Introduction
The vascular endothelial growth factor (VEGF) family
modulates several endothelial cell functions, particularly
angiogenesis and lymphangiogenesis, which are involved in solid
tumor growth, progression and metastasis (1). Higher expression of VEGF and levels of
secretion have been demonstrated in human hematological
malignancies. There is accumulating evidence that some leukemia
cells secrete higher levels of VEGF and also express functional
VEGF receptors (VEGFRs), such as VEGFR2 and VEGFR1, which result in
the induction of an autocrine or paracrine loop. Previous findings
demonstrated that high expression of VEGF can promote the
proliferation and colony formation and inhibit the apoptosis of
leukemic cells, which promotes the progression of hematopoietic
tumors not only by stimulating vascular endothelial growth
(2).
In general, the expression of VEGF is regulated by
some intrinsic and extrinsic factors, with hypoxia and hypoglycemia
being the major stimuli. Hypoxia-inducible factor 1α (HIF-1α) has
been demonstrated to be a key angiogenesis-triggering event,
particularly in solid tumors. Previous studies have described that
hypoxia can enhance invasion and metastasis in chronic myeloid
leukemia (CML) (3), acute lymphatic
leukemia (ALL) (4), chronic
lymphatic leukemia (CLL) cells (5),
possibly by activating VEGF.
Our previous study showed that the HIF-1α-related
pathway was independent of the downregulation of VEGF in HL-60
cells induced by all-trans-retinoic acid (ATRA) (6). Furthermore, another study elucidated
that HIF-1α is an independent prognostic factor (7). Thus, a novel VEGF regulation system
may exist in HL-60 cells.
VEGF plays a crucial pathogenic and prognostic role
in acute myeloid leukemia (AML), including enhanced bone marrow
vascularization (8). However, data
concerning the biological function of VEGF and the molecular
mechanism that modulates VEGF expression and secretion in leukemia
cells, such as HL-60 cells, remain to be elucidated (9). The aim of the present study was to
investigate the role of VEGF and the molecular mechanism for the
transcriptional regulation of VEGF. We hypothesized that VEGF could
trigger proliferation and inhibit differentiation for HL-60 cells,
and some upstream key transcriptional factors may contribute to the
modulation of VEGF expression by binding to its promoters.
Materials and methods
Materials
ATRA was purchased from Sigma-Aldrich Co., Ltd., and
was dissolved in ethanol at 1 mM and stored at −80 or −20°C. These
stocks were diluted with the media to the desired concentrations
immediately before the experiment, keeping the final concentration
of ethanol at 0.1%. All experiments were performed under low-light
conditions to minimize retinoid photoisomerization. Anti-human VEGF
antibody (19003-1-AP, ProteinTech Group, Wuhan, P.R. China),
anti-human stat3 antibody (no. 9132, Cell Signaling Technology,
USA), anti-human c-myc antibody (sc-42, Santa Cruz Biotechnology,
Santa Cruz, CA) and anti-human CD11b antibody (11-0113,
eBiosciences, San Diego, CA) were purchased. Cell counting kit-8
(CCK-8) was purchased from Dojindo Laboratories, Kumamoto,
Japan.
Cell culture
The human promyelocytic leukemia cell line HL-60 was
obtained from the Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences. HL-60 cells were cultured in IMDM
medium (Hyclone, Logan, UT) supplemented with 20% fetal calf serum
(FCS, Hyclone), incubated at 37°C, 5% CO2. To prepare
the cell model, HL-60 cells were seeded at 1×105/ml in
25 cm2 culture flasks in culture medium and supplemented
with 1 μM ATRA. Cells were harvested after treatment and used for
the following experiments.
Differentiation assay
Control and HL-60 cells treated with ATRA
(1×106 cells) were washed with PBS containing 1% FCS and
0.01% sodium azide were incubated for 30 min in FCS at 4°C.
Subsequently, FITC-conjugated anti-human CD11b antibody (1 μg/ml)
was added to the cells and incubated at 25°C for 45 min followed by
washing with PBS. The cells were then fixed in 1% paraformaldehyde
and analyzed on Beckman Coulter Epics XL Flow cytometer. Isotypic
rat IgG was also used to check for nonspecific binding.
Proliferation assay
HL-60 cells were treated with ATRA (1 μM). The cell
proliferation was monitored at 24, 48 and 72 h using the CCK-8
assay according to the instructions, and the absorbance was read at
450 nm using a microplate enzyme-linked immunosorbent assay reader
(Thermo Scientific Multiskan MK3).
Western blot assay
Thirty micrograms of protein extracts from HL-60
cells induced by ATRA or not were used for SDS-PAGE, and then
transferred to PVDF membranes. The membranes were blocked with 5%
skimmed milk in TBST for 1 h and incubated with antibodies against
VEGF, stat3 and c-myc at 1:1,000 dilutions overnight at 4°C. After
HRP-conjugated secondary antibody was added, proteins were detected
using an ECL kit.
RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR) assay
HL-60 cells grown to 50% confluence were incubated
either with or without ATRA at appropriate concentrations for three
days. The cells were then lysed and their total RNAs were isolated
using TRIzol (Invitrogen, USA) according to the manufacturer’s
instructions. For reverse transcription, samples were incubated in
an Eppendorf PCR system at 42°C for 30 min, then at 90°C for 5 min
and at 5°C for 5 min. Reverse transcription mixtures were subjected
to PCR with specific primers. Primer sequences are shown in
Table I. Reactions were incubated
at 94°C for 2 min and were then amplified using temperature
parameters of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30
sec. Amplifications were carried out for 35 cycles, followed by a
7-min extension at 72°C. Results were normalized to GAPDH.
 | Table ISequences of primers. |
Table I
Sequences of primers.
| Gene | Target | Species | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| GAPDH | mRNA | Human |
ACATGTTCCAATATGATTCC |
TGGACTCCACGACGTACTCAG |
| c-myc | mRNA | Human |
GAAGTCATCCTGCCAGTCCC |
CTGAAGTTTGCTGCACCGAC |
| stat3 | mRNA | Human |
GGCTGGTAATTTATATAATCCCT |
ACTAAAAGGCCAATACATTACAA |
| VEGF | mRNA | Human |
CGGGAACCAGATCTCTCACC |
AAAATGGCGAATCCAATTCC |
| SP-1 | mRNA | Human |
AAGAAATGACCTTAGGAACATACCC |
CCGTATATGTCTACACACAGATGAC |
| HIF-1α | mRNA | Human |
CCTATGTAGTTGTGGAAGTTTATGC |
ACTAGGCAATTTTGCTAAGAATG |
| cox-2 | mRNA | Human |
TTACAATGCTGACTATGGCTAC |
CTGATGCGTGAAGTGCTG |
| c-jun | mRNA | Human |
ACGACCTTCTATGACGATGCC |
ATGTGCCCGTTGCTGGAC |
| VHL | mRNA | Human |
TACCGAGGTCACCTTTGGC |
GGAGGCATCGCTCTTTCAG |
| P53 | mRNA | Human |
GTCCTGCTTGCTTACCTCGCTTAGT |
ACCTGATTTCCTTACTGCCTCTTGC |
Specific siRNAs and transfection
siRNA oligonucleotides targeting stat3 (the target
mRNA sequences, 5′-CACA TACAGGCTTAAGCTCTA-3′) and c-myc (the target
mRNA sequences, 5′-CTCGGTGCAGCCGTATTTCTA-3′) were designed and
synthesized by Qiagen along with HiPerFect transfection reagent.
For the transfection assay with siRNA, 2×106 cells were
grown to 80% confluence. The original stock of the siRNA was
re-suspended in siRNA suspension H2O provided by the
manufacturer. The resulting suspension was aliquoted in required
amounts for each experiment and stored at −20°C until it was ready
to use. On the day of transfection, aliquots of siRNA suspension
were diluted by culture medium without serum. The siRNA was then
gently introduced into the cells by mixing with the required amount
of HiPerFect transfection reagent at room temperature to allow
formation of transfection complexes. The cells were harvested 48 h
after transfection for analysis. Parallel experiments with
Mm/Hs-MAPK1 and AllStar negative control provided with the kit were
used as positive and negative controls in our experiment. Culture
cells were harvested for western blotting and RT-PCR analysis.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was carried out in a standard manner,
with minor modifications. HL-60 cells were treated with 1%
formaldehyde for 15 min. The cross-linked chromatin was then
prepared and sonicated to an average size of 300–400 bp before
being immunoprecipitated with antibodies specific to c-myc or
control rabbit IgG at 4°C overnight. After reversal of
cross-linking, the immunoprecipitated chromatin was amplified with
polymerase chain reaction (PCR) and the resulting PCR products were
separated by agarose gel electrophoresis.
Statistical analysis
Data are presented as the means ± SE of three or
four experiments. Analysis was performed using a Student’s t-test.
Values of P<0.05 were considered to indicate a statistically
significant difference.
Results
Effect of VEGF on differentiation and
proliferation of HL-60 cells
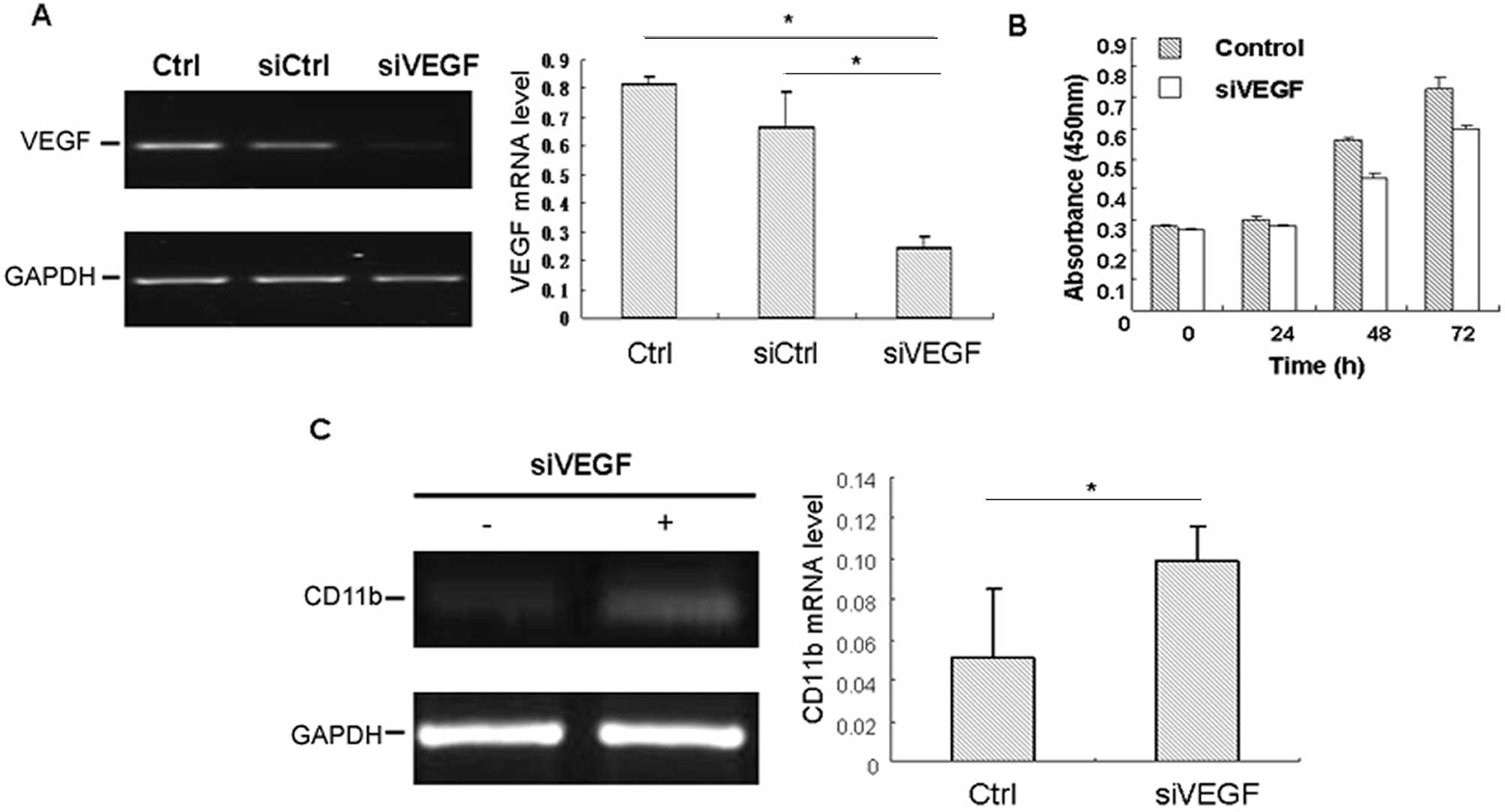
In order to confirm the critical role of VEGF in the
differentiation and proliferation of HL-60 cells, VEGF siRNA
transfection was carried out in HL-60 cells. Treatment with VEGF
siRNA reduced the production of VEGF by 78.2%, as compared with
normal controls (Fig. 1A). In order
to detect the effect of VEGF on the proliferation of HL-60 cells,
the proliferation was detected by CCK-8 assay 48 h after
transfection, and revealed that siRNA knockdown of VEGF could
inhibit the proliferation of HL-60 cells as compared with that of
control cells (Fig. 1B).
Furthermore, the role of VEGF in the regulation of differentiation
of HL-60 cells was also investigated, and the results indicated
that the CD11b expression and positive percent was higher in the
VEGF-siRNA group than in that of various controls (Fig. 1C), which suggested that HL-60 cells
presented evidence of differentiation by silencing the VEGF
expression. These data indicated that VEGF may play an important
role in the progression of proliferation and differentiation in
HL-60 cells.
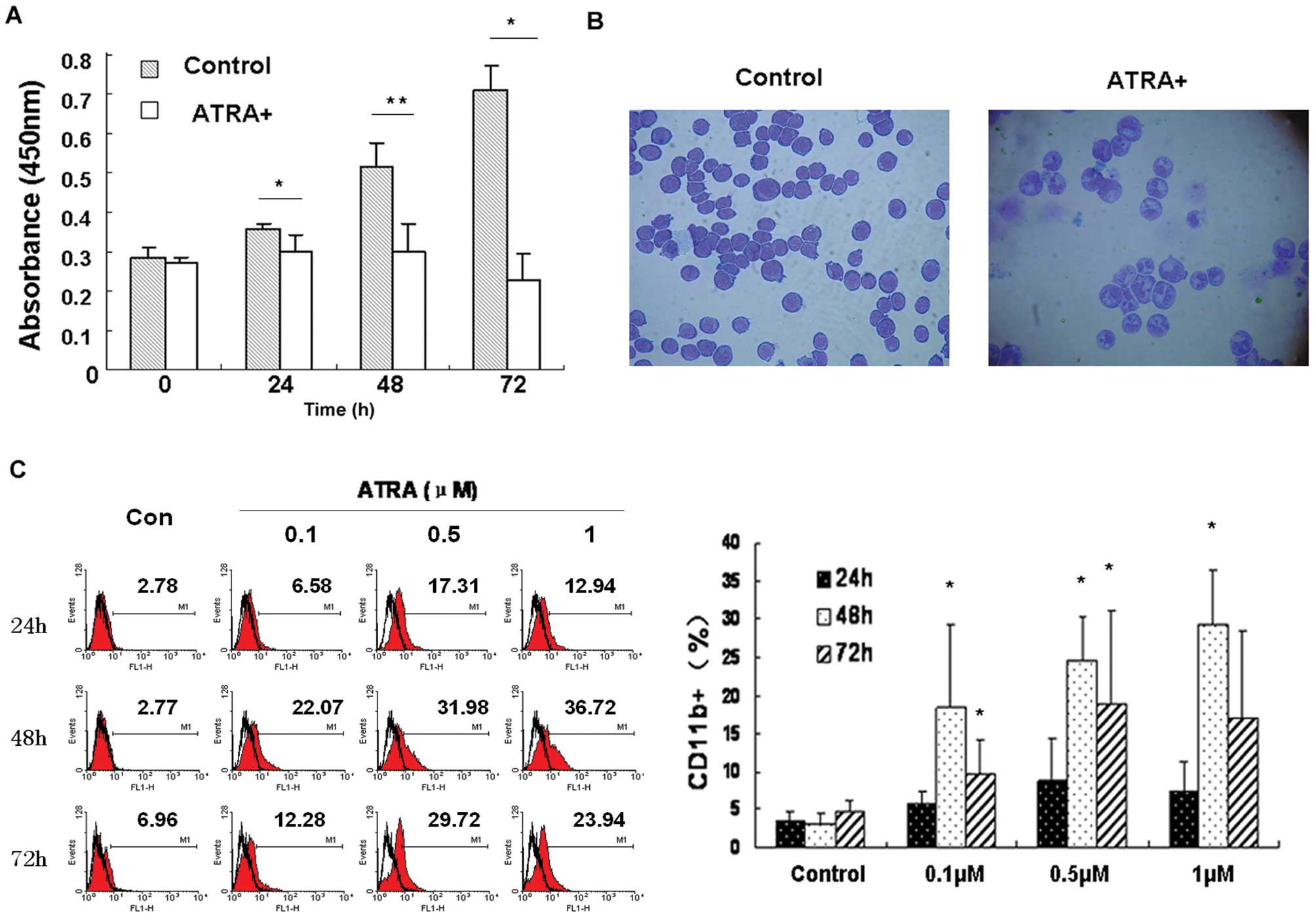
Induction of differentiation and
inhibition of proliferation of HL-60 cells by ATRA
Under experimental conditions of this study, 1 μM
ATRA induced differentiation of HL-60 cells, manifested by
inhibition of cell proliferation, morphological changes and
expression of differentiation marker CD11b. Cell proliferation was
strongly inhibited following ATRA treatment (Fig. 2A). Also, morphological features of
granulocytic differentiation, such as segmented nuclei and
condensed chromatin, were clearly evident in ATRA-treated HL-60
cells (Fig. 2B). Moreover, the
percentage of cells expressing CD11b was higher following ATRA
treatment, as shown in Fig. 2C.
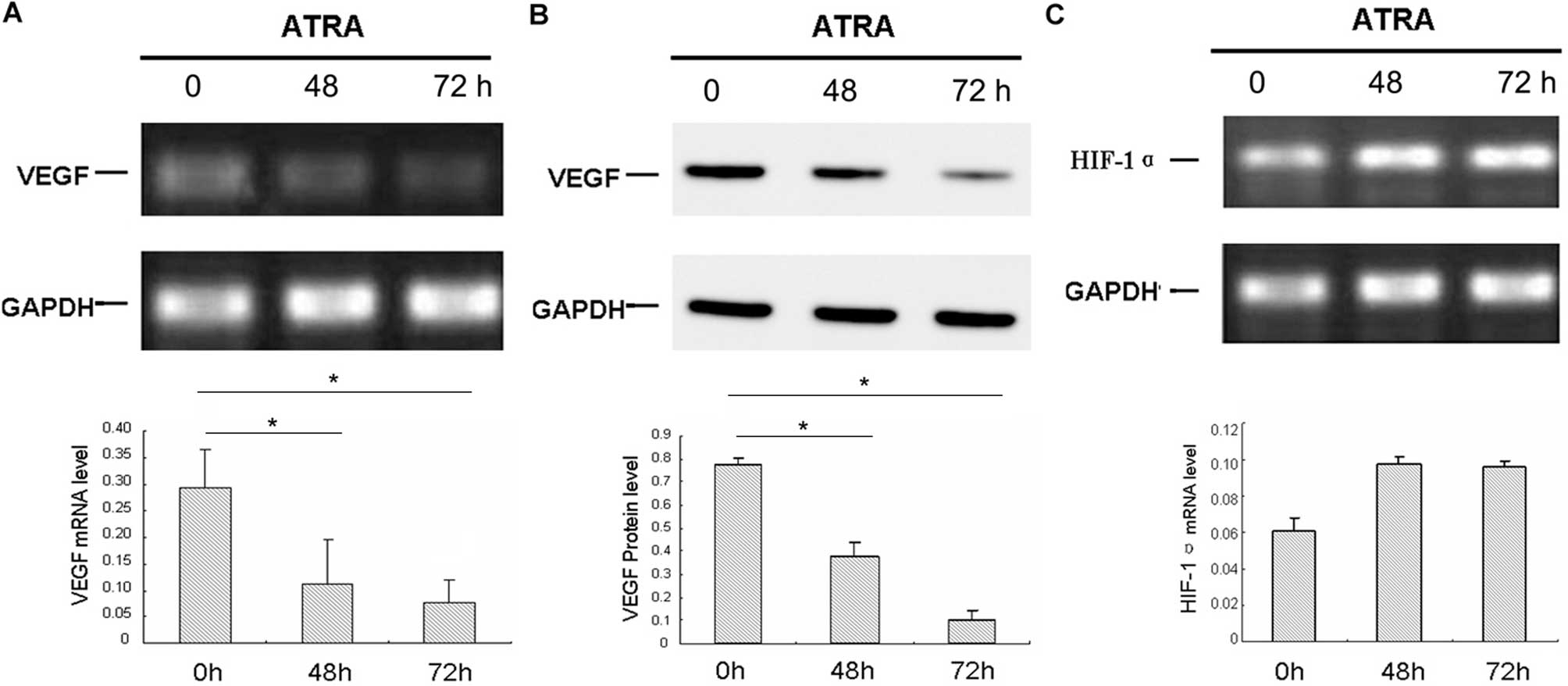
Downregulation of VEGF by ATRA is
Hif-1α-independent in HL-60 cells
As previously reported, 1 μM concentration of ATRA
inhibited the growth of leukemia cells and downregulated the
expression of VEGF (6). The mRNA
and protein levels of VEGF were detected by RT-PCR and western blot
assay, respectively, in HL-60 cells treated with ATRA. As indicated
in Fig. 3A and B, ATRA
downregulated the expression of VEGF in a time-dependent manner.
The mRNA and protein levels of VEGF in HL-60 cells were decreased
significantly in response to treatment with 1 μM ATRA compared with
the control. The housekeeping gene GAPDH was used for
normalization. Hif-1α mRNA was upregulated in HL-60 cells after
exposure to ATRA, and the results indicated that the downregulation
of VEGF by ATRA was not Hif-1α-dependent, which is in contrast to
most solid tumors (Fig. 3C).
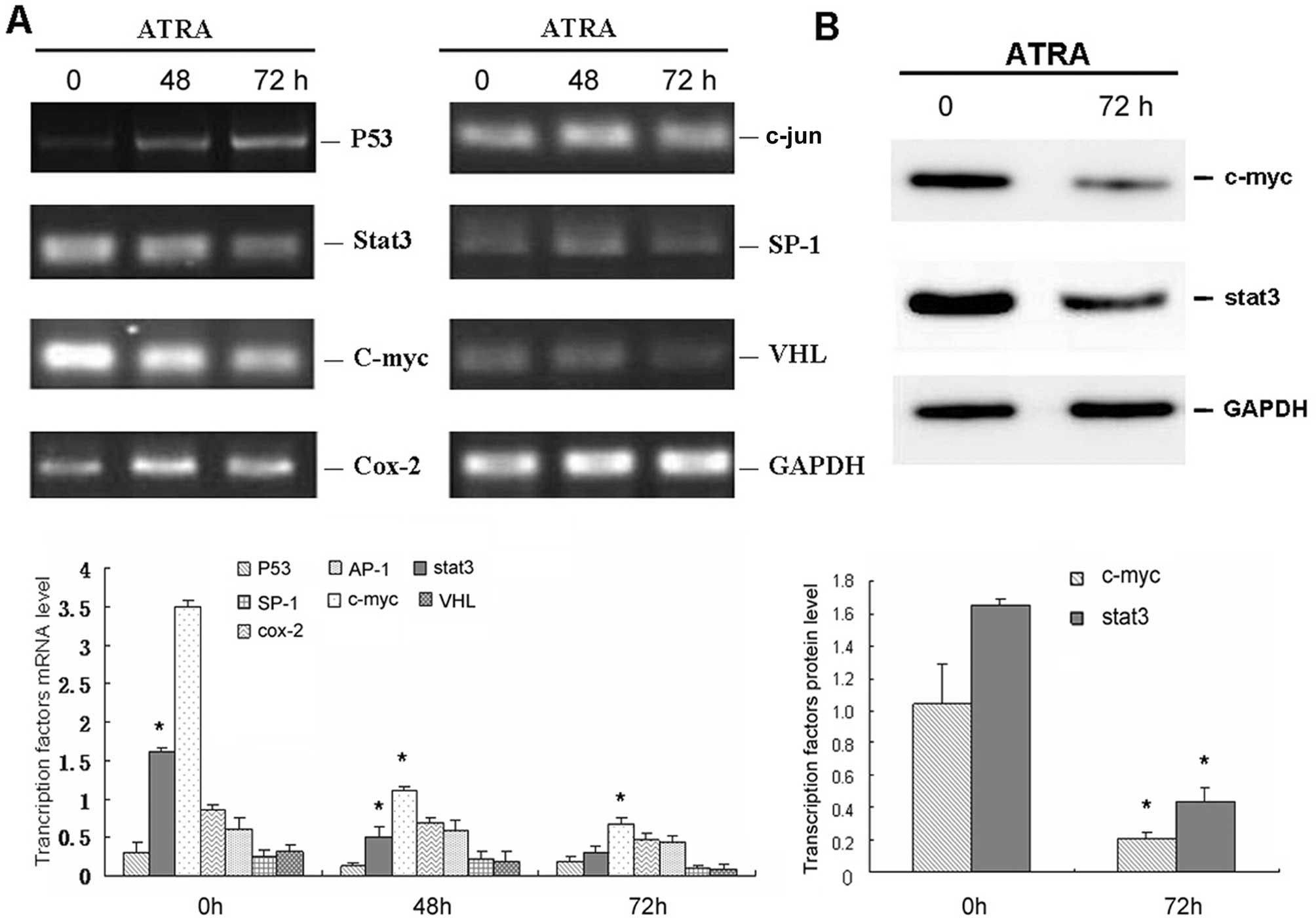
c-myc correlates with the expression of
VEGF in HL-60 cells treated with ATRA
Similar to the mechanism of VEGF observed in solid
tumors, we selected seven transcription factors related to VEGF and
performed RT-PCR to screening factors that may account for VEGF in
HL-60 cells. As demonstrated in Fig.
4A, only stat3 and c-myc were significantly downregulated by
ATRA, which was in agreement with VEGF expression following
exposure to ATRA. Thus, stat3 and c-myc were selected for further
confirmation by western blot assay (Fig. 4B). Consistent with the RT-PCR data,
the expression levels of stat3 and c-myc in HL-60 cells were both
decreased following treatment with ATRA for 72 h.
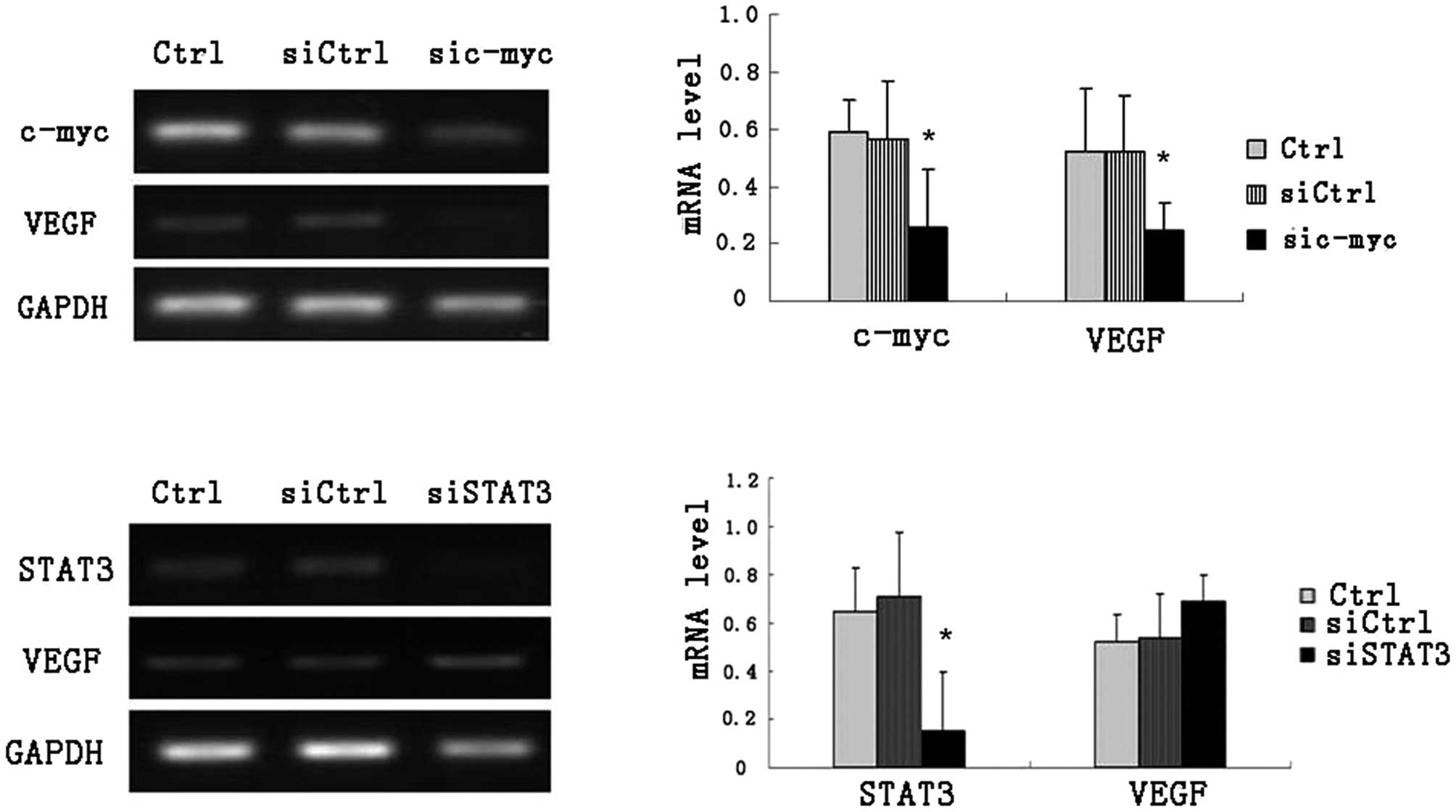
Based on these results, specific siRNAs targeting
stat3 or c-myc were investigated regarding their roles in
regulating VEGF in HL-60 cells. The RT-PCR results showed that VEGF
level could be downregulated significantly by silencing c-myc
expression, other than stat3 siRNA (Fig. 5).
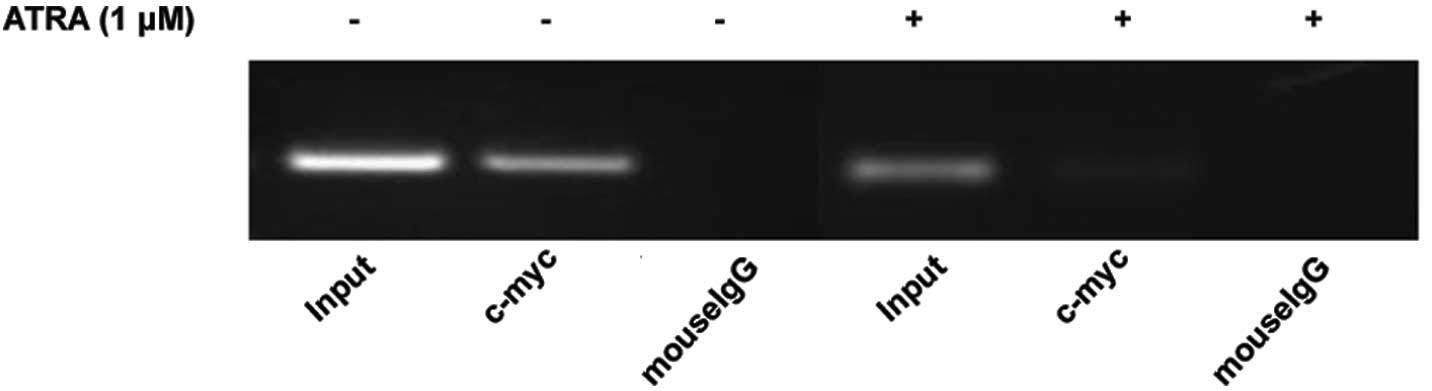
Effect of ATRA on the binding of c-myc to
VEGF promoter in HL-60 cells
Recent studies revealed that c-myc activation is
directly linked to the transcriptional regulation of VEGF by
binding to the VEGF promoter. Based on this, we conducted ChIP
assay. c-myc binding to VEGF promoter was detected (Fig. 6). As expected, ATRA treatment
suppressed the binding of c-myc to VEGF promoter. These data
further support the hypothesis that by downregulating the
expression of c-myc, ATRA could inhibit mRNA and protein expression
of VEGF, which may further influence the proliferation and
differentiation of HL-60 cells.
Discussion
AML is a disease with a poor outcome, and the
overall survival rate of five years is moderate to poor depending
on age and cytogenetics (10). In
1985, ATRA was introduced to APL treatment. Optimization of the
ATRA-based regimens combining ATRA and chemotherapy has further
raised the complete remission (CR) rate up to 90–95%, and a 6-year
DFS up to 86% (±10%) in low-risk patients (11). At diagnosis, an enhanced microvessel
density (MVD) in bone marrow biopsies has been observed, which is
restored to normal levels when a CR has been achieved. Furthermore,
AML bone marrow biopsies display enhanced angiogenesis and
increased VEGF expression. The enhanced bone marrow vascularization
is correlated with an increased expression of VEGF (10). However, the specific contribution of
VEGF to the liquid tumor progression has not been clearly defined.
Several groups have demonstrated that ATRA strongly induces
terminal differentiation of HL-60 cells and reported ATRA treatment
could downregulate VEGF significantly (12). Although VEGF is capable of signaling
through VEGFR and induces endothelial proliferation and migration,
the relationship between VEGF and the differentiative or
proliferative ability of HL-60 remains unclear. In this study, by
applying siRNA especially targeting VEGF, we demonstrated that
HL-60 has a more invasive tumor phenotype inhibiting
differentiation and triggering abnormal proliferation, all of which
suggested that VEGF may participate in other leukemia-related
pathways aside from the fact that it also plays a critical role in
the growth of hematological neoplasm via autocrine or paracrine
mechanism (13).
To further study the molecular mechanism regulating
VEGF, we selected HIF-1α as a cut-point, which appears to be a
major regulator of VEGF gene expression in solid tumors (14,15).
Furthermore, this basic helix-loop-helix transcription factor may
not only contribute to hypoxia-induced VEGF production, but it may
also play a critical role in the oncogene-dependent expression of
VEGF. However, in contrast to the previous results of solid tumors,
our study found that the regulatory mechanism of VEGF in HL-60
cells induced by ATRA is independent of the HIF-1α-related pathway
(6). These facts support the
hypothesis that there do exist some mechanisms regulating VEGF
other than HIF-1α. Through detailed analysis of the VEGF promoter
and its regulatory transcription factors, previous studies have
demonstrated some other critical factors, such as SP-1, c-jun,
cox-2, c-myc and stat3, may be account for, which could also be
disturbed under tumor microenvironment (16–20).
These highlight the complex regulatory network of VEGF. Similar to
the mechanism of VEGF observed in solid tumors, in the present
study, we selected transcription factors concerning VEGF, such as
P53, SP-1, c-jun, von Hippel-Lindau syndrome (VHL), cox-2, c-myc
and stat3, to screen factors that may be directly correlated with
VEGF in HL-60 cells. Following ATRA treatment, the expression of
c-myc and stat3 showed the same tendency as VEGF, which may
indicate that other pathways account for abnormal VEGF expression.
In this regard, we further investigated the roles of stat3 and
c-myc in regulating VEGF using specific siRNAs silencing these two
factors in HL-60 cells separately, and the results indicated that
inhibition of c-myc expression could downregulate VEGF, while the
effect of stat3 on VEGF was not obvious, which was not consistent
with the evidence in solid tumors. In summary, our results
suggested the c-myc may be the upstream regulatory factor of
VEGF.
In a previous study, Dadiani et al(21) found that, in breast cancer, both
c-myc and the activated estrogen receptor α were shown to co-bind
the VEGF promoter in close proximity, indicating a cooperative role
for them in estrogen regulation of VEGF and the ability of c-myc to
partially mimic estrogen regulation of angiogenesis. Mizukami et
al(20) suggested that VEGF may
also be an important target of c-myc in colon cancer, particularly
under hypoxic conditions. In our study, we confirmed that c-myc
could bind to VEGF promoter directly in HL-60 cells by ChIP, and
this interaction may be markedly inhibited by ATRA, which decreased
the expression of c-myc. Furthermore, Yang et al(22) co-delivered the pooled siRNAs
targeting HDM2, c-myc and VEGF and it could effectively and
simultaneously knock down their expressions and significantly
inhibit tumor cell growth in A549 and H460 cells in vitro.
It is possible to establish other pooled siRNAs to inhibit the
abnormal proliferation and promote the differentiation of HL-60
cells.
Collectively, our data demonstrated that in HL-60
human leukemia cells, VEGF play an important role in
differentiation and proliferation, and ATRA promotes
differentiation and inhibits proliferation by suppressing VEGF.
c-myc, but not Hif-1α-dependent downregulation of VEGF, induced by
ATRA contributes to the differentiation of HL-60 cells.
Acknowledgements
This study was supported by the Natural Science
Foundation of China (30771103, 81172792), the Science and
Technology Development Project of Shandong Province (2006GG2302010,
2007GG2002023) and the Natural Science Foundation of Shandong
Province (Y2008C165, ZR2011HL050).
References
|
1
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signaling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Podar K and Anderson KC: The
pathophysiologic role of VEGF in hematologic malignancies:
therapeutic implications. Blood. 105:1383–1395. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mayerhofer M, Valent P, Sperr WR, Griffin
JD and Sillaber C: BCR/ABL induces expression of vascular
endothelial growth factor and its transcriptional activator,
hypoxia inducible factor-1alpha, through a pathway involving
phosphoinositide 3-kinase and the mammalian target of rapamycin.
Blood. 100:3767–3775. 2002. View Article : Google Scholar
|
|
4
|
Perez-Atayde AR, Sallan SE, Tedrow U,
Connors S, Allred E and Folkman J: Spectrum of tumor angiogenesis
in the bone marrow of children with acute lymphoblastic leukemia.
Am J Pathol. 150:815–821. 1997.PubMed/NCBI
|
|
5
|
Ghosh AK, Shanafelt TD, Cimmino A, et al:
Aberrant regulation of pVHL levels by microRNA promotes the
HIF/VEGF axis in CLL B cells. Blood. 113:5568–5574. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang GS, Yang WH, Wen PE, Ren X, Tang TH
and Ren HQ: HIF-1α independent down-regulation of VEGF expression
in HL60 cells after in vitro exposure to ATRA. Blood.
110:42902007.
|
|
7
|
Lidgren A, Hedberg Y, Grankvist K, Torgny
R, Vasko J and Ljungberg B: The expression of hypoxia-inducible
factor 1alpha is a favorable independent prognostic factor in renal
cell carcinoma. Clin Cancer Res. 11:1129–1135. 2005.PubMed/NCBI
|
|
8
|
Schuch G, Machluf M, Bartsch G Jr, et al:
In vivo administration of vascular endothelial growth factor (VEGF)
and its antagonist, soluble neuropilin-1, predicts a role of VEGF
in the progression of acute myeloid leukemia in vivo. Blood.
100:4622–4628. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ter Elst A, Ma B, Scherpen FJ, et al:
Repression of vascular endothelial growth factor expression by the
runt-related transcription factor 1 in acute myeloid leukemia.
Cancer Res. 71:2761–2771. 2011.PubMed/NCBI
|
|
10
|
Weidenaar AC, ter Elst A, Koopmans-Klein
G, et al: High acute myeloid leukemia derived VEGFA levels are
associated with a specific vascular morphology in the leukemic bone
marrow. Cell Oncol. 34:289–296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bajpai J, Sharma A, Kumar L, et al: Acute
promyelocytic leukemia: an experience from a tertiary care centre
in north India. Indian J Cancer. 48:316–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tee MK, Vigne JL and Taylor RN: All-trans
retinoic acid inhibits vascular endothelial growth factor
expression in a cell model of neutrophil activation. Endocrinology.
147:1264–1270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang ES, Teruya-Feldstein J, Wu Y, Zhu Z,
Hicklin DJ and Moore MA: Targeting autocrine and paracrine VEGF
receptor pathways inhibits human lymphoma xenografts in vivo.
Blood. 104:2893–2902. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vaupel P: The role of hypoxia-induced
factors in tumor progression. Oncologist. 9(Suppl 5): 10–17. 2004.
View Article : Google Scholar
|
|
15
|
Ohno H, Shirato K, Sakurai T, et al:
Effect of exercise on HIF-1 and VEGF signaling. J Phys Fitness
Sports Med. 1:5–16. 2012. View Article : Google Scholar
|
|
16
|
Loeffler S, Fayard B, Weis J and
Weissenberger J: Interleukin-6 induces transcriptional activation
of vascular endothelial growth factor (VEGF) in astrocytes in vivo
and regulates VEGF promoter activity in glioblastoma cells via
direct interaction between STAT3 and Sp1. Int J Cancer.
115:202–213. 2005. View Article : Google Scholar
|
|
17
|
Xie K, Wei D, Shi Q and Huang S:
Constitutive and inducible expression and regulation of vascular
endothelial growth factor. Cytokine Growth Factor Rev. 15:297–324.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Colla S, Tagliaferri S, Morandi F, et al:
The new tumor-suppressor gene inhibitor of growth family member 4
(ING4) regulates the production of proangiogenic molecules by
myeloma cells and suppresses hypoxia-inducible factor-1 alpha
(HIF-1alpha) activity: involvement in myeloma-induced angiogenesis.
Blood. 110:4464–4475. 2007. View Article : Google Scholar
|
|
19
|
Lee CC, Chen SC, Tsai SC, et al:
Hyperbaric oxygen induces VEGF expression through ERK, JNK and
c-Jun/AP-1 activation in human umbilical vein endothelial cells. J
Biomed Sci. 13:143–156. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizukami Y, Fujiki K, Duerr EM, et al:
Hypoxic regulation of vascular endothelial growth factor through
the induction of phosphatidylinositol 3-kinase/Rho/ROCK and c-Myc.
J Biol Chem. 281:13957–13963. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dadiani M, Seger D, Kreizman T, et al:
Estrogen regulation of vascular endothelial growth factor in breast
cancer in vitro and in vivo: the role of estrogen receptor alpha
and c-Myc. Endocr Relat Cancer. 16:819–834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Y, Hu Y, Wang Y, Li J, Liu F and
Huang L: Nanoparticle delivery of pooled siRNA for effective
treatment of non-small cell lung cancer. Mol Pharm. Jun
22–2012.(Epub ahead of print).
|