Introduction
Microtubules are a principle component of the
cytoskeleton and play a key role in numerous biological functions
including cell division and organelle transport. The pivotal role
of tubulin in both the formation of the mitotic spindle and
chromosomal separation promoted the surge in the development of
both natural and synthetic microtubule targeting agents (MTAs). One
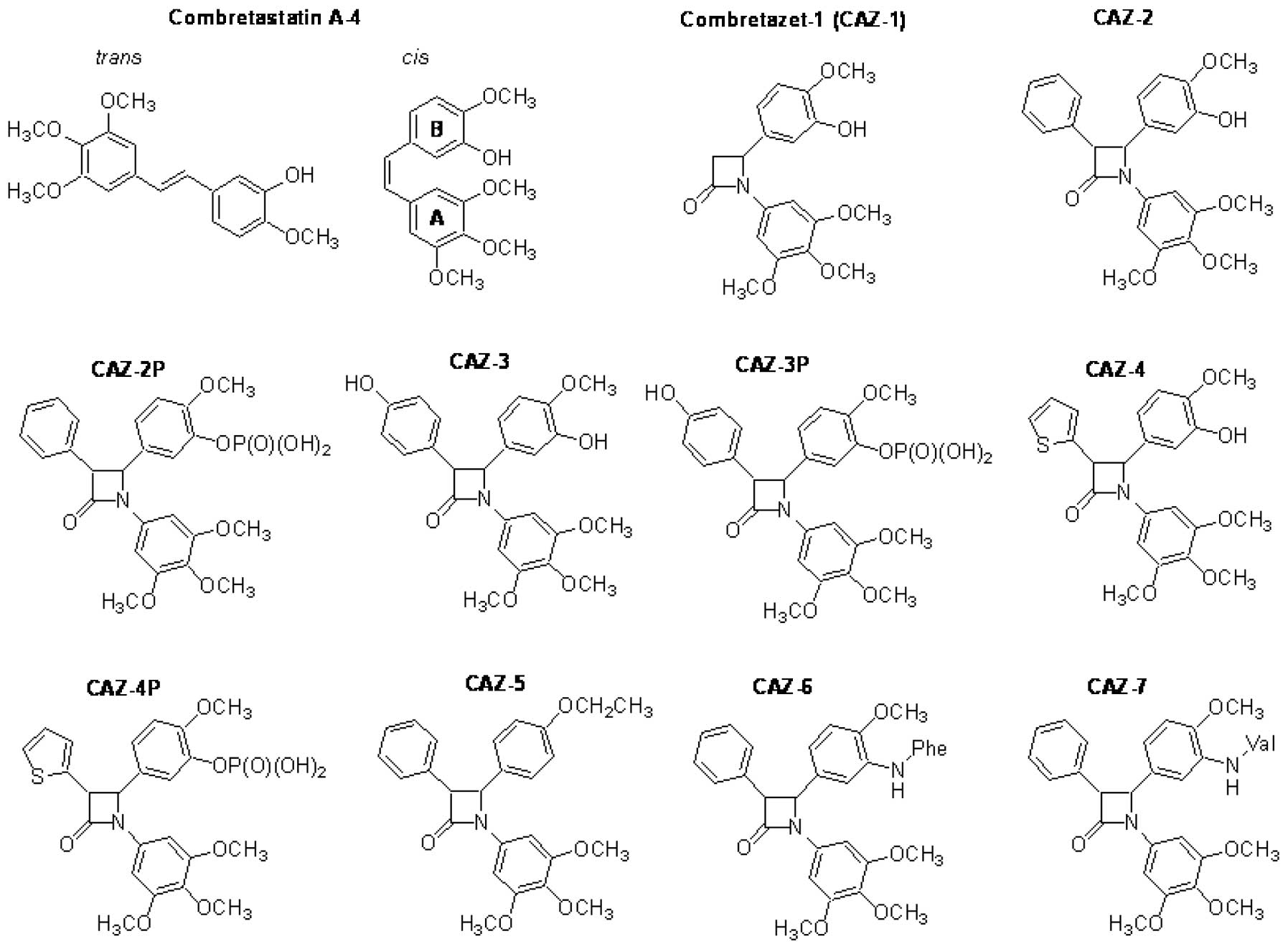
such naturally occurring drug, combretastatin A-4 (CA-4; Fig. 1) was originally described by Pettit
et al(1). The structure of
CA-4 proved readily amenable to chemical manipulation to improve
the stability, solubility and therapeutic index of this class of
MTAs. Over the past two decades a vast array of synthetic CA-4
analogues were designed and synthesised with many surpassing the
stability, solubility and therapeutic efficacy of the parent
compound (2). The clinical success
of the synthetic prodrug of CA-4, combretastatin-A4 phosphate
(CA-4P) in the treatment of anaplastic thyroid carcinoma
(www.clinicaltrials.gov) (3) has maintained an active interest in the
chemical manipulation of CA-4 with the view to further enhancing
the therapeutic efficacy of this lead compound. Furthermore, amino
acid containing prodrugs of CA-4 are also undergoing clinical
evaluation including AVE8062 (www.clinicaltrials.gov) (4). Structural modifications of CA-4 can be
divided into three areas, those involving the manipulation of
either ring A, ring B or those involving the substitution of the
double bond (ethylene bridge structure) connecting both rings
(Fig. 1). These A and B substituted
aromatic rings fit into the A and B pockets of the colchicine
binding site on tubulin. Data collated from numerous structural
activity relationship (SAR) studies confirm that a non-planar
cis conformation is essential for the tubulin binding
properties of CA-4. Furthermore, the majority of studies suggest
that the 3,4,5-trimethoxy-substituted aromatic A-ring should be
conserved to maintain maximum anticancer activity. However,
contrary to this, a recent study conducted by Beale et
al(5) showed that substitution
of the larger meta-methoxy groups of triazole CA-4 derivatives with
smaller halogen atoms yielded more potent CA-4/CA-1 analogues.
Several independent studies have demonstrated that the therapeutic
activity of CA-4 can also be enhanced by the strategic modification
of ring B (6). Apart from
strategies to improve the potency of CA-4 other main areas of
research focused on methods to overcome the solubility issues of
CA-4 and also to prevent the undesired conversion into the inactive
trans isomer (Fig. 1).
Modification of the phenolic group on ring B forming either a
phosphate or an amino acid ester was demonstrated to be an
effective method of improving the solubility of CA-4 whilst
retaining optimum biological activity. Double bond isomerisation
can be prevented by the strategic inclusion of various types of
heterocyclic rings in place of the usual ethylene bridge structure
of CA-4 (7).
In recent years our group has designed and
synthesised an extensive series of azetidinone (β-lactam) CA-4
analogues with a view to overcoming double bond isomerisation by
substituting the ethylene bridge structure for a
1,4-diaryl-2-azetidinone ring. The rigid β-lactam ring scaffold
allows a similar spatial arrangement between the two aromatic rings
as observed in the non-planar cis-conformation of CA-4 while
permanently preventing the undesired conversion to the inactive
trans-configuration (8).
Further studies demonstrated that the inclusion of an aromatic ring
at position 3 of the β-lactam significantly improved the potency of
the series. Hence, a β-lactam substituted at position 3 with a
phenolic ring soon became the core structure for future designs
(9). Solubility issues of the
CA-4-azetidinone analogues were addressed by esterification of the
3′OH group of ring B with phosphates and amino acids (unpublished
data).
However, despite the significant advances made in
recent years in terms of improving the solubility and stability of
CA-4, the lack of therapeutic efficacy as a single agent and the
emergence of resistance to CA-4 has somewhat hindered the clinical
and commercial success of this compound. We recently reported that
CA-432, a lead combretastatin-azetidinone hybrid was 10-fold more
potent than CA-4 in CA-4 refractory HT-29 cells, suggesting a
possible functional advantage of the ethylene bridge-azetidinone
substitution (10). In this study,
we screened selected combretastatin-azetidinone hybrids (hereafter
referred to as combretazets) to further characterise the structure
activity relationship of these compounds in the CA-4 resistant
HT-29 cells. The stability and therapeutic potential of a lead
compound combretazet-3 (CAZ-3) as a single agent in the murine
CT-26 colon cancer model was evaluated.
Materials and methods
Compounds
CA-4 and bafilomycin A1 were purchased from
Sigma-Aldrich (Poole, Dorset, UK). 1,4-Diaryl-2-azetidinone
analogues were synthesised as previously described by Carr et
al(8) CAZ-1, CAZ-2 and CAZ-3
(9), CAZ-4 (7), CAZ-5 (11), CAZ-2P, CAZ-3P, CAZ-4P, CAZ-6 and
CAZ-7 (unpublished data). All general reagents unless stated
otherwise were purchased from Sigma. Bafilomycin A1 was dissolved
in DMSO. CA-4 and all analogues were prepared as a 10-mM stock in
ethanol and stored at −20°C.
Cell culture
CT-26 cells are a chemically
(N-nitroso-N-methylurethane) induced, undifferentiated murine colon
carcinoma fibroblast cell line originating from BALB/c mice. HT-29
and Caco-2 cells originate from a human adenocarcinoma of the colon
and were originally obtained from the European Collection of Cell
Cultures. All cells were grown in DMEM Glutamax media. CT-26 and
HT-29 media were supplemented with 10% foetal bovine serum (FBS)
and Caco-2 were cultivated with 20% FBS. Both CT-26 and Caco-2
media were supplemented with 1% non-essential amino acids (NEAA).
All media contained 100 U/ml penicillin and 100 μg/ml streptomycin.
Cells were maintained at 37°C in 5% CO2 in a humidified
incubator. Cell culture materials were supplied from Gibco,
Invitrogen Corp. (Grand Island, NY, USA). All cells were
sub-cultured 3 times/week by trypsinisation.
Alamar blue cell viability assay
Cell proliferation was analysed using the Alamar
Blue assay (Invitrogen Corp.) according to the manufacturer’s
instructions. Cells were seeded at a density of 5×103
cells/well (CT-26) or 1×104 cells/well (Caco-2, HT-29)
in triplicate in 96-well plates. After 24 h, cells were then
treated with either medium alone, vehicle [1% ethanol (v/v)] or
with serial dilutions of CA-4 or combretazets. After 72 h, Alamar
Blue [10% (v/v)] was added to each well and plates were incubated
for 3–5 h at 37°C in the dark. Fluorescence was read using a
96-well fluorimeter with excitation at 530 nm and emission at 590
nm. Results were expressed as percentage viability relative to
vehicle control (100%). Dose response curves were plotted and
IC50 values (concentration of drug resulting in 50%
reduction in cell survival) were obtained using the commercial
software package Prism (GraphPad Software, Inc., La Jolla, CA,
USA). Experiments were performed in triplicate on at least three
separate occasions.
Cell cycle detection
After treatment, cells were collected then
centrifuged at 800 × g for 10 min and fixed with 70% ethanol
overnight at −20°C. The ethanol was removed by centrifugation at
800 × g for 10 min. The cells were then stained in PBS containing
0.5 mg/ml RNase A and 0.15 mg/ml propidium iodide and then
incubated for 30 min in the dark at 37°C. Cell cycle distribution
was analysed by flow cytometry at 488 nm using the FACSCalibur flow
cytometer (Becton-Dickinson, San Jose, CA). All data were recorded
and analysed using the CellQuest Software (Becton-Dickinson).
Plasma and pH stability studies
Peripheral blood was collected from healthy donors
with informed consent and was made anonymous prior to use. The
plasma was separated by Ficoll-gradient and diluted (1:9) with PBS
pH 7.4 and warmed to 37°C. The pH stability study was carried out
in PBS pH 3.0. Test compounds (1.5 mg/ml) were dissolved in
acetonitrile at time t=0. Plasma solution containing test compound
(250 μl) was added to 2% (w/v) ZnSO4 solution in
acetonitrile: water (1:1) (500 μl). Aliquots were taken at the
specified time intervals, vortexed, centrifuged for 3 min at 9,500
× g before injection onto an HPLC column (Varian Pursuit XRs C18
reverse phase 250×4.6 mm chromatography column) to determine the
stability using a Waters 2487 Dual Wavelength Absorbance Detector,
a Waters 1525 Binary HPLC Pump, a Waters In-Line Degasser AF and a
Waters 717 plus Autosampler. Samples were detected using a
wavelength of 254 nm. All samples were analysed using acetonitrile
(60%):water (40%) with 0.1% (v/v) trifluoroacetic acid over 10 min
and a flow rate of 1 ml/min to evaluate the percentage decline in
the predetermined peak area for each compound. The retention times
for CA-4 and CAZ-3 were 5.9 and 3.7 min, respectively. The
percentage recovery was calculated using the following formula
[(plasma or pH 3.0 peak area/mean aqueous peak area) × 100].
Human microsomal stability study
Microsomal stability was determined using pooled
human liver microsomes (The UK Human Tissue Bank, Leicester, UK).
Ethical approval was obtained from south Cheshire Local Research
Ethics Committee (Chester, UK). Test compound (3 μM) together with
microsome protein (0.5 mg/ml), 1 mM NADPH in 0.1 M phosphate buffer
pH 7.4 was incubated for 0, 5, 15, 30 and 45 min. The negative
control did not contain NADPH. The samples were quenched with
methanol and the protein was precipitated by centrifugation for 20
min at 1,100 × g at 4°C. Supernatants were then analysed by LC/MS.
The In peak area ratio (compound peak area/internal standard peak
area) was plotted against time and the slope of the line determined
to give the elimination rate constant [K = (−1) (slope)]. The half
life (t1/2) and the in vitro intrinsic
clearance (CLint μl/min/mg protein) were calculated by
the following equations; t1/2 = 0.693/K;
CLint = V (0.693)/t1/2 where V,
incubation volume in μl/mg microsomal protein.
Quantification of AVOs with acridine
orange staining using flow cytometry
Autophagy is characterised by the formation and
promotion of acidic vesicular organelles (AVOs). The formation of
acidic compartments was quantified by flow cytometric analysis of
acridine orange stained cells (10). Acridine orange stains the cytoplasm
green and the nucleus a dim red, whereas acidic compartments
fluoresce bright red. The intensity of the red fluorescence is
proportional to the amount of acidity. Following treatment, cells
were stained with acridine orange 1 μg/ml for 15 min at 37°C.
Bafilomycin A1 (5 nM) was dissolved in DMSO and added to the cells
45 min prior to the addition of acridine orange. Cells were then
trypsinised and collected in phenol-red free medium. Green (510–530
nm) and red (650 nm) fluorescence emission from 104
cells illuminated with blue (488 nm) excitation light was measured
with a CyAn ADP Flow Cytometry Analyzer (Beckman Coulter, Nyon,
Switzerland). The red:green fluorescence ratio for individual cells
was calculated using FlowJo software (Tree Star, Inc., San Carlos,
CA).
In vivo studies
Tumour growth was initiated by subcutaneous
injection of a CT-26 cell suspension (106 cells) into
the right flank of 6–8 week old female Balb/c mice. The experiments
were conducted on Day 7 when tumours reached a maximum diameter
range of 3.6–6.3 mm. Mice were randomly divided into two groups of
five. The treatment group received a single intraperitoneal (i.p.)
injection of 40 mg/kg CAZ-3 dissolved in ethanol:cremophore:PBS
[10%:10%:80% (v/v)] and the control group received one i.p.
injection of vehicle only. Tumour growth was measured every second
day with a sterile vernier callipers. The long (L) and short (S)
axes were recorded and tumour volume (V) was calculated using the
following equation V = (S2xL)/2. Mice were culled by
CO2 asphyxiation at the experimental end point. Ethical
approval was obtained from the Research Ethical Approval Committee,
Trinity College Dublin. The study was performed under the license
number: B100/4275 granted by Department of Health and Children,
Hawkins House, Dublin 2, Ireland.
Results
The effects of CA-4 and selected
combretazets on the viability of colon cancer-derived cell
lines
The synthetic combretazets were designed and
synthesised with a view to improve the stability, therapeutic
efficacy and aqueous solubility of the parent compound CA-4, hence
many in the series contained a phenolic group, phosphate ester or
an amino group (Fig. 1). All
combretazets analysed were effective in the nanomolar range in
drug-sensitive CT-26 and Caco-2 cells and were more potent than
CA-4 in CA-4 refractory HT-29 cells (Table I). Compound CAZ-2 is identical to
CA-4 with the exception of the azetidinone-ethylene bridge
substitution and demonstrated an 8-fold increase in activity in
HT-29 cells confirming a functional advantage of the ethylene
bridge-azetidinone substitution in overcoming combretastatin
resistance. Compounds containing a B-ring meta-hydroxy group
(CAZ-1, CAZ-2 and CAZ-4) or a phosphate (CAZ-2P, CAZ-3P and CAZ-4P)
conjugate were the least active of the series in the combretastatin
refractory HT-29 cells. Deletion (CAZ-5) or substitution of the
B-ring meta-hydroxy group with an amine conjugated amino acid
(CAZ-6 and CAZ-7) significantly increased activity of the series
with IC50 values in the nanomolar range in
combretastatin refractory HT-29 cells. However, CAZ-3 with a B-ring
meta-hydroxy group was the exception to this observation. CAZ-3 was
more potent than CA-4 in all three adenocarcinoma-derived cell
lines tested. Hence, CAZ-3 was selected for further biological
analysis.
 | Table IEvaluation of CA-4 and selected
combretazets in adenocarcinoma-derived colon cancer cells. |
Table I
Evaluation of CA-4 and selected
combretazets in adenocarcinoma-derived colon cancer cells.
| CT-26 | Caco-2 | HT-29 |
|---|
|
|
|
|
|---|
| Compound | IC50
(nM) | RI | IC50
(nM) | RI | IC50
(nM) | RI |
|---|
| CA-4 | 5.7 | 1.0 | 41.2 | 1.0 | 9020 | 1.0 |
| CAZ-1 | 11.8 | −2.1 | 236.0 | −5.7 | 1011 | +8.1 |
| CAZ-2 | 13.46 | −2.4 | 113.6 | −2.8 | 909 | +9 (11) |
| CAZ-3 | 4.25 | +1.4 | 15.3 | +5.6 | 50 | +180.0 |
| CAZ-4 | 165.0 | −28.9 | 28.3 | +1.5 | 3510 | +2.6 |
| CAZ-5 | 136.9 | −24.1 | ND | ND | 30 | +300.0 |
| CAZ-6 | 118.0 | −20.7 | 120.7 | −2.9 | 70 | +128.9 |
| CAZ-7 | 245.3 | −43.0 | 704.2 | −17.1 | 260 | +34.7 |
| CAZ-2P | 75.17 | −13.2 | 96.7 | −2.4 | 440 | +20.5 |
| CAZ-3P | 8.0 | −1.4 | 109.3 | −2.7 | 119 | +7.6 |
| CAZ-4P | 9.7 | −1.7 | ND | ND | 4670 | +1.93 |
The effects of CA-4 and selected
combretazets on the cell cycle in CT-26 cells
CT-26 cells were selected for further analysis given
that all combretazets were effective in the nanomolar range in this
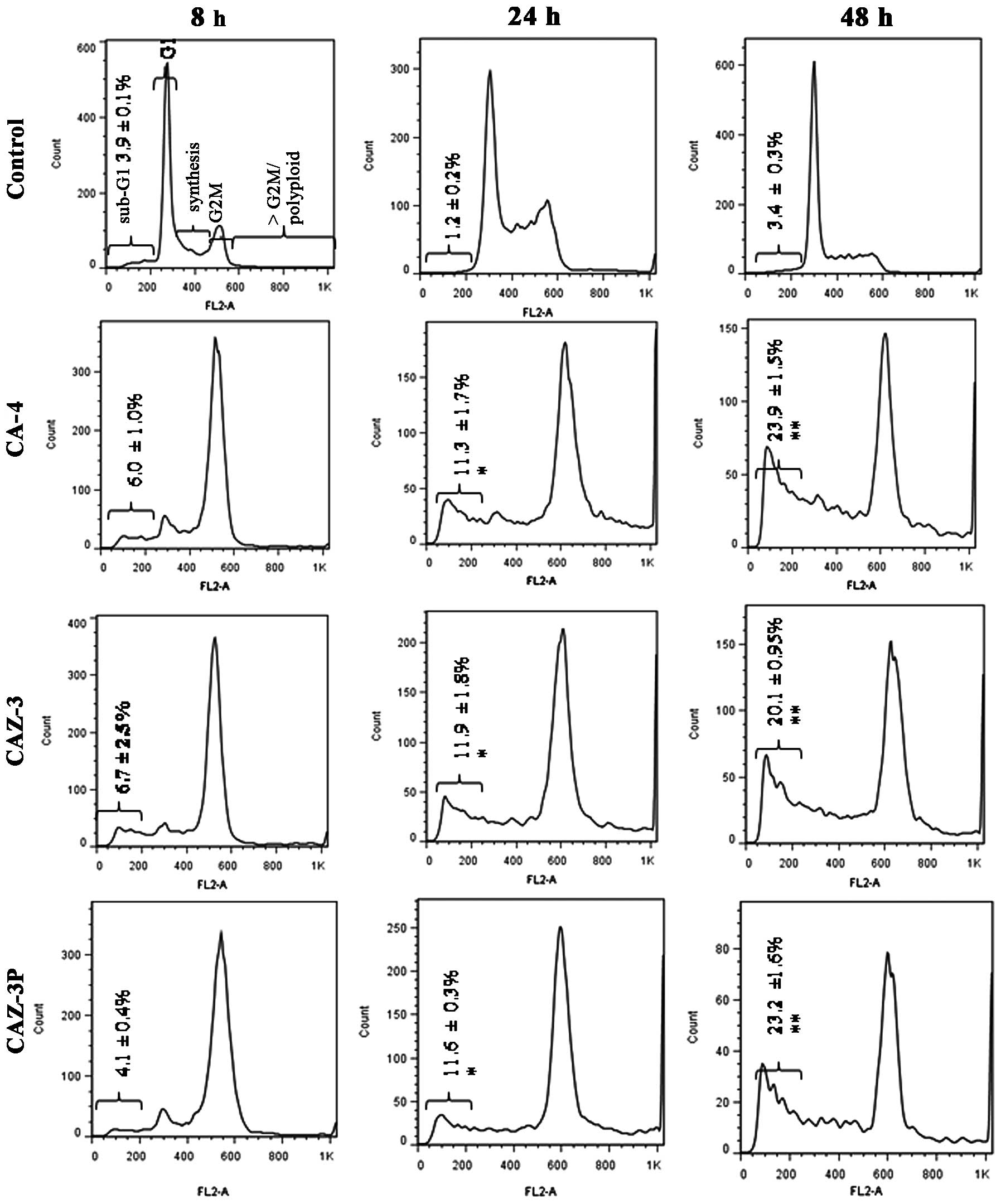
cell line. The effects of CA-4, CAZ-3 and its corresponding prodrug
CAZ-3P on the cell cycle and cell death were assessed by flow
cytometric analysis of propidium iodide stained CT-26 cells. The
percentage of cell death was estimated by the quantification of the
pre-G1 peak. As shown in Fig. 2, all compounds tested produced an
early G2M cell cycle arrest at 8 h followed by a
significant time-dependent increase in cell death.
Induction of autophagy by CAZ-3
Our recent findings demonstrated that CA-4 and CAZ-2
(CA-432) induced autophagy in adenocarcinoma-derived colon cells as
confirmed by acridine orange staining of vesicle formation,
electron microscopy and increased expression of LC3-II (10). Hence, the effect of CAZ-3 on
autophagic vesicle formation was evaluated by flow cytometric
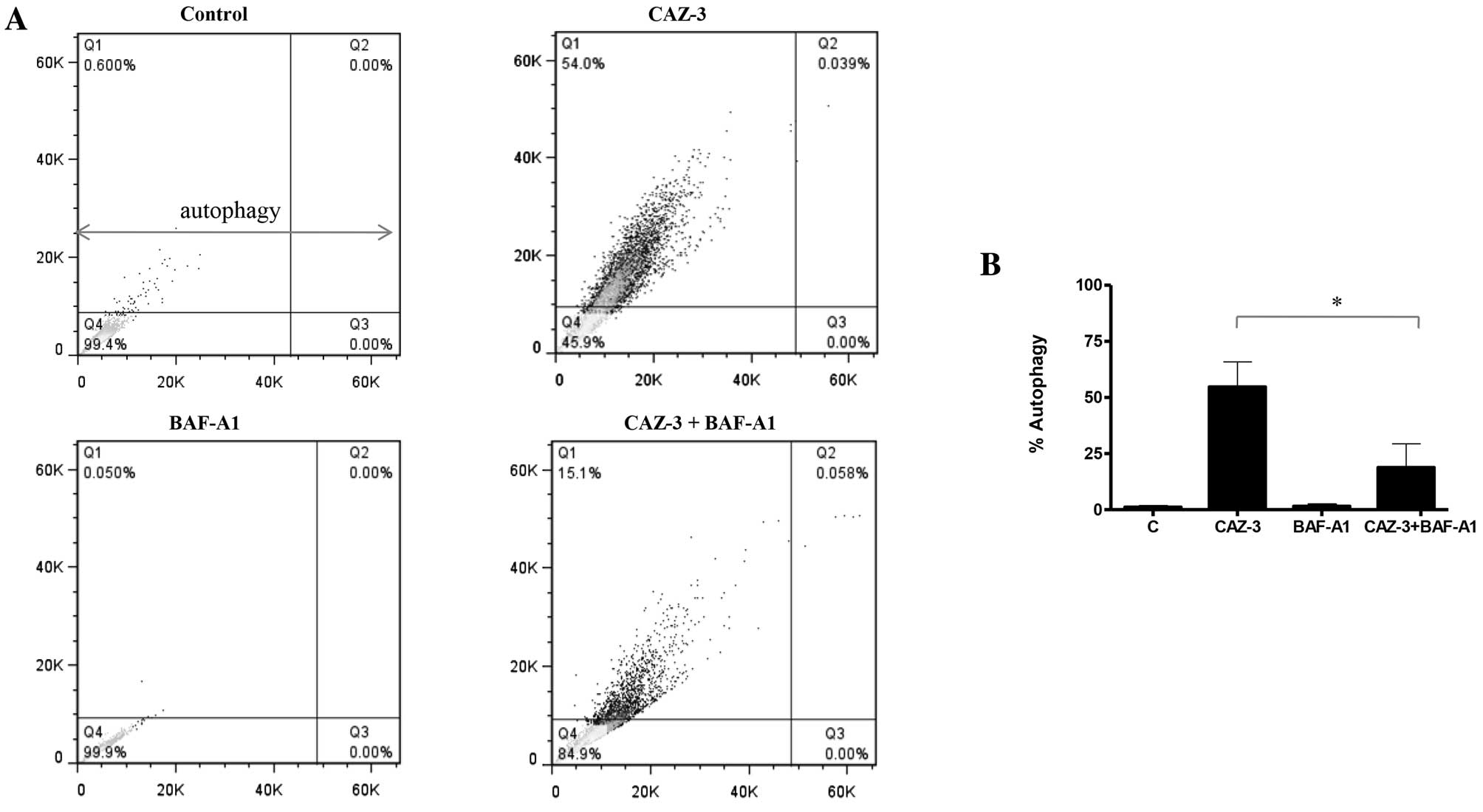
analysis of acridine orange stained cells. Fig. 3 indicates that like other
combretastatins CAZ-3 also induced autophagy in adenocarcinoma
cells. Furthermore, CAZ-3 also induced autophagy in HT-29 and
Caco-2 adenocarcinoma-derived colon cancer cells (data not shown).
Numerous studies have demonstrated a dependence of the
acidification of cellular organelles on the vacuolar H+
ATPase using the specific inhibitor bafilomycin A1. Similarly,
pretreatment of CT-26 cells with bafilomycin A1 significantly
inhibited CAZ-3 induced autophagy (Fig.
3).
CAZ-3 is more stable than CA-4 in both
human plasma and microsomes
The stability of CA-4 and CAZ-3 in acidic media and
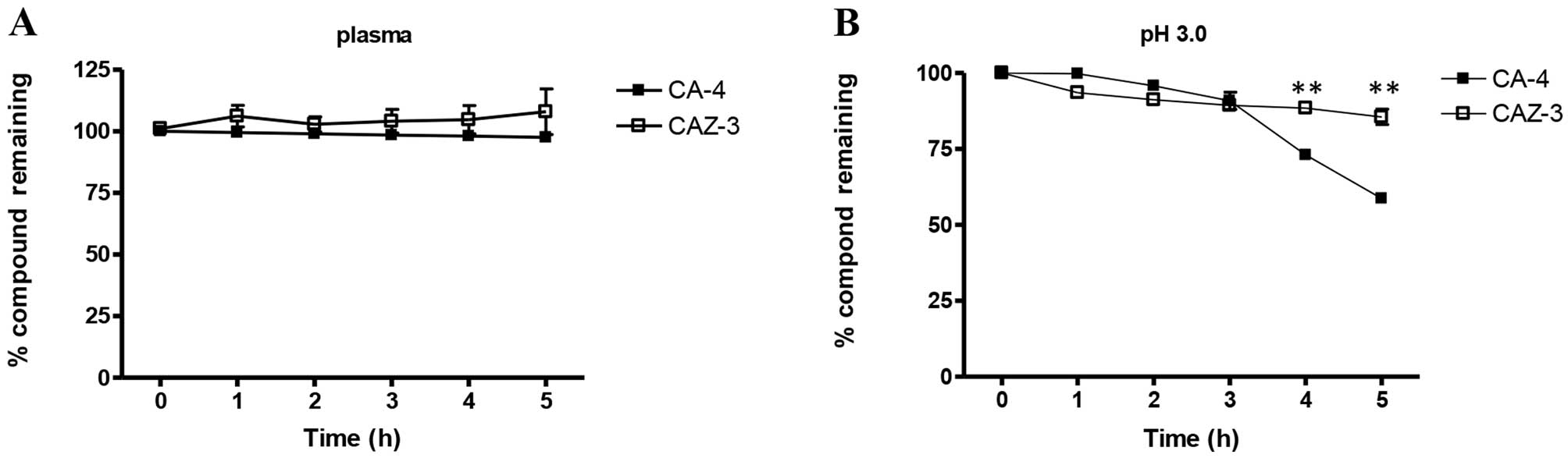
in human plasma was next determined by HPLC. Both compounds were
stable in plasma at physiological pH 7.3 for up to 5 h (Fig. 4A) and remained stable up to 24 h
(data not shown). Under acidic conditions (pH 3.0) CAZ-3 is more
stable than CA-4 (Fig. 4B). These
findings are in agreement with other studies demonstrating
instability of CA-4 in acidic media (12). Microsome stability was determined
using pooled human liver microsomes (Table II). The β-lactam bridge improved
the metabolic stability of CAZ-3 as compared with CA-4 by doubling
the in vitro clearance time.
| Table IICalculation of intrinsic clearance
values (CLint) for CA-4 and CAZ-3 in human
microsomes. |
Table II
Calculation of intrinsic clearance
values (CLint) for CA-4 and CAZ-3 in human
microsomes.
| Compound | CLint
(μl/min/mg protein) |
t1/2 (min) |
|---|
| CA-4 | 157.0±18.9 | 8.83 |
| CAZ-3 | 76.7±21.4 | 18.1 |
CAZ-3 significantly inhibited the growth
of CT-26 cells grafted to mice
To study the effects of CAZ-3 on tumour growth we
selected the CT-26 murine model of colon carcinoma, a model
frequently used to test the efficacy of CA-4 and its synthetic
analogues (13). Furthermore,
experimental models involving xenografts of human tumours in a
mouse host may lack some of the critical tumour host interactions.
In the antitumour efficacy experiment mice received a single IP
injection of 40 mg/kg on Day 7 when tumours were on average 5 mm in
diameter. On Day 7 there was no significant difference between
control and CAZ-3 treated groups. By Days 9 (data not shown) and 11
(Table III), CAZ-3 significantly
inhibited tumour growth. Both a rough coat and diarrhoea were
observed in 100% of CAZ-3 treated mice.
| Table IIISuppression of tumour growth by
CAZ-3. |
Table III
Suppression of tumour growth by
CAZ-3.
| Group | Tumour volume (Day
7) | Tumour volume (Day
11) | Mortality |
|---|
| Control | 37.08±8.326 | 263.2±62.13 | 0/5 |
| CAZ-3 | 42.45±11.45 | 77.62±19.24 | 1/5 |
| Single IP 40
mg/kg | NS |
<0.05a | |
Discussion
Several water soluble CA-4 analogues including CA-4P
(ZYBRESTAT), CA-1P (OXi4503) and AC7700 (AVE 8062) are currently
undergoing clinical trials as vascular targeting agents (www.clinicaltrials.gov) (14,15).
However, these compounds all contain the isomerisable olefinic bond
which may hinder the continued clinical success of the compounds.
To date there is no cis-stable CA-4 analogue undergoing
clinical trials and hence there is a demand for pre-clinical data
on potent cis-restricted CA-4 analogues. The combretazets
are a novel class of synthetic combretastatin and azetidinone
(β-lactam) hybrids that function through a combretastatin-like
mechanism. Overall the combretastatins and the combretazets are
structurally and functionally similar. Both classes exhibit a
similar spatial arrangement between the two phenyl A and B rings
but differ in the bridge structure connecting the rings. The
strategic ethylene bridge-azetidinone substitution produced
cis-stable analogues with improved chemical stability and
ease of synthesis. Extensive biochemical analysis demonstrated that
the ethylene bridge-azetidinone substitution did not influence the
biological properties of CA-4 (9,10,16,17).
In more detail, both classes of drugs inhibit the polymerisation of
tubulin inducing a range of cellular responses including;
G2M cell cycle arrest, autophagy, mitotic catastrophe,
caspase-dependent cell death and caspase-independent cell death. In
this report, we demonstrate that further substitutions to the
aromatic ring at position 3 of the azetidinone yielded a superior
compound with enhanced stability and potency against combretastatin
refactory adenocarcinoma-derived cells and tumours without altering
the biological properties of CA-4. The lead compound CAZ-3
inhibited the polymerisation of tubulin (9), induced a G2M cell cycle
arrest and a time-dependent increase in cell death
(sub-G1) in the colon adenocarcinoma-derived CT-26 cells
in a similar manner to its phosphate prodrug counterpart (CAZ-3P)
and CA-4. As recently observed with CA-4 (10), CAZ-3 also induced autophagy in CT-26
adenocarcinoma cells. Autophagy is a highly regulated
self-catabolic process which can facilitate a prolonged cell
survival in spite of adverse stress by generating energy via
lysosomal degradation of cytoplasmic constituents (18). Furthermore, we have previously
demonstrated that manipulation of autophagy can enhance the
therapeutic potential of CAZ-2 (10).
The adenocarcinoma-derived HT-29 cells are
inherently resistant to CA-4. The mechanism of innate resistance
remains undefined. Recent studies rule out multidrug resistance
protein-1 (MRP-1) mediated resistance to CA-4 in HT-29 cells
(12). MRP-1 is a member of the
ATP-binding cassette family of polytopic membrane transporters and
is responsible for conferring resistance to a broad range of
chemotherapeutic drugs (19).
However, the authors demonstrate a role for MRP-1 in mediating
resistance to some oxazole CA-4 derivatives (12). Data obtained from SARs from numerous
independent studies on CA-4 analogues provide some insight into the
possibility of structural modification of CA-4 as a means of
overcoming CA-4 resistance. In this report we demonstrate that a
substitution of the ethylene bridge with a β-lactam ring (CAZ-1)
increased activity in HT-29 cells compared to CA-4 by 8-fold. This
finding would suggest that cis-trans isomerisation is
not solely responsible for CA-4 resistance in HT-29 cells but may
contribute in part to CA-4 resistance in these cells. This
mechanism of resistance may be overcome by synthetic analogues
featuring an ethylene bridge substitution with various types of
heterocyclic rings yielding stable analogues which do not
isomerise. We also report that deletion or substitution of the
B-ring meta-hydroxy group with an amine conjugated amino acid in
conjunction with introduction of a 3-position aromatic ring
significantly enhances the activity of the series by up to 300-fold
in HT-29 cells as compared to CA-4. These findings are in agreement
with a recent report by Schobert et al(12) demonstrating improved activity of
oxazole bridged CA-4 analogues by substitution of the B-ring
phenolic group with H, fluoro or amino groups. Also, substitution
of the ethylene bridge with a sulfone group coupled with the
substitution of B-ring with a 5-amino-6-methoxyquinoline moiety
yielded a novel compound with activity in the low nanomolar range
in HT-29 cells (IC50=16 nM) (20). Ring B 4-ethoxyphenyl 1,5-diaryl
substituted 1,2,3,4-tetrazoles also displayed potent activity in
HT-29 cells (21). Taken together
these findings highlight the potential of B-ring meta-hydroxy group
substitutions or deletions in cis-stable analogues of CA-4
as a means of overcoming innate resistance to CA-4. However, our
lead compound CAZ-3, a cis-restricted CA-4 analogue with a
B-ring meta-hydroxy group demonstrated potent nanomolar activity in
HT-29 cells. Molecular modelling studies highlighted a novel site
of interaction between the para-phenolic 3-position of compound
CAZ-3 with the colchicine binding site of tubulin (9). This unique tubulin binding
characteristic is shared with compound CAZ-5 via the B-ring ethoxy
group but not with CA-4 and the other listed β-lactams (11). The additional hydrophobic contact of
CAZ-3 and CAZ-5 with tubulin may facilitate the observed potent
antiproliferative effects observed in the CA-4 resistant HT-29
cells and offer a novel means of overcoming CA-4 resistance.
Based on promising in vitro data we proceeded
to evaluate the therapeutic efficacy of CAZ-3 in the mouse CT-26
model of colon adenocarcinoma. As a single agent CA-4 failed to
reduce the growth of CT-26 tumours in vivo(13). Here we report that a single
injection of CAZ-3 (40 mg/kg) significantly inhibited the growth of
CT-26 tumours in vivo. Furthermore, CAZ-3 reduced the tumour
levels to 34% of control untreated tumour size. This value is below
the T/C value of 42% which is defined as the minimum level of
activity required by the National Cancer Institute criteria.
However, despite an excellent tumour response rate, a single i.p.
injection of CAZ-3 at 40 mg/kg gave adverse side effects such as
rough coat, loss of appetite and diarrhoea along with a mortality
rate of 1/5. The single maximum tolerated dose for CA-4P was 360
mg/m2 in rats and 100 mg/m2 in dogs (22). Serious diarrhoea has been reported
elsewhere in animals given an i.p. injection of CA-4 at 100 mg/kg
(23). CA-4P (100 mg/kg) had a
mortality rate of 25% in rats (http://arno.unimaas.nl/show.cgi?fid=7247). In 90% of
patients CA-4P is well tolerated at 52 mg/m2(22). Given that CAZ-3 is intrinsically
more stable than CA-4 and has a slower intrinsic clearance time,
CAZ-3 may be more active in vivo as well as in vitro
and thus may require significantly lower dosing rates. Optimising
the dosing schedule and/or administration route may yield more
favourable results.
In conclusion, we have presented preclinical data on
a novel series of cis-stable combretastatin analogues. We
demonstrate that our lead compound CAZ-3 is effective against CA-4
resistant colon cancer-derived cells in vitro and in
vivo. Further studies are warranted to characterise the
metabolites of CAZ-3 and optimise dosing schedules with a view to
improving the therapeutic potential of this novel class of
cis-stable vascular targeting agents.
Acknowledgements
We would like to thank Health Research Board Ireland
for funding the project. We would also like to thank Sally Lee at
Cyprotex Discovery, Ltd., (Macclesfield, UK) for carrying out the
microsomal stability studies.
References
|
1
|
Pettit GR, Singh SB, Hamel E, Lin CM,
Alberts DS and Garcia-Kendall D: Isolation and structure of the
strong cell growth and tubulin inhibitor combretastatin A-4.
Experientia. 45:209–211. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marrelli M, Conforti F, Statti GA, Cachet
X, Michel S, Tillequin F and Menichini F: Biologicalpotential and
structure-activity relationships of most recently developed
vascular disrupting agents: an overview of new derivatives of
natural combretastatin a-4. Curr Med Chem. 18:3035–3081. 2011.
View Article : Google Scholar
|
|
3
|
Dowlati A, Robertson K, Cooney M, Petros
WP, Stratford M, Jesberger J, Rafie N, Overmoyer B, Makkar V,
Stambler B, Taylor A, Waas J, Lewin JS, McCrae KR and Remick SC: A
phase I pharmacokinetic and translational study of the novel
vascular targeting agent combretastatin a-4 phosphate on a
single-dose intravenous schedule in patients with advanced cancer.
Cancer Res. 62:3408–3416. 2002.
|
|
4
|
Delmonte A and Sessa C: AVE8062: a new
combretastatin derivative vascular disrupting agent. Expert Opin
Investig Drugs. 18:1541–1548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beale TM, Myers RM, Shearman JW,
Charnock-Jones SD, Brenton JD, Gergely FV and Ley SV: Antivascular
and anticancer activity of dihalogenated A-ring analogues of
combretastatin A-4. Med Chem Commun. 1:202–208. 2010. View Article : Google Scholar
|
|
6
|
Shan Y, Zhang J, Liu Z, Wang M and Dong Y:
Developments of combretastatin A-4 derivatives as anticancer
agents. Curr Med Chem. 18:523–538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O’Boyle NM, Greene LM, Bergin O, Fichet
JB, McCabe T, Lloyd DG, Zisterer DM and Meegan MJ: Synthesis,
evaluation and structural studies of antiproliferative
tubulin-targeting azetidin-2-ones. Bioorg Med Chem. 19:2306–2325.
2011.PubMed/NCBI
|
|
8
|
Carr M, Greene LM, Knox AJ, Lloyd DG,
Zisterer DM and Meegan MJ: Lead identification of conformationally
restricted beta-lactam type combretastatin analogues: synthesis,
antiproliferative activity and tubulin targeting effects. Eur J Med
Chem. 45:5752–5766. 2010. View Article : Google Scholar
|
|
9
|
O’Boyle NM, Carr M, Greene LM, Bergin O,
Nathwani SM, McCabe T, Lloyd DG, Zisterer DM and Meegan MJ:
Synthesis and evaluation of azetidinone analogues of combretastatin
A-4 as tubulin targeting agents. J Med Chem. 53:8569–8584.
2010.PubMed/NCBI
|
|
10
|
Greene LM, O’Boyle NM, Nolan DP, Meegan MJ
and Zisterer DM: The vascular targeting agent Combretastatin-A4
directly induces autophagy in adenocarcinoma-derived colon cancer
cells. Biochem Pharmacol. 84:612–624. 2012. View Article : Google Scholar
|
|
11
|
O’Boyle NM, Carr M, Greene LM, Keely NO,
Knox AJ, McCabe T, Lloyd DG, Zisterer DM and Meegan MJ: Synthesis,
biochemical and molecular modelling studies of antiproliferative
azetidinones causing microtubule disruption and mitotic
catastrophe. Eur J Med Chem. 46:4595–4607. 2011.PubMed/NCBI
|
|
12
|
Schobert R, Effenberger-Neidnicht K and
Biersack B: Stable combretastatin A-4 analogues with sub-nanomolar
efficacy against chemoresistant HT-29 cells. Int J Clin Pharmacol
Ther. 49:71–72. 2011.PubMed/NCBI
|
|
13
|
Ohsumi K, Nakagawa R, Fukuda Y, Hatanaka
T, Morinaga Y, Nihei Y, Ohishi K, Suga Y, Akiyama Y and Tsuji T:
Novel combretastatin analogues effective against murine solid
tumors: design and structure-activity relationships. J Med Chem.
41:3022–3032. 1998. View Article : Google Scholar
|
|
14
|
Hinnen P and Eskens FA: Vascular
disrupting agents in clinical development. Br J Cancer.
96:1159–1165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Patterson DM, Zweifel M, Middleton MR,
Price PM, Folke LK, Stratford MR, Ross P, Halford S, Peters J,
Balkissoon J, Chaplin DJ, Padhani AR and Rustin GJ: Phase I
clinical and pharmacokinetic evaluation of the vascular-disrupting
agent OXi4503 in patients with advanced solid tumors. Clin Cancer
Res. 18:1415–25. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Greene LM, Nathwani SM, Bright SA, Fayne
D, Croke A, Gagliardi M, McElligott AM, O’Connor L, Carr M, Keely
NO, O’Boyle NM, Carroll P, Sarkadi B, Conneally E, Lloyd DG, Lawler
M, Meegan MJ and Zisterer DM: The vascular targeting agent
combretastatin-A4 and a novel cis-restricted β-lactam analogue,
CA-432, induce apoptosis in human chronic myeloid leukemia cells
and ex vivo patient samples including those displaying multidrug
resistance. J Pharmacol Exp Ther. 335:302–313. 2010.
|
|
17
|
Greene LM, Carr M, Keeley NO, Lawler M,
Meegan MJ and Zisterer DM: BubR1 is required for the mitotic block
induced by combretastatin-A4 and a novel cis-restricted β-lactam
analogue in human cancer cells. Int J Mol Med. 27:715–723.
2011.PubMed/NCBI
|
|
18
|
Rubinsztein DC, Codogno P and Levine B:
Autophagy modulation as a potential therapeutic target for diverse
diseases. Nat Rev Drug Discov. 11:709–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leslie EM, Deeley RG and Cole SP:
Toxicological relevance of the multidrug resistance protein 1, MRP1
(ABCC1) and related transporters. Toxicology. 167:3–23. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee HY, Chang JY, Nien CY, Kuo CC, Shih
KH, Wu CH, Chang CY, Lai WY and Liou JP: 5-Amino-2-aroylquinolines
as highly potent tubulin polymerization inhibitors. Part 2 The
impact of bridging groups at position C-2. J Med Chem.
54:8517–8525. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Romagnoli R, Baraldi PG, Salvador MK,
Preti D, Aghazadeh Tabrizi M, Brancale A, Fu XH, Li J, Zhang SZ,
Hamel E, Bortolozzi R, Basso G and Viola G: Synthesis and
evaluation of 1,5-disubstituted tetrazoles as rigid analogues of
combretastatin A-4 with potent antiproliferative and antitumor
activity. J Med Chem. 55:475–488. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rustin GJ, Galbraith SM, Anderson H,
Stratford M, Folkes LK, Sena L, Gumbrell L and Price PM: Phase I
clinical trial of weekly combretastatin A4 phosphate: clinical and
pharmacokinetic results. J Clin Oncol. 21:2815–2822. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eikesdal HP, Schem BC, Mella O and Dahl O:
The new tubulin-inhibitor combretastatin A-4 enhances thermal
damage in the BT4An rat glioma. Int J Radiat Oncol Biol Phys.
46:645–652. 2000. View Article : Google Scholar : PubMed/NCBI
|