Introduction
All-trans retinoic acid (ATRA) is a member of
the retinoid family which includes substances structurally or
funtionally related to retinol (vitamin A). ATRA exerts a profound
effect on the survival, growth and differentiation of many types of
cells during prenatal and adult life (1). Since 1989, when ATRA was introduced as
a targeted therapy against acute promyelocytic leukemia (APL), it
has been used extensively as an antitumor agent for many types of
tumors (2). Several molecular
mechanisms of ATRA have been identified in the induction of tumors
undergoing cell cycle arrest and apoptosis by activating RAR and
RXR (3–5). ATRA regulates the fate of cells also
by other pathways, including p53 (6–10).
These studies revealed that ATRA induces cell cycle arrest and
apoptosis by regulating the transcriptional activity of p53 and/or
stability of its protein. However, it remains unclear whether p53
take part in ATRA-induced cell death in glioma cells.
Axin is a negative regulator of axis formation in
the development of mouse embryos. Its deficiency leads to axis
duplication (11). Overexpression
of Axin blocked embryo axis formation in Xenopus and caused
apoptosis in transgenic mice and certain cell lines (12,13).
Accumulating data show that Axin emerges as a major scaffold for
many pathways, including Wnt signaling, JNK mitogen-activated
protein kinase signaling and transforming growth factor (TGF) β
signaling (14–16). Notably, a previous study revealed
that Axin participates in ATRA-induced differentiation of embryonic
carcinoma cells (17), suggesting
that Axin plays an important role in retinoid-mediated cell fate.
Furthermore, we previously found that ATRA activates the
transcription of Axin in glioma cell lines (18). Studies have demonstrated that Axin
forms a complex with p53 and induces p53-mediated apoptosis by
stimulating p53 transcriptional activity (19,20).
In the present study, the expression of p53 was
found to be highly upregulated by ATRA treatment in the glioma C6
cell line. Ectopic expression of Axin induced cell cycle arrest and
apoptosis and activated the expression of p53. Furthermore, the
knockdown of Axin blocked ATRA-induced cell cycle arrest and
apoptosis with downregulation of p53. These findings indicate that
ATRA activated the intrinsic upregulation of p53 by a novel
mechanism involved in the activation of Axin.
Materials and methods
Cell cultures and transfection
The rat C6 glioma cell line was cultured in
Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented
with 10% (vol/vol) heat-inactivated fetal bovine serum in a
humidified atmosphere with 5% CO2. The glioma C6 cells
were transfected with pIRES2-EGFP-Axin and pIRES2-EGFP using
Lipofectamine™ 2000 reagent (Invitrogen) according to the
manufacturer’s protocol. After an 8-h transfection, cells were
cultured with DMEM for 24 h.
Growth curve assay
For the growth curve assay (MTT methods), C6 cells
were plated in a 96-well plate at a density of 1×103
cells/well, and incubated in a humidified 5% CO2 at 37°C
and transfected with Axin siRNAs for 8 h. After 24, 48 and 72 h,
methyl thiazolyl tetrazolium (MTT) (20 μl/well) was added to the
cells. After a 4-h incubation, the supernatants were removed, and
150 μl dimethyl sulfoxide (DMSO) was added to each well and swirled
for 10 min to solubilize the crystals. The absorbance was measured
at 490 nm.
Analysis of apoptosis
TUNEL method was used for analysis of apoptosis
using an apoptosis detection kit (Maixin, China) according to the
manufacturer’s protocol. Briefly, cell slices were washed with PBS
and digested with proteinase K for 3 min. Then, slices were labeled
with TdT and DIG-d-UTP for 2 h at 37°C, and blocked with the
blocking reagents at room temperature for 30 min. The
anti-Dig-antibody was applied to the cells and incubated at 37°C
for 30 min. After washing with PBS, slices were incubated with SABC
at 37°C for 1 h, and visualized with DAB, and counterstained with
hematoxylin. The apoptotic cells in five different fields were
counted, and the apoptotic index was calculated as the ratio of
apoptotic cells over total cells.
Flow cytometric analysis
Cells (1×106) were washed twice with PBS
and pelleted at 1,000 × g for 5 min. Cells were trypsinized and
fixed with 70% ethanol and washed with PBS and resuspended in PBS
containing RNAase A and propidium iodide (PI) in the dark for 10
min at 4°C. The DNA contents of the stained nuclei were analyzed on
a Becton-Dickinson FACScan flow cytometer. The distribution of DNA
content categorized cells as being in the G1 and S phases. The
cells with DNA content less than G1 were categorized in pre-G1
(hypodiploid cells) and determined as apoptotic phase cells.
Immunohistochemical staining
The cultured cells were incubated with 5% normal
goat serum (Sigma) and incubated overnight at 4°C with a mouse
monoclonal antibody for p53 (Sigma). Following washing with PBS,
cells were incubated at room temperature with biotinylated goat
anti-mouse/rabbit IgG followed by streptavidin enzyme conjugate
(Zhongshan, China) at room temperature. The reaction product was
visualized by diaminobenzidine tetrahydrochloride (DAB). All
sections were counterstained with hematoxylin. Quantification of
the percentage of immunoreactive cells was determined by capturing
images from random fields.
RNA interference
RNA interference (RNAi) was employed in this study
to inhibit Axin expression and analyze the effect of Axin on
ATRA-inhibited cell growth and the expression of p53. Duplex RNAi
oligos with a two-nucleotide overhang at the end of the sequence
were designed and synthesized at GenePharm Co. The sequence of RNA
interference was as follows (Axin1–2253): sense sequence,
5′-GUAUCGUUGUG GCCUACUATT-3′ and antisense sequence, 5′-UAGUAGGCC
ACAACGAUACTG-3′. The negative control siRNA has no known specific
effects on gene expression: sense sequence,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense sequence,
5′-ACGUGACACGUUCGGAGAATT-3′. C6 cells were transfected with siRNAs
at a final concentration of 100 nM using Lipofectamine 2000 reagent
according to the manufacturer′s instructions. At 8 h
post-transfection, cells were cultured by administration of 2.5 μM
of ATRA for 24 h. The interference efficiency exceeded 60% by
comparison with the negative control.
RT-PCR
Total RNA was isolated using TRIzol (Invitrogen)
according to the manufacturer’s instructions. Single-stranded cDNA
was synthesized from 1 μg of total RNA using the PrimeScript™ RT
reagent kit following the protocol recommended by the manufacturer
(Takara). PCR primers were as follows: Axin forward,
5′-AGGGTCTGGAACAGGGAA-3′ and reverse, 5′-GGATAGCGTGTCAGCATCA-3′;
p53 forward, 5′-GCGTTGCTCTGATGGTGA-3′ and reverse,
5′-CAGCGTGATGATGGTAAGGA-3′; β-actin forward, 5′-TC
ACCCACACTGTGCCCATCTA-3′ and reverse, 5′-CATC
GGAACCGCTCATTGCCGATAG-3′. A template-free negative control was
included in each experiment. The PCR cycle was preceded by an
initial denaturation of 5 min at 94°C followed by a final extension
of 10 min at 72°C. Each PCR regime was followed by 30 cycles at
94°C for 30 sec, 58°C for 30 sec, and 72°C for 45 sec. Band
intensity was quantified by Bandscan software. The gray values were
expressed in relation to that of the control and presented as means
± SD of three independent experiments.
Western blot analysis
For western blot analysis, cells were lysed in 2X
SDS loading buffer. Total extracts were obtained in the
supernatant. Approximately 20 μl of the samples was resolved on 10%
SDS-PAGE, and transferred to PVDF membranes. For western blot
analysis a mouse monoclonal antibody for p53 (Sigma), a mouse
polyclonal antibody for Axin (Santa Cruz Biotechnology) and a
rabbit polyclonal antibody for β-actin (Sigma) were used to detect
the corresponding proteins. Peroxidase-conjugated goat
anti-mouse/rabbit secondary antibody (Sigma) was used, and then the
proteins were detected using an enhanced chemiluminescence reagent
(Pierce). Band intensity was quantified by Bandscan software.
Protein expression was normalized by the quantity of β-actin and
presented as means ± SD of three independent experiments.
Statistical evaluation
Data are reported as means ± SD of three independent
experiments. One-way ANOVA was used to assess statistical
significance between the means. Significant differences were
established at P<0.05.
Results
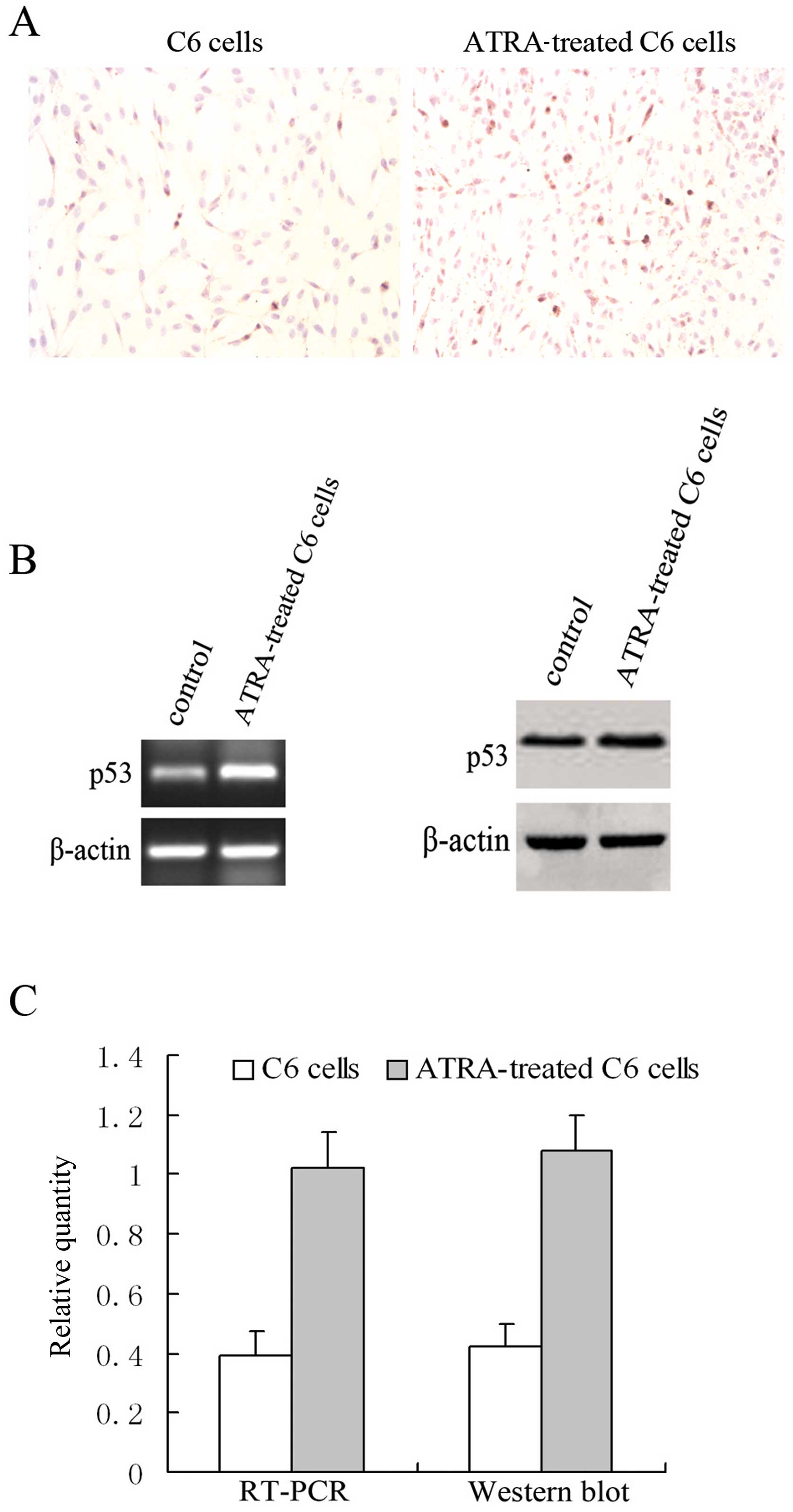
ATRA activates the expression of p53
We previously cultured glioma C6 and U251 cell lines
with or without ATRA for 24 h, and the levels of mRNA and protein
of Axin were examined by RT-PCR and western blotting, respectively.
A marked increase in Axin was observed after ATRA treatment in the
cell lines accompanied with significant growth inhibition of the
glioma cells (18). These results
indicates that ATRA affects tumor cell survival via activation of
Axin. To ascertain whether p53 is involved in ATRA-induced cell
cycle arrest and apoptosis in C6 cells, immunohistochemistry was
employed to investigate the expression of p53 in ATRA-treated C6
cells. ATRA treatment increased the expression of p53 protein from
9.8±1.2 to 19.6±2.2% (Fig. 1A). We
then investigated the levels of p53 mRNA and protein by RT-PCR and
western blotting, respectively (Fig.
1B). The level of total p53 protein was significantly elevated
in correspondence with an increase in its mRNA after ATRA treatment
(Fig. 1C). These data suggest that
the activation of p53 participates in ATRA-induced cell cycle
arrest and apoptosis in C6 cells.
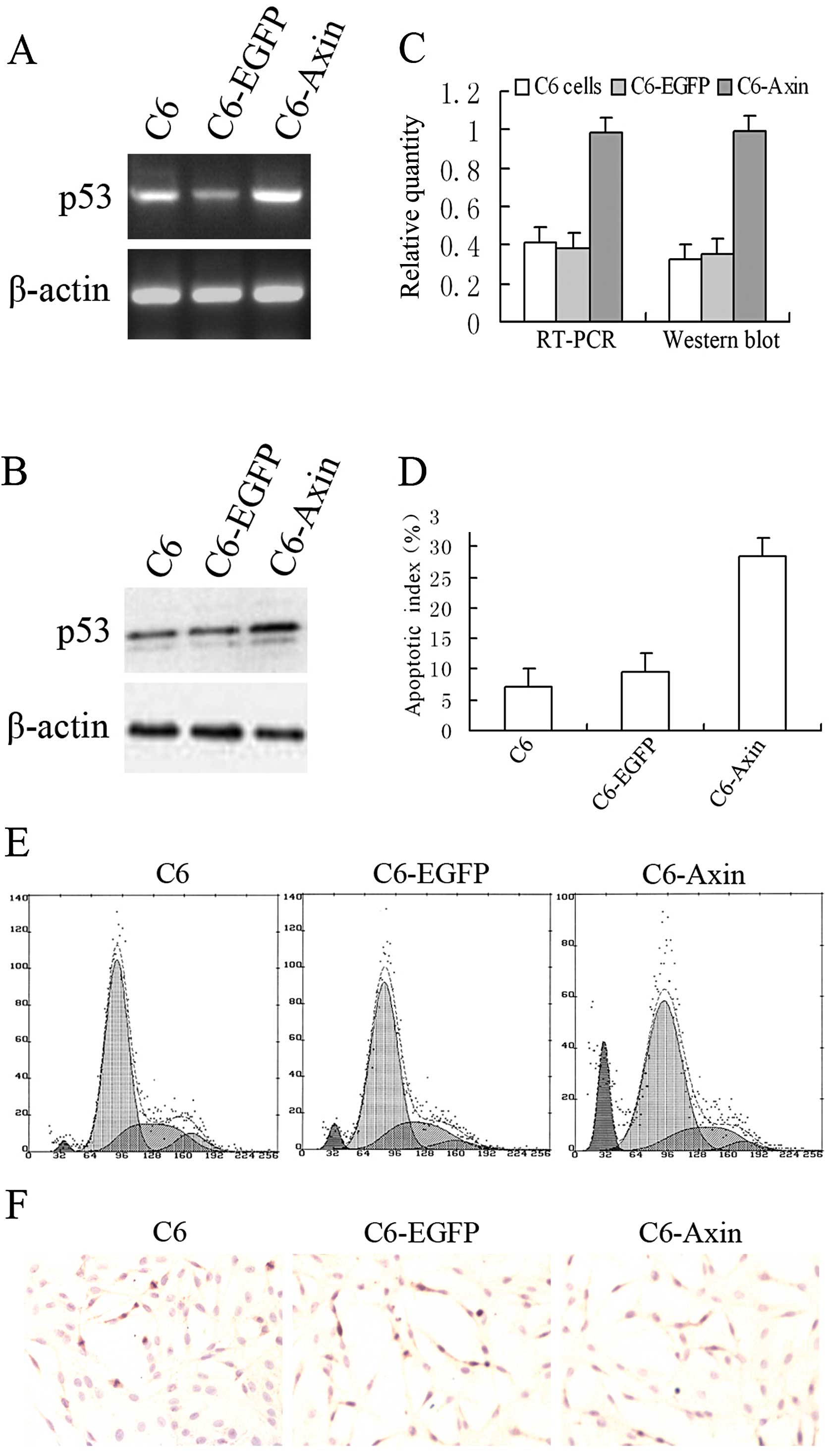
Overexpression of Axin activates the
expression of p53 and induces G1/S phase arrest and apoptosis
To identify whether overexpression of Axin induces
the expression of p53, rat glioma C6 cells were transiently
transfected with rat wild-type Axin. RT-PCR and western blotting
were performed to detect the levels of p53 mRNA and protein
(Fig. 2A and B). The levels of p53
mRNA and its protein in cells transfected with rAxin were increased
compared to the levels in the cells transfected with the empty
vector (Fig. 2C). These results
confirmed that the overexpression of Axin was required for the
activation of p53. To identify whether overexpression of Axin
induces cell cycle arrest and apoptosis, flow cytometry (Fig. 2E) and TUNEL analysis (Fig. 2D and F) were used to determine the
DNA content and apoptotic index, respectively. Flow cytometric
analysis showed that the percentages of C6 cells transiently
transfected with the empty vector in pre-G1, G1 and S phases were
4.8±1.4, 63.6±2.8 and 25.8±2.4%, respectively, while the
percentages of C6 cells transfected with rAxin in pre-G1, G1 and S
phases were 16.0±2.3, 74.8±3.2 and 11.6±1.7%, respectively. The
apoptotic index of C6 cells transfected with Axin (28.4±3.3%) was
significantly increased compared to the C6 cells transfected with
the empty vector (9.6±2.4%) and the C6 cells (6.8±1.8%). These
results revealed that overexpression of Axin inhibits cell
proliferation and induces apoptotic cell death.
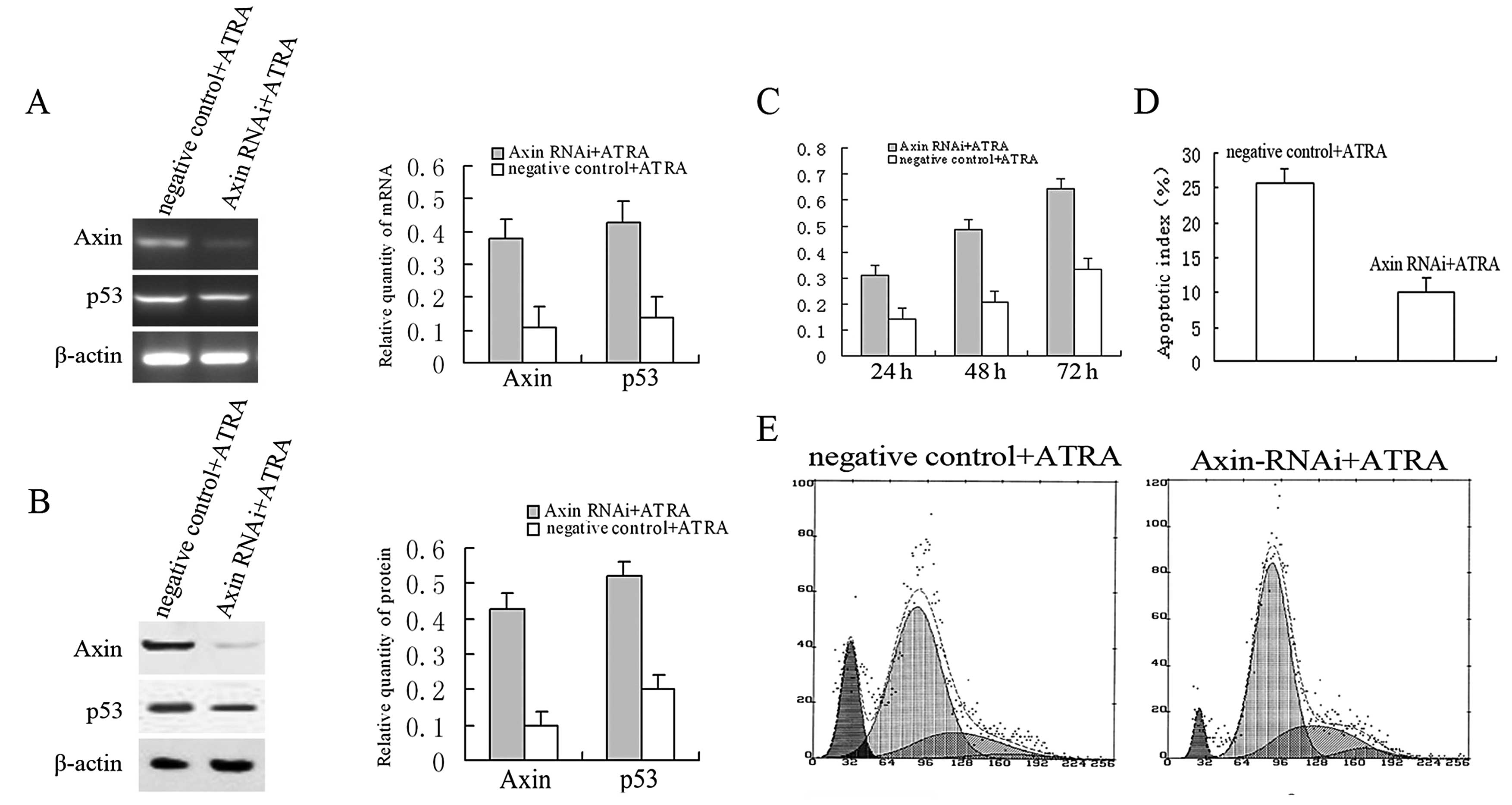
Axin RNAi attenuates ATRA-induced G1/S
arrest and apoptosis accompanied by inhibition of the expression of
p53
We performed RNA interference to further investigate
the role of Axin in ATRA-induced cell cycle arrest and apoptotic
cell death of C6 cells. Axin mRNA (Fig.
3A) was barely detected, and its protein expression (Fig. 3B) was downregulated significantly in
cells transfected with the RNAi sequence against Axin following
ATRA treatment. These data demonstrated that Axin-RNAi effectively
silenced the ATRA-induced increase in Axin. Interestingly,
Axin-RNAi caused only a moderate decrease in p53 mRNA and its
protein following ATRA treatment (Fig.
3A and B). These data revealed that the inactivation of p53 is
associated with the silencing of Axin and implies that there are
other mechanisms participating in ATRA-induced activation of p53.
We next examined the effect of silencing Axin expression on
ATRA-mediated cell growth. As compared with the control, the
percentages of cells in the pre-G1 and G1 phases of the cell cycle
were decreased (12.6±2.3 and 14.9±2.8%, respectively), while the
percentage of cells in the S phase was increased (15.6±2.1%)
(Fig. 3E). Forty-eight hours after
transfection, the viability of the Axin RNAi-transfected C6 cells
decreased by ~60% compared with the control according to the MTT
assay (Fig. 3C). TUNEL analysis
revealed that the apoptotic index of cells with knockdown of Axin
significantly decreased compared to the negative control (from
25.8±2.5 to 10.1±2.3%) (Fig. 3D).
These data reinforced the finding that Axin is an efficient
regulator of ATRA-induced G1/S phase arrest and apoptosis, and
silencing of its expression compromised the susceptibility to the
effects of ATRA.
Discussion
The aggressiveness of cancer cells results from
their proliferative advantage over their normal counterparts and
from their inability to undergo apoptosis. Consistent with previous
research (21–24), our results demonstrated that ATRA
induced cell cycle arrest as well as apoptosis in glioma cells. The
present study for the first time demonstrated that ATRA-mediated
cell cycle arrest and apoptosis were associated with the activation
of p53 resulting from the activation of Axin and this activation
may be a general mechanism contributing to ATRA-inhibited tumor
cell growth.
The tumor suppressor protein p53 is a nuclear
phosphoprotein that can potently regulate the growth of mammalian
cells (25). Activation of p53
results in altered transcription of a wide variety of genes that
are involved in many aspects of cell metabolism, cell cycle
regulation and apoptosis. Moreover, the introduction of a wild-type
p53 expression vector into tumor cells including glioma cells
suggests its ability to halt the cell cycle, trigger apoptosis, and
prevent undue cell proliferation (26–29).
ATRA regulation of the fate of cells by p53 has been reported
(30). These studies revealed that
ATRA induces cell cycle arrest and apoptosis by regulating the
transcriptional activity of p53 and/or stability of its protein.
However, whether p53 participates in ATRA-mediated growth control
in glioma cells remains unclear. Current evidence showed that G1/S
arrest and apoptosis induced by ATRA were associated with the
activity of p53 in the C6 glioma cell line.
Previous research revealed that Axin participates in
ATRA-mediated differentiation of embryonic carcinoma cells
(17) and overexpression of Axin
causes apoptotic cell death in transgenic mice and certain cell
lines (12,13). Recently, our group found that the
activation of Axin was required for ATRA-inhibited cellular
proliferation in glioma cell lines (18). Here, we found that overexpression of
Axin by transiently transfecting C6 cells with rAxin inhibited the
G1/S phase progression in addition to inducing apoptotic cell
death. These results were further confirmed by Axin gene silencing.
Herein, we propose that the activation of Axin, as well as the
activation of p53, inhibited cell growth by cell cycle arrest and
apoptotic cell death in the C6 glioma cell line following ATRA
treatment.
Axin has been reported to stimulate p53 and
p53-dependent transcriptional activity by the formation of a
complex with p53 (19). This raised
an intriguing possibility that ATRA-activated expression of Axin
may inhibit tumor cell growth and activate the expression of p53.
In the present study, we provide evidence that ATRA increases the
expression of p53 through the activation of Axin. As mentioned
above, p53, widely expressed in normal adult tissues, tumors, and
embryos, is ubiquitously involved in growth arrest and/or apoptosis
via a multitude of molecular pathways involving transactivation of
target genes and direct signaling events (31–33).
Base on these results, it is legitimate to suggest that Axin is an
important regulator of ATRA-induce growth arrest and apoptosis of
C6 glioma cells by mediating the p53-dependent cell death pathway,
and this may be a general mechanism contributing to ATRA-mediated
cell cycle arrest and apoptotic cell death.
Acknowledgements
The present study was supported by grants from the
State Key Laboratory of Cancer Biology, China (CBSKL 2005004) and
the Science and Technology Department of Shaanxi Province, China
(2011JM4005). We thank Dr Zhizhong Wang at the Department of
Epidemiology of The Fourth Military Medical University for
assistance with the statistical analysis.
References
|
1
|
Zauli G, Visani G, Bassini A, et al:
Nuclear translocation of protein kinase C-alpha and -beta isoforms
in HL-60 cells induced to differentiate along the granulocytic
lineage by all-trans retinoic acid. Br J Haematol.
93:542–550. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yung WK, Lotan R and Lee P: Modulation of
growth and epidermal growth factor receptor activity by retinoic
acid in human glioma cells. Cancer Res. 49:1014–1019.
1989.PubMed/NCBI
|
|
3
|
Idres N, Benoît G, Flexor MA, Lanotte M
and Chabot GG: Granulocytic differentiation of human NB4
promyelocytic leukemia cells induced by all-trans retinoic
acid metabolites. Cancer Res. 6:700–705. 2001.PubMed/NCBI
|
|
4
|
Launay S, Gianni M, Diomede L, et al:
Enhancement of ATRA-induced cell differentiation by inhibition of
calcium accumulation into the endoplasmic reticulum: cross-talk
between RARα and calcium-dependent signaling. Blood. 101:3220–3228.
2003.PubMed/NCBI
|
|
5
|
Chambon P: A decade of molecular biology
of retinoic acid receptors. FASEB J. 10:940–954. 1996.PubMed/NCBI
|
|
6
|
Mrass P, Rendl M, Mildner M, et al:
Retinoic acid increases the expression of p53 and proapoptotic
caspases and sensitizes keratinocytes to apoptosis: a possible
explanation for tumor preventive action of retinoids. Cancer Res.
64:6542–6548. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Um SJ, Kim EJ, Hwang ES, et al:
Antiproliferative effects of retinoic acid/interferon in cervical
carcinoma cell lines: cooperative growth suppression of IRF-1 and
p53. Int J Cancer. 85:416–423. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng A, Mäntymaa P, Säily M, et al: p53
pathway in apoptosis induced by all-trans-retinoic acid in
acute myeloblastic leukaemia cells. Acta Haematol. 103:135–143.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ronca F, Yee KS and Yu VC: Retinoic acid
confers resistance to p53-dependent apoptosis in SH-SY5Y
neuroblastoma cells by modulating nuclear import of p53. J Biol
Chem. 274:18128–18134. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Curtin JC, Dragnev KH, Sekula D, Christie
AJ, Dmitrovsky E and Spinella MJ: Retinoic acid activates p53 in
human embryonal carcinoma through retinoid receptor-dependent
stimulation of p53 transactivation function. Oncogene.
20:2559–2569. 2001. View Article : Google Scholar
|
|
11
|
Zeng L, Fagotto F, Zhang T, et al: The
mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling
pathway that regulates embryonic axis formation. Cell. 90:181–192.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Satoh S, Daigo Y, Furukawa Y, et al: AXIN1
mutations in hepatocellular carcinomas, and growth suppression in
cancer cells by virus-mediated transfer of AXIN1. Nat Genet.
24:245–250. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsu W, Shakyo R and Costantini F: Impaired
mammary gland and lymphoid development caused by inducible
expression of Axin in transgenic mice. J Cell Biol. 155:1055–1064.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peifer M and Polakis P: Wnt signaling in
oncogenesis and embryogenesis - a look outside the nucleus.
Science. 287:1606–1609. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo W, Ng WW, Jin LH, Ye Z, Han J and Lin
SC: Axin utilizes distinct regions for competitive MEKK1 and MEKK4
binding and JNK activation. J Biol Chem. 278:37451–37458. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu W, Rui H, Wang J, et al: Axin is a
scaffold protein in TGF-β signaling that promotes degradation of
Smad7 by Arkadia. EMBO J. 25:1646–1658. 2006.
|
|
17
|
Lyu J, Costantini F, Jho EH and Joo CK:
Ectopic expression of Axin blocks neuronal differentiation of
embryonic carcinoma P19 cells. J Biol Chem. 278:13487–13495. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu J, Zhang F, Zhao D, et al:
ATRA-inhibited proliferation in glioma cells is associated with
subcellular redistribution of β-catenin via up-regulation of Axin.
J Neurooncol. 87:271–277. 2008.PubMed/NCBI
|
|
19
|
Rui Y, Xu Z, Lin S, et al: Axin stimulates
p53 functions by activation of HIPK2 kinase through multimeric
complex formation. EMBO J. 23:4583–4594. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li QX, Wang X, Wu X, et al: Daxx
cooperates with the Axin/HIPK2/px53 complex to induce cell death.
Cancer Res. 67:66–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karmakar S, Banik NL and Ray SK:
Combination of all-trans retinoic acid and
paclitaxel-induced differentiation and apoptosis in human
glioblastoma U87MG xenografts in nude mice. Cancer. 112:596–607.
2008.
|
|
22
|
Papi A, Bartolini G, Ammar K, Guerra F,
Ferreri AM, Rocchi P and Orlandi M: Inhibitory effects of retinoic
acid and IIF on growth, migration and invasiveness in the U87MG
human glioblastoma cell line. Oncol Rep. 18:1015–1021.
2007.PubMed/NCBI
|
|
23
|
Karmakar S, Banik NL, Patel SJ and Ray SK:
Combination of all-trans retinoic acid and taxol regressed
glioblastoma T98G xenografts in nude mice. Apoptosis. 12:2077–2087.
2007.
|
|
24
|
Zhang R, Banik NL and Ray SK: Combination
of all-trans retinoic acid and interferon-gamma suppressed
PI3K/Akt survival pathway in glioblastoma T98G cells whereas
NF-kappaB survival signaling in glioblastoma U87MG cells for
induction of apoptosis. Neurochem Res. 32:2194–2202. 2007.
|
|
25
|
Vogelstein B and Kinzler KW: p53 function
and dysfunction. Cell. 70:523–526. 1992. View Article : Google Scholar
|
|
26
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Timiryasova TM, Chen B, Haghighat P and
Fodor I: Vaccinia virus-mediated expression of wild-type p53
suppresses glioma cell growth and induces apoptosis. Int J Oncol.
14:845–854. 1999.PubMed/NCBI
|
|
28
|
Cirielli C and Inyaku K:
Adenovirus-mediated wild-type p53 expression induces apoptosis and
suppresses tumorigenesis of experimental intracranial human
malignant glioma. J Neurooncol. 43:99–108. 1999. View Article : Google Scholar
|
|
29
|
Merzak A, Raynal S, Rogers JP, Lawrence D
and Pilkington GJ: Human wild type p53 inhibits cell proliferation
and elicits dramatic morphological changes in human glioma cell
lines in vitro. J Neuro Sci. 127:125–133. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shin DM, Xu XC, Lippman SM, et al:
Accumulation of p53 protein and retinoic acid receptor β in
retinoid chemoprevention. Clin Cancer Res. 3:875–880. 1997.
|
|
31
|
Li PF, Dietz R and von Harsdorf R: p53
regulates mitochondrial membrane potential through reactive oxygen
species and induces cytochrome c independent apoptosis blocked by
Bcl-2. EMBO J. 18:6027–6036. 1999. View Article : Google Scholar
|
|
32
|
Lee SW, Fang L, Igarashi M, Ouchi T, Lu KP
and Aaronson SA: Sustained activation of Ras/Raf/mitogen-activated
protein kinase cascade by the tumor suppressor p53. Proc Natl Acad
Sci USA. 97:8302–8305. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yamaguchi A, Tamatani M, Matsuzaki H, et
al: Akt activation protects hippocampal neurons from apoptosis by
inhibiting transcriptional activity of p53. J Biol Chem.
271:31929–31936. 1996.
|