured in DMEM (Mediatech, VA, USA) containing 10% FBS, 100 U/ml
penicillin and 100 μg/ml streptomycin (Gibco, Carlsbad, CA, USA) in
a humidified incubator at 37°C with 5% CO2.
RT-PCR assay
Total RNA was extracted using TRIzol (Invitrogen,
Carlsbad, CA, USA) and concentrations of RNA were determined using
a NanoDrop 2000 Spectrophotometer (Thermo Scientific, USA). Reverse
transcription for synthesizing cDNA was carried out using the
QuantiTect Reverse Transcription kit (Qiagen, Valencia, CA, USA).
PCR amplification (35 cycles of 95°C for 20 sec, 58°C for 30 sec
and 72°C for 20 sec) was conducted in a total volume of 25 μl using
the GoTaq Hot Start Green Master Mix (Promega, Madison, WI, USA).
Following PCR amplification, 25 μl of the samples were separated
via electrophoresis on a 1.5% agarose gel. The primers used for PCR
amplification were: mPRα: 5′-CCTGCTGT GTGATCTTAG-3′ and
5′-CGGAAATAGAAGCGCCAG-3′ (19),
18-S: 5′-GTTGGTTTTCGGAACTGAGGC-3′ and 5′-GTC GGCATCGTTTATGGTCG-3′
(20).
Immunoblotting assay
Western blot assays were performed as previously
described (18). Following
treatment with or without P4 and/or diverse pathway inhibitors, the
growth-arrested cells were lysed with 500 μl ice-cold lysis buffer
(50 mM HEPES, 5 mM EDTA, 50 mM sodium chloride, pH 7.4), 1% Triton
X-100, protease inhibitors (10 μg/ml aprotinin, 1 mM
phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin) and phosphatase
inhibitors (50 mM sodium fluoride, 1 mM sodium orthovanadate, 10 mM
sodium pyrophosphate). Cell lysates (30 μg) were separated using
SDS-PAGE and transferred to nitrocellulose membranes, blocked for 1
h in TBS buffer containing 5% non-fat dry milk and 0.1% Tween-20
and incubated overnight with primary antibodies at proper
dilutions. Following incubation with secondary antibodies, proteins
of interest were detected by ECL chemiluminescence. Image J
(http://rsb.info.nih.gov/ij/) was used
for image analysis and quantitative data were normalized with the
reference proteins (i.e., GAPDH or α-tubulin) or calculated as
ratios of phosphor protein/total protein when the reference
proteins were the same.
Localization of P4-BSA-FITC binding
sites
Cells were cultured in chamber slides and exposed to
100 nM P4-3-(o-carboxymethyl) oxime-BSA-FITC (P4-BSA-FITC) for 30
min in serum-depleted medium. Cells were then washed with PBS
buffer, fixed with 10% buffered formalin, counter-stained with DAPI
and observed under a confocal microscope (Olympus FV1000, Tokyo,
Japan) using an oil objective lens (x60).
Wound closure migration assay
Cells (5×105/well) were seeded in a
24-well plate, cultured to reach confluence and then scraped with a
sterile micropipette tip to create a denuded zone (gap) with a
constant width (W0). After removing cell debris with
repeated PBS rinses, fresh serum-free DMEM medium with or without
P4 (30 ng/ml) and/or other testing reagents was added. Anti-mPRα
antibody (1:200) and/or anti-mPRα blocking peptide (1:100) was
added 2 h prior to P4 treatment. PP1 (10 μM) was added 1 h prior to
P4 treatment. The cells migrated at various speeds toward the
middle axis from both edges of the scraped gaps, depending on the
treatment of the aforementioned testing reagents, when they were
incubated continually for 16 h. Following incubation, the width of
the gap (T16h) was measured by Image J. The rate of
wound closure (WC) was calculated by the following equation: WC = 1
- (W16h /W0) × 100% (21); regarding control cells, migration
inhibiting rate of treated cells (MIR) = 1 -
(WCtreatmen/WCcontrol) × 100%.
Invasion assay
Cell invasion was assayed using the BD BioCoat™
Matrigel™ Invasion Chamber (BD Biosciences, MD, USA) (22). Cells (4×104 cells/well)
were seeded in the upper chamber of a 24-well BD transwell coated
with Matrigel and cultured with DMEM medium containing 1% FBS.
Following treatment with P4 at 30 ng/ml for 24 h with or without
PP1 treatment at 10 μM for 1 h, the complete medium was applied to
the lower chamber as chemoattractant. Cells were then incubated for
an additional 16 h and the cells in the upper surface of the
chamber membrane were then carefully removed with a cotton swab.
Cells that invaded to the lower surface of the membrane were fixed
with 10% buffered formalin and stained with hematoxylin solution.
The number of invaded cells (IC) from 20 random microscopic fields
(magnification, ×200) was counted. Invasion inhibition rate (IIR)
was calculated as follow: IIR = 1 -
(ICtreatment/ICcontrol) × 100%.
Statistical analysis
Data are expressed as the means ± standard error
(SE) and statistical differences between mean values were
determined by the Student’s paired two-tailed t-test, followed by
the Fisher’s protected least significance difference (PLSD).
P<0.05 was considered to indicate statistically significant
differences.
Results
mPRα expression in lung adenocarcinoma
A549 cells
We set 18-S mRNA and α-tublin as internal
references, and breast cancer MB231 and MB231 w/mPRα cells as
negative and positive control. We found that expression of mPRα was
positive both at the transcriptional and translational levels in
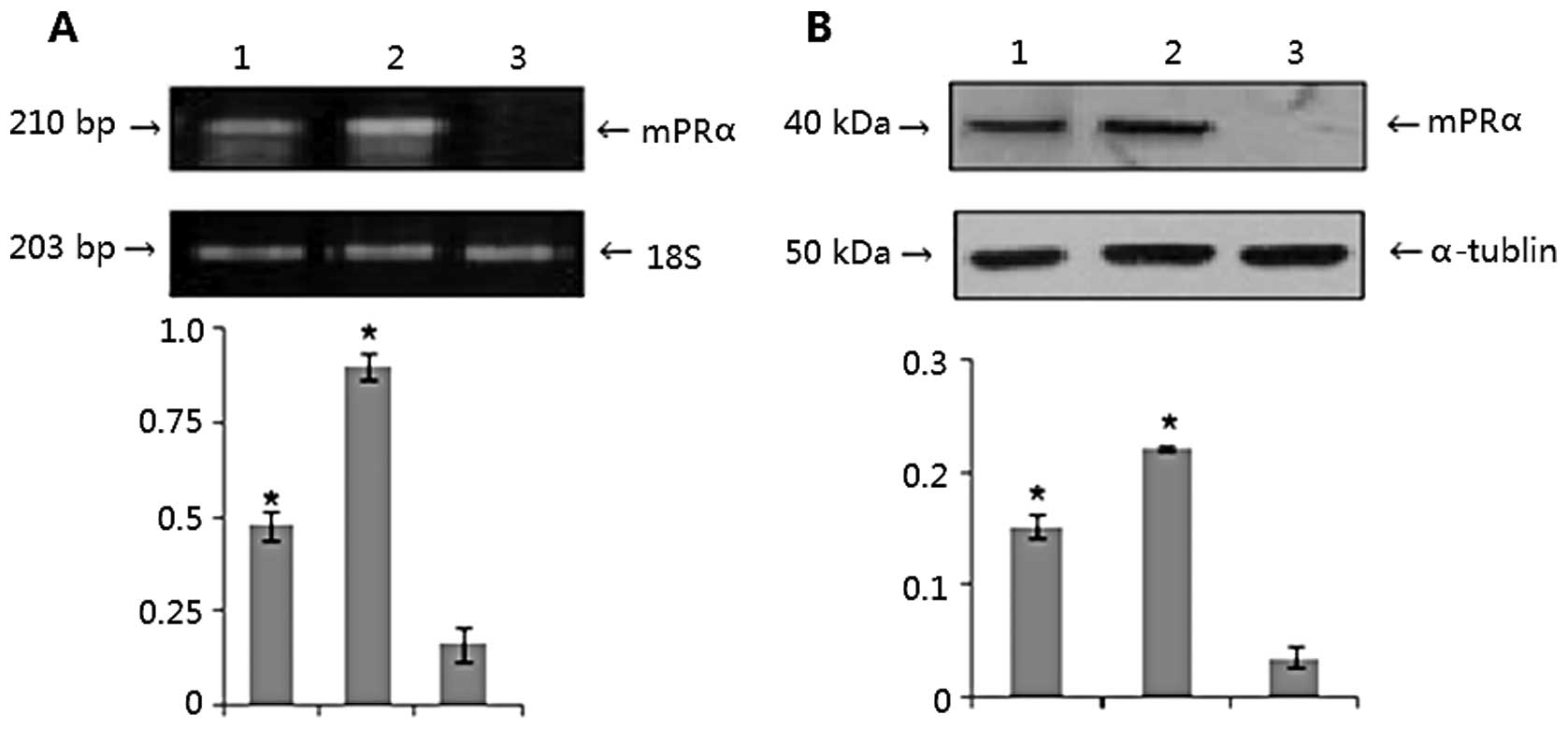
lung adenocarcinoma A549 cells. As shown in Fig. 1A, the designated PCR band for mPRα
in A549 cells was clearly observed at a moderate level (lane 1),
distinct bands were found for the mPRα cDNA transfected MB231
w/mPRα cells (lane 2), however no band for parental MB231 cells was
identified (lane 3; P<0.05). Using cell lysates isolated from
those cells, an identical pattern of mPRα protein expression was
documented by western blot assays (Fig.
1B) (P<0.05).
Positioning of mPRα on A549 cells and its
binding characteristics
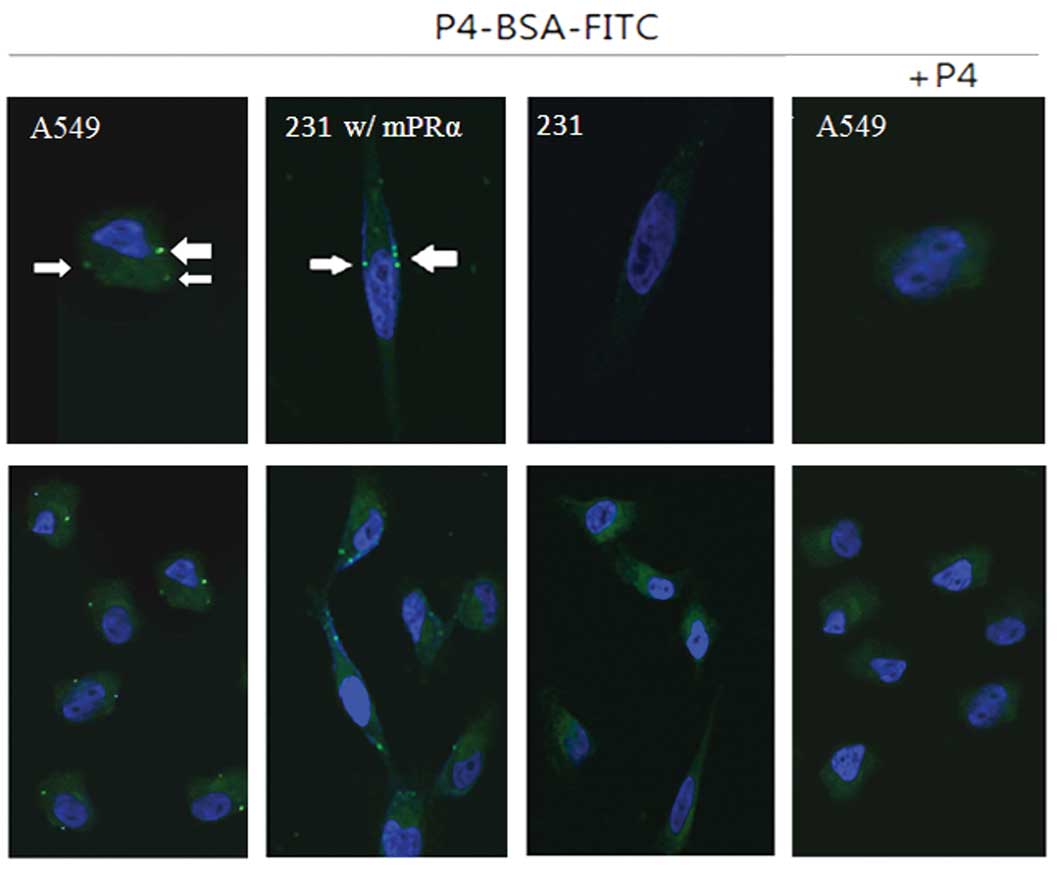
To determine if the mPRα protein is translocated to
the membrane compartment of A549 cells, we performed in
vitro binding tests using a cell-impermeable P4 conjugate
(P4-BSA-FITC). After a 30-min incubation, we observed clear
fluorescent signals in the membrane of A549 cells (Fig. 2, white arrows). Similar fluorescent
signals were also found in the membrane of MB231 w/mPRα cells, but
not in parent MB231 cells. To further demonstrate the binding
specificity, we co-incubated A549 cells with P4-BSA-FITC conjugate
and excessive un-conjugated free P4. As shown in Fig. 2, no fluorescent signals were
observed in A549 cells.
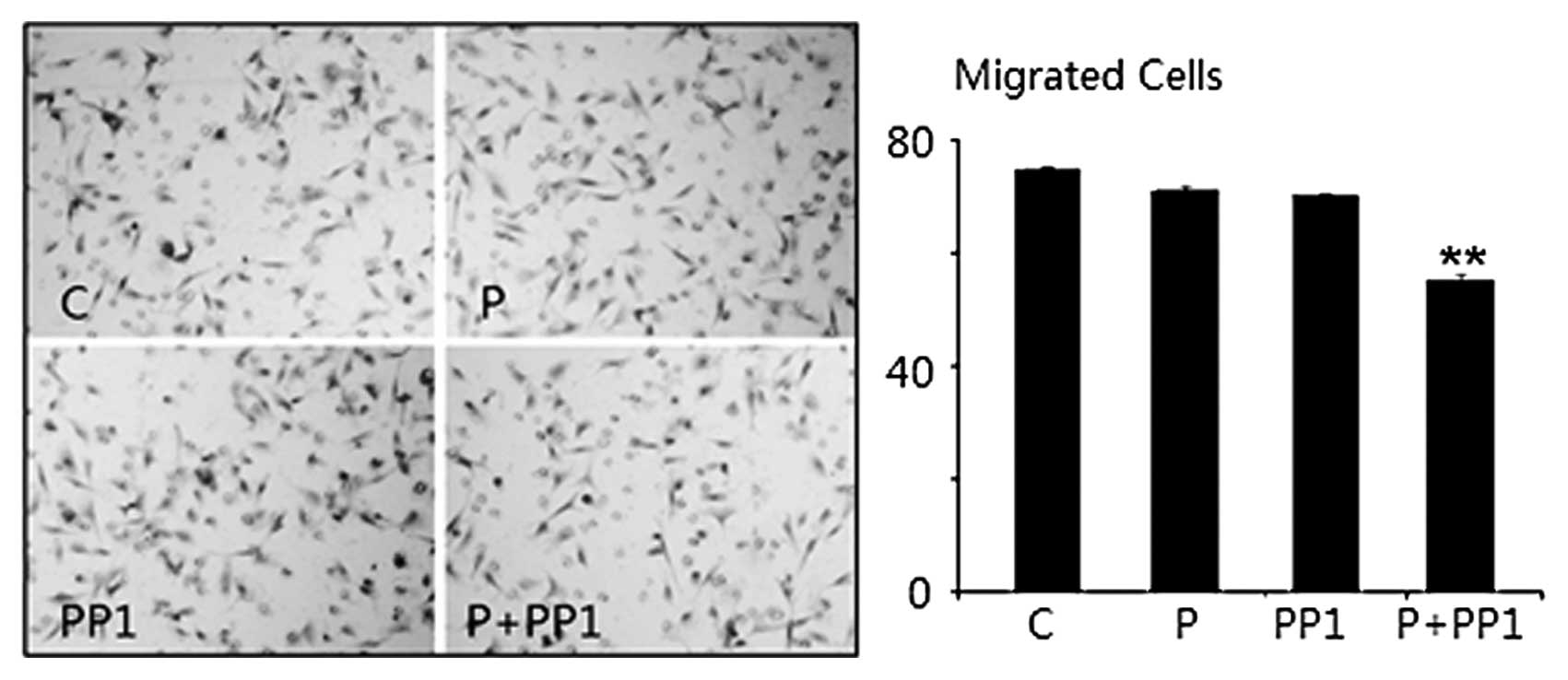
Cell migration of A549 cells in response
to treatment of P4 and/or PP1
Further experiments were performed to determine the
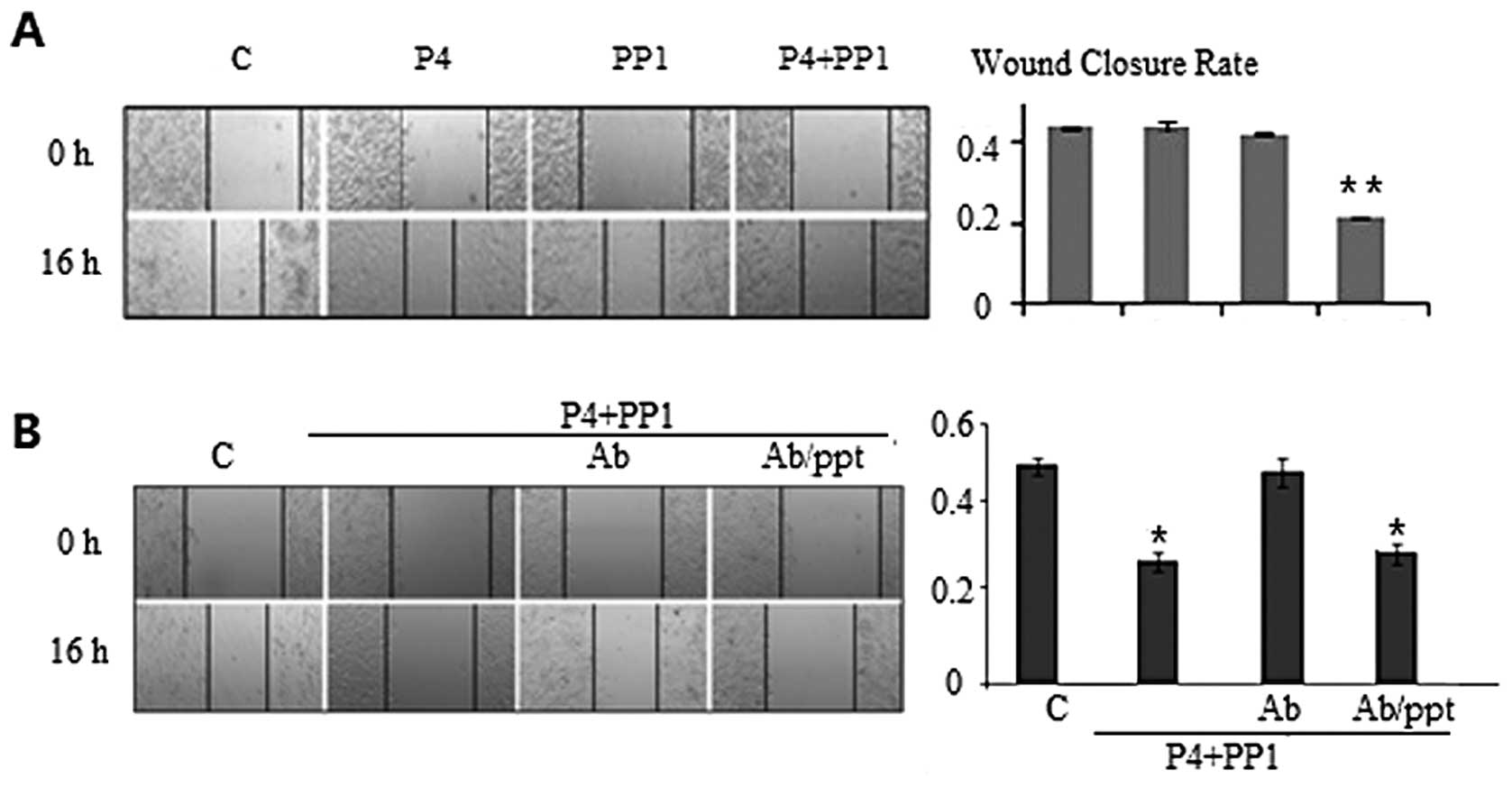
effect of P4 treatment on cell migration. Using a WC assay, we
found that the WC of A549 cells was slower, although it was only
marginally significant, when the cells were treated with P4 (30
ng/ml) for 16 h as compared to the cells without P4 treatment
(45.4±1.8 vs. 47.3±1.7%, MIR 4.0%, PWC=0.51). The WC
rate for cells treated by PP1 alone was minimally inhibited
(45.6±0.7 vs. 47.3±1.7%, MIR 3.4%, PWC=0.63), which was
comparable to cells treated with P4 (P4 vs. PP1 P=0.46).
Co-incubation with P4 and PP1 resulted in a WC rate that was
significantly slower in A549 cells, compared to control (23.0±1.1
vs. 47.3±1.7%, MIR 51.3%, PWC<0.001) or to P4 or PP1
treatment alone (PWC=0.02 and 0.02). These results
indicated a synchronous inhibitory effect of P4+PP1 in the cell
migration of mPRα+A549 cells (Fig.
3A).
In order to further clarify the role of P4→mPRα
signaling in cell migration, we pre-incubated A549 cells with
anti-mPRα antibody to block the binding of P4 to mPRα receptor 1 h
prior to P4+PP1 treatment. The inhibitory effects of P4+PP1 on cell
migration were abrogated (WC 45.8±1.9 vs. 46.5±2.2%, MIR 1.5%,
PWC=0.82), indicating the mPR receptor plays a key role
in P4+PP1-induced cell migratory inhibition. When the cells were
pre-incubated with anti-mPRα antibody and excess anti-mPRα blocking
peptide, the inhibitory effects of P4+PP1 on the cell migration of
A549 cells were restored (25.8±1.4 vs. 46.5±2.2%, MIR 44.5%,
PWC<0.001) (Fig.
3B).
Neither nPR nor PGRMC1 plays a key role
in the mediation of P4+PP1 inhibitory effects on the cell migration
of A549 cells
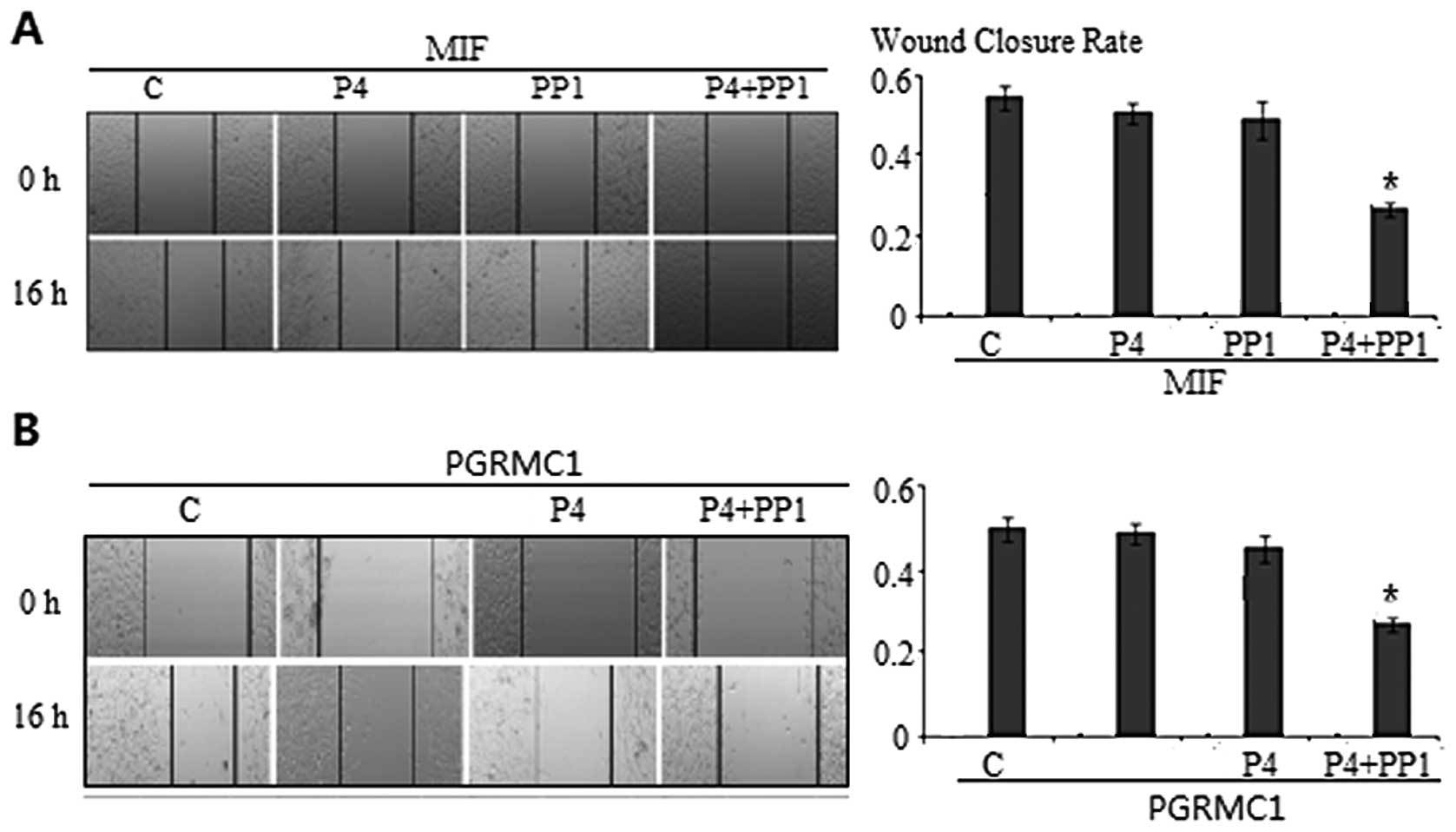
The expression of PR in PR negative cancer cells may
be induced by P4 treatment, although the extent of induction is
very low. To clarify if induction of endogenous PR has a role in
P4-induced cell migration inhibition, we pre-incubated A549 cells
with MIF, an nPR antagonist, prior to P4 and/or PP1 treatment. WC
rates were not affected (P4 vs. MIF+P4, PP1 vs. MIF+PP1, P4+PP1 vs.
MIF+P4+PP1 were 47.4±0.2 vs. 46.0±1.4%, 45.6±0.6 vs. 44.9±2.8% and
23.0±0.2 vs. 24.0±1.1%, respectively, all PWC values
>0.05) (Fig. 4A).
In addition to mPRα, PGRMC1 has been implicated in
membrane-initiated progesterone signaling. It is unclear whether
mPRα functions alone or if it requires PGRMC1. We then
pre-incubated A549 cells with PGRMC1 antibody to block or interfere
with the function of PGRMC1 receptor 1 h prior to P4 and/or PP1
treatment. The WC rates, in the presence of anti-PGRMC1 antibody,
demonstrated no change on the cell migration pattern as compared to
those induced by P4+PP1 (WC 26.0±0.1 vs. 47.4±1.3%, P=0.02).
Treatment of anti-PGRMC1 antibody alone had no effect on cell
migration (47.0±1.1 vs. 47.4±1.3%, P=0.78) (Fig. 4B).
Cell invasion of A549 cells in response
to treatment of P4 and/or PP1
As cancer invasion in vivo is a three
dimensional process involving transendothelial migration and
penetration through extracellular matrix, we considered that a 3D
cell invasion model would further delineate the role of P4 and/or
PP1 on the metastatic potential of A549 cells. To confirm the role
of P4 and PP1 on the cell migration of A549 cells, a cell invasion
assay was performed. Following P4 and/or PP1 treatment for 16 h,
the number of cells that invaded into the lower chamber of Matrigel
(IC) was decreased as compared to control (53±2 vs. 78±1 cells, IIR
32.1%, P<0.001), but treatment with either P4 or PP1 alone was
ineffective (76±2 and 74±3 cells, IIR were 3.2 and 7.2%,
PIC values were 0.83 and 0.92) (Fig. 5).
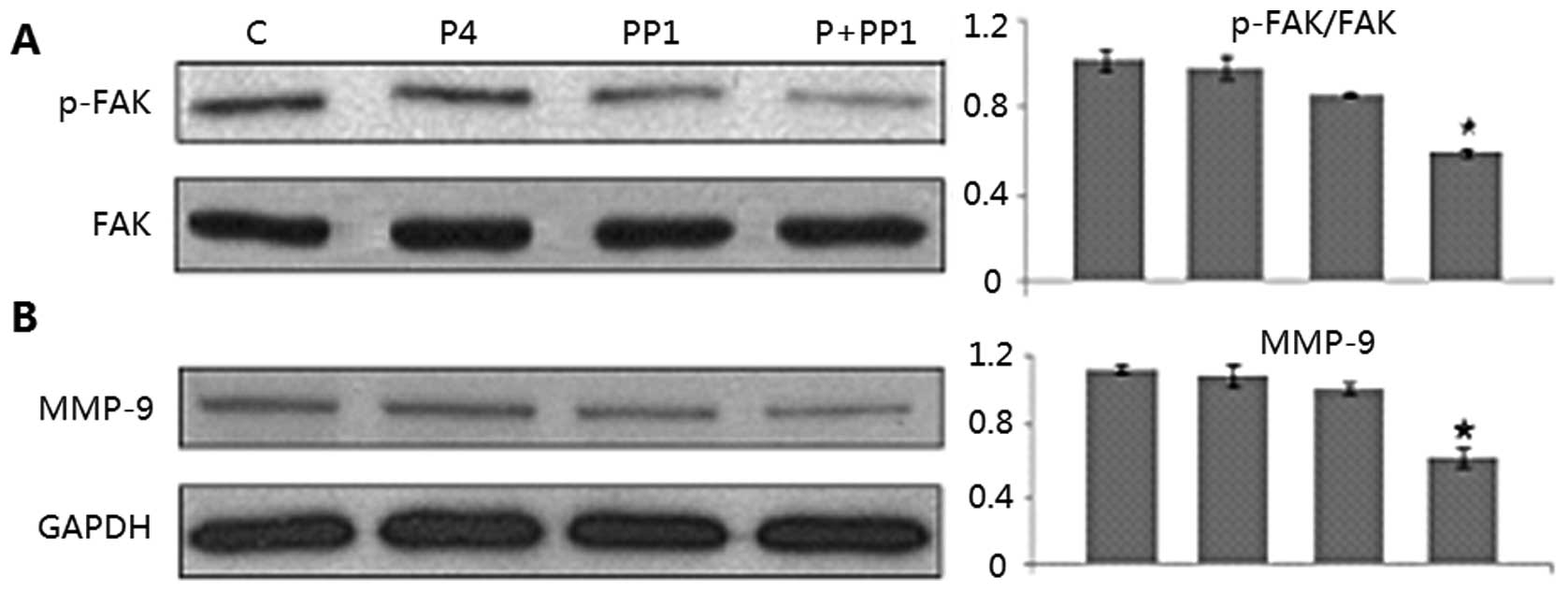
Molecular pathways involved in the
P4+PP1-induced cell migration inhibition of A549 cells
Based on the results of the cell migration assays, a
synergistic effect of P4 and PP1 on cell migration and invasion of
A549 cells was suggested. Moreover, P4 has been reported to signal
via Src family kinases for the formation of focal adhesion complex
via focal adhesion kinase (FAK, a key component for tumor
metastasis) phosphorylation at Tyr (397). To confirm the molecular
mechanisms underlying this action, we evaluated the phosphorylation
of FAK using western blot assay. It was found that the level of
phosphor-FAK in A549 cells was significantly inhibited by P4+PP1
treatment (54.2 vs. 100%, P=0.01), but not by P4 or PP1 treatment
alone (97.3, 88.9 vs. 100%, all P-values >0.05). We also
investigated the effect of P4 and/or PP1 on the expression of other
selected cancer metastasis relevant proteins such as MMP-9. The
expression levels of MMP-9 (58.3 vs. 100%, P=0.01) were markedly
reduced by the P4+PP1 combination treatment in A549 cells, but
again not by P4 or PP1 individual treatments as compared to
controls (all P-values >0.05) (Fig.
6).
Discussion
mPRα expression and positioning in the
lung adenocarcinoma A549 cell line
In 1985, Beattie et al reported that hormone
receptors were significantly higher in lung tissue than in normal
tissue and proposed that lung cancer is a hormone-dependent tumor
(23). Several studies have shown
that estrogen and progesterone receptors are expressed in normal
lung tissue, lung cancer and paraneoplastic tissues, as well as in
lung cancer cell lines. However, nPR expression rates in lung
cancer fluctuated in various laboratories, its association between
pathological characteristics, clinical stage, lymph node metastasis
or clinical characteristics did not reach a unanimous conclusion
(24–26). mPRα is a new protein found in cancer
and has not yet been widely investigated in lung cancer. Thomas
et al also detected cell surface positioning of mPRα using
antibodies against the extracellular amino-terminal of mPRα in
mPRα-transfected MB231 cells; subsequently, flow cytometry and
immunofluorescence staining showed the same cell membrane location
(27). Our study is the first to
detect mPRα mRNA and protein expressions in the lung adenocarcinoma
A549 cells. Using the progestin binding experiment, we detected
clear fluorescent signals from P4-BSA-FITC in the cell membrane of
A549 cells, but not in the cytoplasm and nucleus. This suggests
that the mPRα is positioned on the cell membrane of A549 cells and
that mPRα is combined with progesterone.
In order to further explore the binding of mPRα with
progesterone, we co-cultured P4-BSA-FITC with excessive free P4 in
A549 cells, considering that excessive free P4 can replace
P4-BSA-FITC to bind with P4. At this time, no green fluorescent
signals were detected in the cell membrane, cytoplasm or nucleus of
A549 cells, suggesting that the original cell binding P4-BSA-FITC
was competitively replaced by excessive free P4 and eluted. These
results confirmed the specific binding of mPRα with P4-BSA-FITC in
the membrane. Therefore, the fluorescence signal in the cell
membrane is neither non-specific fluorescence, nor is it due to
binding with BSA; it is the specific binding of P4 with mPRα
occurring in the cell surface. Pang and Thomas used an mPRα small
interfering RNA (mPRα siRNA) to interfere with mPRα expression in
MB468 cells and found reduced radioactive [3H]-labeled
progesterone binding to the cell membrane by a laser microscope
(28). These studies further
support the specific binding of mPRα and P4 in the plasma membrane,
instead of combined with progesterone nuclear receptor or other
steroid hormone receptors, which is consistent with our study.
However, Krietsch et al reported that recombinant mPRα of
several vertebrate species was not present on the plasma membranes
of transfected cells, but was localized in the endoplasmic
reticulum, which is inconsistent with our findings (29). Foster et al later confirmed
the membrane localization of mPRα using immuno-gold transmission
electron microscopy. Stimulation of M11 cells with P4 (100 nM)
resulted in internalization of mPRα from the plasma membrane to the
cytoplasm in 10 min and subsequent partial translocation back to
the cell surface in 20 min using RT-PCR, immunofluorescence and
immuno-gold electron transmission microscopy (30). This internalization and recycling of
mPRα may provide an explanation of mPRα inside the cell plasma. The
accurate positioning of mPRα requires further exploration.
mPRα and its role in P4+PP1-enhanced cell
migration and invasion inhibition
Cell migration plays a vital role in several
biological processes, such as immune response, wound healing,
embryogenesis and cancer metastasis. During cell migration, a
series of cellular events, such as substrate sensing, adhesion
formation, dynamic cytoskeletal reorganization and cell membrane
rearrangements, occur in a strictly regulated manner (31). Limited knowledge, however, is
available on the mechanisms by which P4 modulates cancer cell
migration and invasion. Our study demonstrated that P4 inhibited,
rather than enhanced, cell migration of mPRα-positive A549 cells
slightly, and, notably, when co-incubating with P4+PP1, cell
migration was inhibited significantly. Since PP1 treatment alone
inhibited cell migration only at a moderate level which was
comparable to P4, we hypothesized that combination treatment with
both can synchronize the molecular signal magnitude and vigorously
inhibit cell migration in vitro. Similarly, synchronizing
results were also obtained from assays in which cell invasion was
inhibited by P4+PP1, but not by P4 or PP1 treatment alone.
The role of P4 in cancer development has attracted
substantial interest, but the mechanisms remain controversial. It
is believed that the physiological action of P4 is mediated through
either nuclear PR or membrane-bound receptors. A549 cells were
reported to be nPR positive in previous research (32). To exclude the possibility that nPR
could mediate the effect of P4 in A549 cells, we applied a
pre-incubation of MIF, a P4 antagonist. This procedure had no
impact on the effects of P4 and/or PP1 on A549 cell migration,
indicating nPR is not involved in inhibiting cell migration.
Additionally, 2 h prior to P4 treatment, the addition of
specificity mPRα antibodies eliminated inhibition of A549 cell
migration; adding a further specific binding mPRα antibody blocking
peptide could restore P4+PP1’s synergetic inhibition of the
migration of lung adenocarcinoma A549 cells. PGRMC1 is required for
some aspects of P4 signaling in estrogen receptor-negative breast
tumors through an unidentified mechanism (33,34).
In this study, we demonstrated that the cell migration patterns
were not affected by incubating A549 cells with P4 and/or PP1 in
the presence or absence of anti-PGRMC1 antibody, which suggested
that PGRMC1 and its signaling pathways are not involved in the role
of P4 and PP1 on cell migration. These data indicated that mPRα
served as a key mediator of P4 in regulating migration of A549 lung
cancer cells.
Molecular pathways involved in
P4+PP1/mPRα signaling
Src has been reported to be a starting point for a
number of biochemical cascades and exerts a profound effect on
focal adhesion systems and cytoskeleton reorganization, thereby
influencing cancer cell migration and invasion as well as other
tumor progression-related events such as EMT (35). PP1 has been identified as a powerful
inhibitor of Src family members, which binds to the ATP domain of
Src and does not affect Src expression (36,37).
PP1 inhibits Src-mediated tumor cell migration, invasion and
metastasis. However, Src inhibitor alone does not appear to achieve
therapeutic effects in clinical trials. Finn et al
demonstrated that the Src family inhibitor dasatinib alone showed
only limited anticancer effects in a phase II clinical trial of
triple negative breast cancer patients (38); combination with chemotherapy
medication may improve the therapeutic effects (39). Our results also suggested that
P4+PP1 combined treatment significantly inhibited the migration and
invasion of lung adenocarcinoma A549 cells; P4 or PP1 alone showed
limited effects. Therefore, we consider that P4 and PP1 collaborate
and synergistically expand molecular signaling cascade in the
inhibition of lung adenocarcinoma A549 cell migration and
invasion.
FAK is a downstream signaling component to control
cell motility. Through multifaceted and diverse molecular
connections, FAK regulates cell movement by influencing the
cytoskeleton, structures of cell adhesion sites and membrane
protrusions (40). FAK is highly
expressed in lung cancer. All metastatic tumor tissues were found
with high levels of FAK expression (41,42).
Further research is being performed to investigate PR-mediated P4
impact on FAK. Hsu et al found PR can mediate P4’s rapid
nongenomic effect to inhibit Src/FAK phosphorylation in mice aortic
smooth muscle cells, strengthen RhoA degradation, thus inhibiting
the migration of smooth muscle cells (43). In this A549 cell model, which is
depleted of nPR but has expressed mPRα, P4 and PP1 treatment alone
affected the status of FAK minimally; however, combination
treatment with both induced significant dephosphorylation of FAK.
These results indicated that individual treatment with P4 or PP1
might not be powerful enough to inhibit cell migration and
inactivate FAK, and combination treatment with both is essential
for FAK inactivation.
Matrix metalloproteinases (MMPs) have been
implicated in several aspects of tumor progression, such as
invasion through basement membrane and interstitial matrices,
angiogenesis and tumor cell growth. Expression of MMP-9 is strictly
monitored in physical conditions. In malignant cells, the balance
is destroyed which results in elevated expression of MMP-9 and
enhanced metastatic abilities. Studies showed that MMP-9 is highly
expressed in NSCLC and serves as a pivotal step in the process of
cancer metastasis (44–46). Hung et al found that Skp2
stable transfectants from A549 cells exhibited increased migratory
and invasive abilities by upregulated expression of MMP-9 (47). By contrast, Xu et al
demonstrated that osthole suppresses migration and invasion of A549
human lung cancer cells through inhibition of MMP-9 (48). In the present study, we found that
in response to P4 or PP1 treatment alone, the expression of MMP-9
in A549 cells exhibited minimal changes; however, treatment with
both induced significant reduction in MMP-9 expression, a similar
pattern as that of FAK dephosphorylation, supporting this
pro-metastatic protein as the downstream effector of P4→Src/FAK
pathway mediated by mPRα.
In conclusion, our study first detected the
expression and positioning of mPRα in the cell membrane of lung
adenocarcinoma A549 cells. We also identified an mPRα-mediated
pathway that involves Src/FAK and a downstream cell signaling
component MMP-9. This cascade of molecular pathways can be
inhibited by the concurrent use of P4 and PP1. Our results provide
insight into the combinational use of an Src inhibitor and hormone
agonist for the treatment of lung cancer and metastatic lung
adenocarcinoma in particular.
References
|
1
|
Yang M and Pyo MY: Molecular epidemiology
of lung cancer in female passive smokers. J Environ Sci Health C
Environ Carcinog Ecotoxicol Rev. 23:75–97. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stabile LP, Davis AL, Gubish CT, et al:
Human non-small cell lung tumors and cells derived from normal lung
express both estrogen receptor alpha and beta and show biological
responses to estrogen. Cancer Res. 62:2141–2150. 2002.PubMed/NCBI
|
|
3
|
Kuiper GG, Enmark E, Pelto-Huikko M,
Nilsson S and Gustafsson JA: Cloning of a novel receptor expressed
in rat prostate and ovary. Proc Natl Acad Sci USA. 93:5925–5930.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clarke CL and Sutherland RL: Progestin
regulation of cellular proliferation. Endocr Rev. 11:266–301. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee WS, Harder JA, Yoshizumi M, Lee ME and
Haber E: Progesterone inhibits arterial smooth muscle cell
proliferation. Nat Med. 3:1005–1008. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Groshong SD, Owen GI, Grimison B, et al:
Biphasic regulation of breast cancer cell growth by progesterone:
role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1).
Mol Endocrinol. 11:1593–1607. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Musgrove EA, Hunter LJ, Lee CS, Swarbrick
A, Hui R and Sutherland RL: Cyclin D1 overexpression induces
progestin resistance in T-47D breast cancer cells despite p27(Kip1)
association with cyclin E-Cdk2. J Biol Chem. 276:47675–47683. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Santen RJ, Manni A, Harvey H and Redmond
C: Endocrine treatment of breast cancer in women. Endocr Rev.
11:221–265. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thigpen JT, Brady MF, Alvarez RD, et al:
Oral medroxyprogesterone acetate in the treatment of advanced or
recurrent endometrial carcinoma: a dose-response study by the
Gynecologic Oncology Group. J Clin Oncol. 17:1736–1744.
1999.PubMed/NCBI
|
|
10
|
Lee YT: Better prognosis of many cancers
in female: a phenomenon not explained by study of steroid
receptors. J Surg Oncol. 25:255–262. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Su JM, Hsu HK, Chang H, et al: Expression
of estrogen and progesterone receptors in non-small-cell lung
cancer: immunohistochemical study. Anticancer Res. 16:3803–3806.
1996.PubMed/NCBI
|
|
12
|
Check JH, Sansoucie L, Chern J and Dix E:
Mifepristone treatment improves length and quality of survival of
mice with spontaneous lung cancer. Anticancer Res. 30:119–122.
2010.PubMed/NCBI
|
|
13
|
Marquez-Garban DC, Mah V, Alavi M, et al:
Progesterone and estrogen receptor expression and activity in human
non-small cell lung cancer. Steroids. 76:910–920. 2011.PubMed/NCBI
|
|
14
|
Tiutiunova AM, Chirvina ED, Mironenko TV,
Kartashov SZ and Luntovskaia VA: Hormonal balance in women with
lung cancer and its changes after combined treatment. Vopr Onkol.
32:26–30. 1986.(In Russian).
|
|
15
|
Zhu Y, Rice CD, Pang Y, Pace M and Thomas
P: Cloning, expression, and characterization of a membrane
progestin receptor and evidence it is an intermediary in meiotic
maturation of fish oocytes. Proc Natl Acad Sci USA. 100:2231–2236.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu Y, Bond J and Thomas P:
Identification, classification, and partial characterization of
genes in humans and other vertebrates homologous to a fish membrane
progestin receptor. Proc Natl Acad Sci USA. 100:2237–2242. 2003.
View Article : Google Scholar
|
|
17
|
Thomas P: Characteristics of membrane
progestin receptor alpha (mPRalpha) and progesterone membrane
receptor component 1 (PGMRC1) and their roles in mediating rapid
progestin actions. Front Neuroendocrinol. 29:292–312. 2008.
View Article : Google Scholar
|
|
18
|
Zuo L, Li W and You S: Progesterone
reverses the mesenchymal phenotypes of basal phenotype breast
cancer cells via a membrane progesterone receptor mediated pathway.
Breast Cancer Res. 12:R342010. View
Article : Google Scholar
|
|
19
|
Dosiou C, Hamilton AE, Pang Y, et al:
Expression of membrane progesterone receptors on human T
lymphocytes and Jurkat cells and activation of G-proteins by
progesterone. J Endocrinol. 196:67–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ciavatta VT, Kim M, Wong P, et al: Retinal
expression of Fgf2 in RCS rats with subretinal microphotodiode
array. Invest Ophthalmol Vis Sci. 50:4523–4530. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dhanesuan N, Sharp JA, Blick T, Price JT
and Thompson EW: Doxycycline-inducible expression of
SPARC/Osteonectin/BM40 in MDA-MB-231 human breast cancer cells
results in growth inhibition. Breast Cancer Res Treat. 75:73–85.
2002. View Article : Google Scholar
|
|
22
|
Desai B, Ma T and Chellaiah MA:
Invadopodia and matrix degradation, a new property of prostate
cancer cells during migration and invasion. J Biol Chem.
283:13856–13866. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Beattie CW, Hansen NW and Thomas PA:
Steroid receptors in human lung cancer. Cancer Res. 45:4206–4214.
1985.PubMed/NCBI
|
|
24
|
Wu CT, Chang YL, Shih JY and Lee YC: The
significance of estrogen receptor beta in 301 surgically treated
non-small cell lung cancers. J Thorac Cardiovasc Surg. 130:979–986.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fasco MJ, Hurteau GJ and Spivack SD:
Gender-dependent expression of alpha and beta estrogen receptors in
human nontumor and tumor lung tissue. Mol Cell Endocrinol.
188:125–140. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaiser U, Hofmann J, Schilli M, et al:
Steroid-hormone receptors in cell lines and tumor biopsies of human
lung cancer. Int J Cancer. 67:357–364. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thomas P, Pang Y, Dong J, et al: Steroid
and G protein binding characteristics of the seatrout and human
progestin membrane receptor alpha subtypes and their evolutionary
origins. Endocrinology. 148:705–718. 2007. View Article : Google Scholar
|
|
28
|
Pang Y and Thomas P: Progesterone signals
through membrane progesterone receptors (mPRs) in MDA-MB-468 and
mPR-transfected MDA-MB-231 breast cancer cells which lack
full-length and N-terminally truncated isoforms of the nuclear
progesterone receptor. Steroids. 76:921–928. 2011.
|
|
29
|
Krietsch T, Fernandes MS, Kero J, et al:
Human homologs of the putative G protein-coupled membrane progestin
receptors (mPRalpha, beta, and gamma) localize to the endoplasmic
reticulum and are not activated by progesterone. Mol Endocrinol.
20:3146–3164. 2006. View Article : Google Scholar
|
|
30
|
Foster H, Reynolds A, Stenbeck G, Dong J,
Thomas P and Karteris E: Internalisation of membrane progesterone
receptor-alpha after treatment with progesterone: Potential
involvement of a clathrin-dependent pathway. Mol Med Rep. 3:27–35.
2010.
|
|
31
|
Schafer C, Born S, Mohl C, et al: The key
feature for early migratory processes: dependence of adhesion,
actin bundles, force generation and transmission on filopodia. Cell
Adh Migr. 4:215–225. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ishibashi H, Suzuki T, Suzuki S, et al:
Progesterone receptor in non-small cell lung cancer - a potent
prognostic factor and possible target for endocrine therapy. Cancer
Res. 65:6450–6458. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu Y, Hanna RN, Schaaf MJ, Spaink HP and
Thomas P: Candidates for membrane progestin receptors - past
approaches and future challenges. Comp Biochem Physiol C Toxicol
Pharmacol. 148:381–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Losel RM, Besong D, Peluso JJ and Wehling
M: Progesterone receptor membrane component 1 - many tasks for a
versatile protein. Steroids. 73:929–934. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guarino M: Src signaling in cancer
invasion. J Cell Physiol. 223:14–26. 2010.
|
|
36
|
Hanke JH, Gardner JP, Dow RL, et al:
Discovery of a novel, potent, and Src family-selective tyrosine
kinase inhibitor. Study of Lck- and FynT-dependent T cell
activation. J Biol Chem. 271:695–701. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Karni R, Mizrachi S, Reiss-Sklan E, Gazit
A, Livnah O and Levitzki A: The pp60c-Src inhibitor PP1 is
non-competitive against ATP. FEBS Lett. 537:47–52. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Finn RS, Bengala C, Ibrahim N, et al:
Dasatinib as a single agent in triple-negative breast cancer:
results of an open-label phase 2 study. Clin Cancer Res.
17:6905–6913. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tryfonopoulos D, Walsh S, Collins DM, et
al: Src: a potential target for the treatment of triple-negative
breast cancer. Ann Oncol. 22:2234–2240. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mitra SK, Hanson DA and Schlaepfer DD:
Focal adhesion kinase: in command and control of cell motility. Nat
Rev Mol Cell Biol. 6:56–68. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Owens LV, Xu L, Craven RJ, et al:
Overexpression of the focal adhesion kinase (p125FAK) in invasive
human tumors. Cancer Res. 55:2752–2755. 1995.PubMed/NCBI
|
|
42
|
Siesser PM and Hanks SK: The signaling and
biological implications of FAK overexpression in cancer. Clin
Cancer Res. 12:3233–3237. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hsu SP, Chen TH, Chou YP, et al:
Extra-nuclear activation of progesterone receptor in regulating
arterial smooth muscle cell migration. Atherosclerosis. 217:83–89.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Herbst RS, Yano S, Kuniyasu H, et al:
Differential expression of E-cadherin and type IV collagenase genes
predicts outcome in patients with stage I non-small cell lung
carcinoma. Clin Cancer Res. 6:790–797. 2000.PubMed/NCBI
|
|
45
|
Martinella-Catusse C, Nawrocki B, Gilles
C, Birembaut P and Polette M: Matrix-metalloproteinases in
bronchopulmonary carcinomas. Histol Histopathol. 14:839–843.
1999.PubMed/NCBI
|
|
46
|
Aleshin A and Finn RS: SRC: a century of
science brought to the clinic. Neoplasia. 12:599–607.
2010.PubMed/NCBI
|
|
47
|
Hung WC, Tseng WL, Shiea J and Chang HC:
Skp2 overexpression increases the expression of MMP-2 and MMP-9 and
invasion of lung cancer cells. Cancer Lett. 288:156–161. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu XM, Zhang Y, Qu D, Feng XW, Chen Y and
Zhao L: Osthole suppresses migration and invasion of A549 human
lung cancer cells through inhibition of matrix metalloproteinase-2
and matrix metallopeptidase-9 in vitro. Mol Med Report.
6:1018–1022. 2012.PubMed/NCBI
|