Introduction
Apoptin, a small viral protein, was originally
identified in the chicken anemia virus (1). Research has shown that apoptin
specifically induced apoptosis in a broad range of different human
cancer cell lines derived from various cancer types but not in
their normal cell counterparts (2).
Furthermore, apoptosis induced by apoptin is p53-independent
(3,4). Yet, the underlying molecular mechanism
of the roles of apoptin in inducing apoptosis in cancer cell lines
is largely unknown.
Previous studies have demonstrated that
apoptin-induced cancer cell apoptosis is tightly associated with
its cancer-specific phosphorylation and subcellular localization
(5). In the last decade, several
apoptin interacting partner proteins have been identified, such as
the N-myc-interacting protein (NMI) (6), promyelocytic leukemia protein (PML)
(7), anaphase promoting
complex/cyclosome subunit 1 (APC/C) (8), death effector domain associated factor
(DEDAF) (9), acid ceramidase
(10), importin-β1 (11) and peptidylprolyl isomerase-like 3
(Ppil3) (12). It has been
demonstrated that these proteins play crucial roles in apoptin’s
selective toxicity in different cancer cell lines. Maddika et
al(13) reported that apoptin
interacts with the SH3 domain of the p85 regulatory subunit of
phosphoinositide 3-kinase (PI3K) through its proline-rich sequence
(amino acid 81–86) and activates PI3K. Furthermore, PI3K downstream
effector kinase Akt1 is also activated by apoptin and interacts
with it. Subsequently, apoptin and activated Akt1 are translocated
to the nucleus (14). Nuclear Akt1
then activates CDK2 by both direct and indirect mechanisms, and the
cyclin A-CDK2 complex directly phosphorylates apoptin at Thr-108
and contributes to the regulation of its subcellular location
(15). Identification of these
apoptin-interacting proteins extends the knowledge of apoptin
cancer-specific toxicity. The search for, the identification of,
and the characterization of novel apoptin-interacting partner
proteins are important to further elucidate the molecular mechanism
underlying the cancer-specific toxic effects of apoptin, thereby
extensively identifying the detailed mechanisms of its anticancer
action.
HSPA9 (mortalin/mthsp70/Grp75/PBP74/mot-2) is a heat
uninducible member of the hsp70 protein family (16). It is a 74-kDa protein and resides in
mitochondria, endoplasmic reticulum, the plasma membrane and
cytoplasmic vesicles (17,18). The function of HSPA9 is related to
proliferation, functional maintenance and stress response in cancer
cells (16,19–22).
Wadhwa et al(23) found that
HSPA9 overexpression may enhance cellular growth or proliferation
and prolong the cellular lifespan. Expression study of HSPA9 in
cancer cells has revealed that it is commonly upregulated in
cancers (23), suggesting that this
protein is involved in many cellular processes.
In the present study, prokaryotic native His-apoptin
fusion protein purified with Ni-NTA affinity chromotography was
used as a bait for capturing apoptin-interacting proteins in both a
hepatoma carcinoma cell line (HepG2) and a human fetal liver cell
line (L-02) by using a pull-down assay. The captured proteins were
separated by two-dimensional gel electrophoresis and identified by
mass spectrum. Our data indicated that HSPA9, a major protein
captured from HepG2 cells by pull-down assay, was overexpressed in
the HepG2 cells. This resulted in partial apoptin retention in the
cytoplasm which significantly inhibited the apoptosis rate of HepG2
cells from 41.2 to 31.7% induced by apoptin, while the
downregulation of HSPA9 by siRNA interference significantly
enhanced the apoptosis of HepG2 cells.
Materials and methods
Plasmid construction
The plasmid pcDNA3-apoptin was previously
constructed by our laboratory group and preserved (stored at −20°C)
(24). In brief, to construct
pcDNA3-HA-apoptin, a sequence derived from influenza
haemagglutinin (HA) was inserted into pcDNA3-apoptin (25). The apoptin fragment was excised from
pcDNA3-apoptin with EcoRI and XhoI and then inserted
into the EcoRI and XhoI enzyme cutting sites of the
pET-28a (+)-vector (Invitrogen, USA). First-strand cDNA was
synthesized from 8 μg of total mRNA (purified from the HepG2 cell
line) by using a reverse transcriptase-polymerase chain reaction
(RT-PCR) kit (Takara, Shiga, Japan) according to the manufacturer’s
instructions. The HSPA9 fragment was amplified by PCR, and
then inserted into the BamHI and XhoI enzyme cutting
sites of the pcDNA4-vector (Invitrogen). The HSPA9 primers
(commercially synthesized by Shanghai Biotech Bioscience and
Technology Co., Ltd., Shanghai, China) employed in the PCR reaction
were as follows: sense, 5′-TGGATCCATGATAAGTGCCAGCCGAGCTGCAGCAG C-3′
and antisense, 5′-CTCGAGCTGTTTTTCCTCCTTTTG ATCTTCC-3′.
Expression and purification of native
His-apoptin
Escherichia coli (E. coli) strain
BL21(DE3) transformed with pET28a(+)-apoptin was cultured at
37°C overnight, and the E. coli solution was diluted at a
volume ratio of 1:50 in 1 liter of Luria-Bertani medium containing
kanamycin (50 μg/ml), followed by incubation at 37°C. The
absorbance (A)600 was adjusted to 0.6–0.8, and 1 mM of
isopropyl-β-D-thiogalactopyranoside was added to the culture
system. The culture temperature was then changed to 24°C and
further cultured for 5 h, followed by centrifugation at 5,000 × g
at 4°C for 10 min. Resuspension was carried out in equilibrium
buffer (containing 50 mM sodium phosphate, 300 mM NaCl, 1 mM PMSF,
pH 8.0). Subsequently, the E. coli solution was sonicated
and centrifugated at 10,000 × g at 4°C for 30 min and filtered
through a 0.22-μm pore-size filter. The imidazole concentration of
the lysate was adjusted to 150 mM. The lysate was applied to a
Ni-NTA resin (Qiagen) affinity column (2 ml) which was equilibrated
with 5-fold column volumes of equilibration buffer (containing 50
mM sodium phosphate pH 8.0, 300 mM NaCl, 150 mM imidazole and 1 mM
PMSF) and washed with 10-fold column volume equilibration buffer
until the A280 was stable. The protein was eluted with
5-fold column volumes of elution buffer (containing 50 mM sodium
phosphate pH 8.0, 300 mM NaCl, 500 mM imidazole and 1 mM PMSF).
Eluting protein-fractions was screened by SDS-PAGE. Protein
fractions containing His-apoptin were collected and dialyzed at 4°C
for 20 h in solution containing 50 mM sodium phosphate and 300 mM
NaCl, pH 7.5.
In vitro pull-down assay
HepG2 or L-02 cells were maintained in RPMI-1640
medium supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml
streptomycin and 2 mmol/l L-glutamine in a saturated humidity, in
an atmosphere of 5% CO2 at 37°C. Cells were washed twice
with ice-cold PBS and lysed by repetitive freeze-thawing in binding
buffer (26) (containing 20 mM
HEPES, pH 7.4, 20 mM MgCl2, 150 mM NaCl, and standard
inhibitors of proteases and phosphatases) (3). Then the cell lysates were centrifuged
(18,000 × g) at 4°C for 20 min. Purified fusion His-apoptin (500
μg/column) was immobilized on 0.5 ml Ni-NTA resin, and incubated
with 4 mg total proteins of HepG2 or L-02 cells at 4°C for 1.5 h,
and gently shaken on a rotating platform. The mixed liquor was
centrifuged and washed 5 times with the binding buffer to avoid
nonspecific binding. Apoptin-interacting proteins were eluted with
elution buffer (containing 50 mM sodium phosphate, pH 8.0, 300 mM
NaCl, 500 mM imidazole and 1 mM PMSF), and dialyzed at 4°C
overnight. Interacting proteins underwent freeze drying.
Two-dimensional gel electrophoretic
analysis of apoptin-interacting proteins
Two-dimensional gel electrophoresis was performed
according to the manufacturer’s technical guidelines (GE
Healthcare). Briefly, the protein samples were solved by
rehydration buffer (containing 8 M urea, 2% CHAPS, 1% DTT, 0.5% IPG
buffer and 0.002% bromophenol blue). NL IPG strips, 18 cm in
length, pH 4.0–7.0 (GE Healthcare), were applied in
first-dimensional isoelectrofocusing and then transferred to 12.5%
SDS-PAGE for second-dimensional electrophoresis. The gels were
visualized by modified silver staining. The maps were analyzed by
ImageMaster 2D Platinum 6.0 software (GE Healthcare). The target
protein spots were cut from the gel, and then identified by Mass
Spectrometry (Thermo Scientific).
Co-immunoprecipitation and
immunoblotting
Transfection was carried out using Lipofectamine™
2000 (Invitrogen) according to the manufacturer’s instructions.
HepG2 cells with 70% confluency were transiently co-transfected
with pcDNA3-HA-apoptin and pcDNA4-HSPA9, and the pcDNA3-vector and
pcDNA4-vector were set up as controls, respectively. HepG2 cells
were washed twice with ice-cold PBS 24 h after transfection, and
then lysed with ice-cold lysis buffer (containing 50 mm Tris, pH
7.5, 1% Nonidet P-40, 150 mM NaCl, 1 mM
Na3VO4, 2 mM EGTA and protease inhibitor
cocktail) (14) for 30 min on ice,
and then centrifuged (16,000 × g) at 4°C for 20 min.
Co-immunoprecipitation was performed using the indicated
antibodies, and the immune complex was captured using protein
A-agarose beads (Sigma). After washing for 3 times with cell lysis
buffer, bead-bound proteins were subjected to immunoblot
analysis.
Transient transfection of small
interfering RNA (siRNA)
siRNA purchased from GenePharma (Shanghai, China)
was introduced into HepG2 cells by Lipofectamine 2000. The target
sequence was 5′-AAA CTC TAG GAG GTG TCT TTA-3′ (27). Briefly, a total of 7 μg siRNA, mixed
with 3 μl of Lipofectamine 2000, was added to HepG2 cells in 1.5 ml
of DMEM medium, in accordance with the manufacturer’s instructions.
Transfection medium was replaced with fresh DMEM medium 12 h after
incubation. Scrambled siRNA served as the control. Immunoblot assay
was conducted to detect the expression levels of HSPA9. The β-actin
served as an internal loading control.
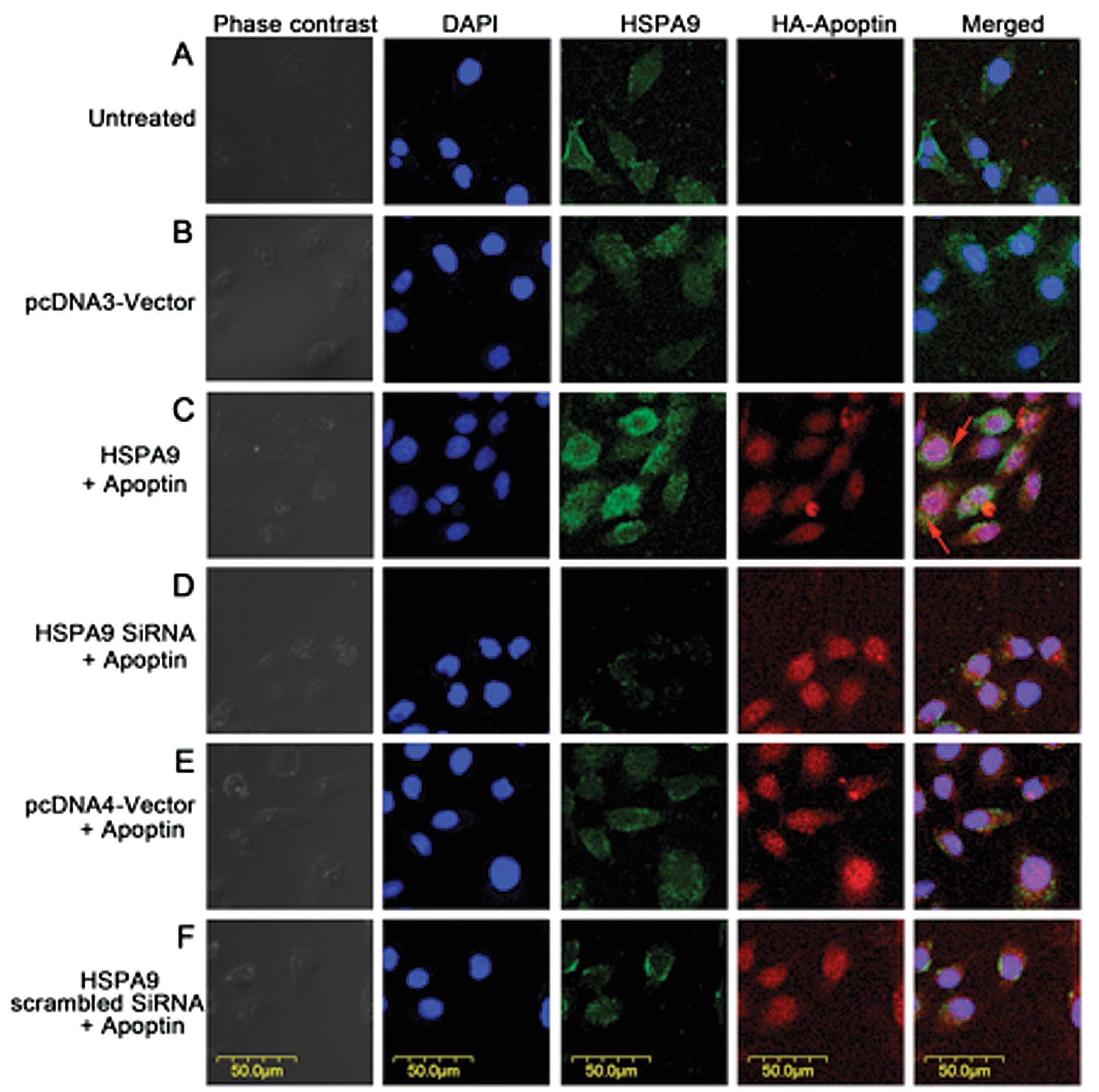
Immunofluorescence
Cells cultured on coverslips in 3.5-cm dishes the
day before transfection underwent the following preparations prior
to immunofluorescence observation. Plasmids or siRNA were used
according to Fig. 4. Twenty-four
hours after transient transfection, cells were fixed with 4%
paraformaldehyde, permeabilized in 0.5% Triton X-100, blocked in 5%
bovine serum albumin (BSA). HA-apoptin was then stained with rat
anti-HA monoclonal antibody (1:500) (Tiangen Biotech Co., Ltd.,
Beijing, China), followed by staining with RBITC-conjugated goat
anti-rat IgG secondary antibody (KPL, Gaithersburg, MD, USA). HSPA9
was stained with rabbit anti-HSPA9 monoclonal antibody (1:200)
(Cell Signaling Technology, Inc.), followed by staining with
FITC-conjugated goat anti-rabbit IgG secondary antibody (KPL).
Coverslips were then stained with DAPI, mounted on slides and
observed using an Olympus Fv500 confocal microscope (Olympus).
Apoptosis assay
Cells were cultured on coverslips in 3.5-cm dishes
the day before transfection. Twenty-four hours after transfection,
apoptosis was assessed by cytofluorometric analysis using an
Annexin V-fluorescein-5-isothiocyanate (FITC) and propidium iodide
(PI) apoptosis detection kit (KeyGen).
Statistical analysis
Experiments were repeated a minimum of three times
unless otherwise noted. Experimental data are expressed as the
means ± SD. The significance was determined by Student’s t-test for
comparisons between two groups using SPSS 12.0 software. A level of
P<0.05 or 0.01 was considered to indicate a statistically
significant result.
Results
In vitro pull-down assay identifies HSPA9
as an apoptin-interacting protein
To identify human cellular proteins that interact
with apoptin, we performed a pull-down assay with purified bait
protein of the native His-apoptin (Fig.
1A). Apoptin-interacting proteins in both hepatoma carcinoma
HepG2 cells and human fetal liver L-02 cells were first seperated
by 12% SDS-PAGE (Fig. 1B). Many
protein bands were differentially captured from the HepG2 and L-02
cells. Then two-dimensional gel electrophoresis was conducted for
further separation of the captured proteins (Fig. 1C and D). The proteic maps were
analyzed by ImageMaster 2D Platinum 6.0. The differential protein
spots captured from HepG2 and L-02 cells were excised in gel, and
then identified by mass spectrum. HSPA9 was a major interacting
protein with apoptin in vitro among the identified proteins
in the HepG2 cells by mass spectrum, as HSPA9 is much less abundant
in the L-02 cells compared with HepG2 cells (23). In our study, we did not detect HSPA9
as an interacting protein with apoptin in L-02 cells in
vitro (Fig. 1D).
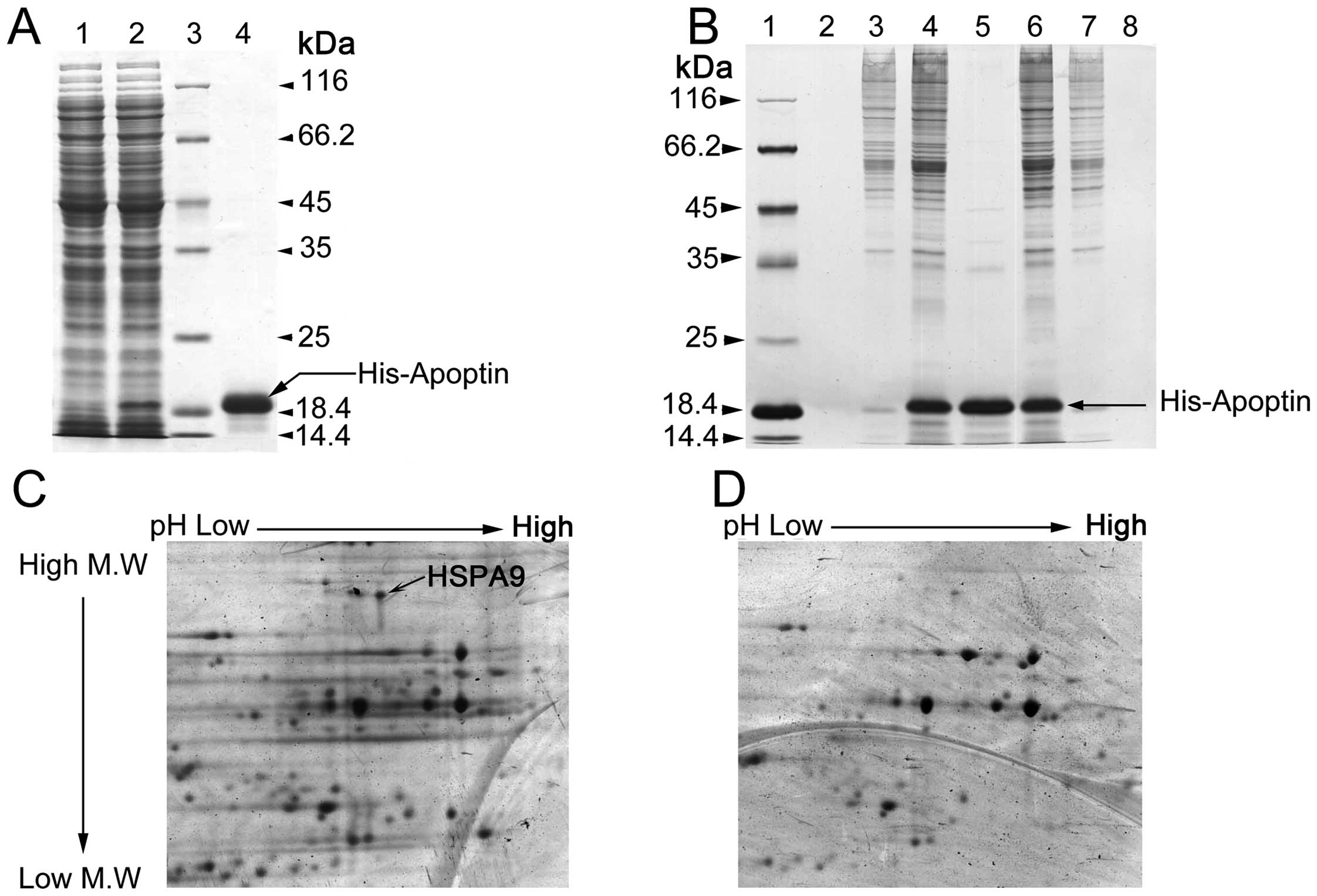 | Figure 1Interaction of HSPA9 and apoptin in
vitro. (A) Prokaryotic native His-apoptin fusion protein was
purified with Ni-NTA affinity chromatography (Coomassie Blue
staining). Lane 1, pET-28a(+)-vp3 E. coli BL21 (DE3) PlysS
total proteins without IPTG induction. Lane 2, pET-28a(+)-vp3 E.
coli BL21 (DE3) PlysS total proteins with 1 mM IPTG treatment.
Lane 3, Protein standard marker. Lane 4, His-apoptin fusion protein
purified by Ni-NTA His-Bind® resin affinity column. (B)
The proteins interacting with apoptin in vitro were captured
by His pull-down assay using His-apoptin as a bait protein (silver
staining). Lane 1, protein standard marker. Lane 2, nonspecific
affinity proteins (HepG2) were eluted by elution buffer. Lane 3,
HepG2 cellular total proteins and Ni-NTA His-Bind resin nonspecific
adhesive protein. Lane 4, interacting proteins with 6His-VP3 in
HepG2 cellular total proteins. Lane 5, purified 6His-VP3 proteins.
Lane 6, His-apoptin captured interacting proteins in L-02 total
proteins. Lane 7, nonspecific adhesive protein with Ni-NTA His-Bind
resin in L-02 total proteins. Lane 8, nonspecific proteins eluted
by elution buffer using affinity proteins (Ni column solidified
6His-VP3 incubated with L-02 cellular total proteins). (C and D)
Two-dimensional electrophoretogram showing the proteins interacting
with His-VP3 and 6His-VP3 (modified silver staining). (C)
Two-dimensional electrophoretogram-separated apoptin-interacting
proteins of His-VP3 capured from HepG2 cells (black arrow indicates
HSPA9 in the two-dimensional electrophoretogram). (D)
Two-dimensional electrophoretogram-separated apoptin-interacting
proteins of 6His-VP3 captured from L-02 cells. M.W., molecular
weight. |
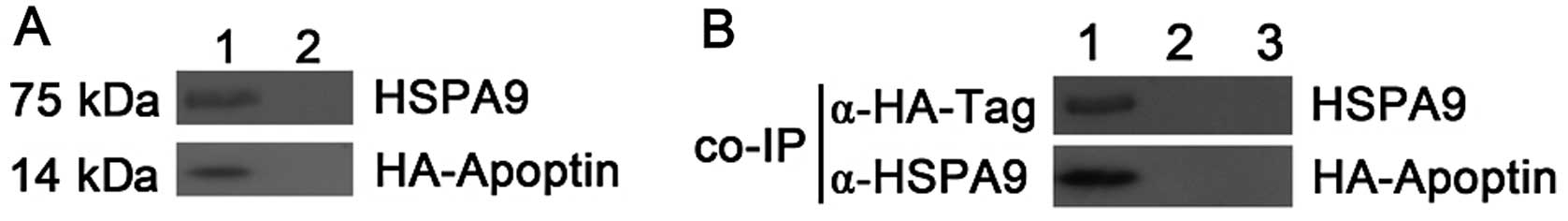
HSPA9 interaction with apoptin in HepG2
cells
To confirm the interaction observed in the pull-down
assay, the binding of HSPA9 to apoptin was tested in HepG2 cells by
co-immunoprecipitation. HepG2 cells were transiently co-transfected
with pcDNA3-HA-VP3 and pcDNA4-HSPA9, respectively. pcDNA3-vector
and pcDNA4-vector were transfected into HepG2 cells, respectively,
which served as controls in the co-immmunoprecipitation test.
pcDNA4-HSPA9- or pcDNA3-HA-apoptin-transfected HepG2 cells showed
that the overexpression of HSPA9 or HA-apoptin did not adhere to
the beads in an unspecific manner. These results suggest that HSPA9
indeed co-precipitated with apoptin (Fig. 2).
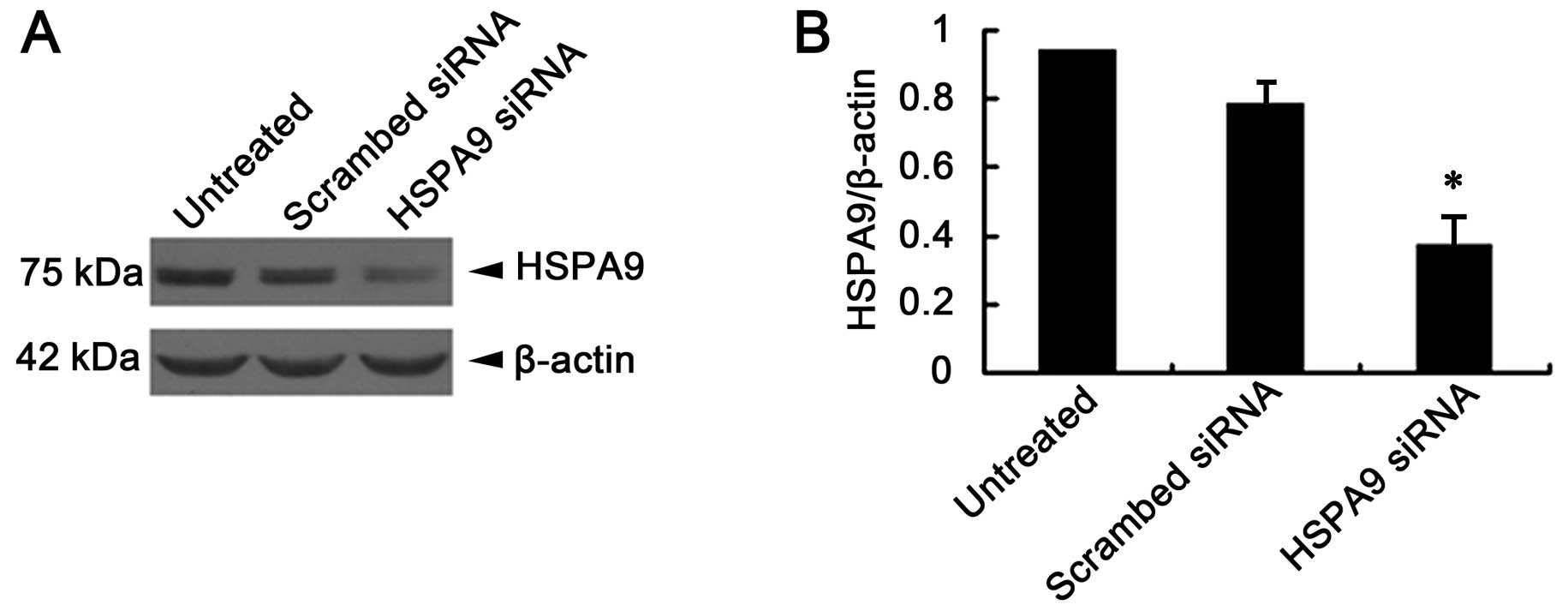
siRNA suppresses HSPA9 expression levels
in HepG2 cells
Twenty-four hours following transfection with siRNA,
HepG2 cell lysates were prepared. The HSPA9 protein expression in
HepG2 cells was detected by immunoblotting. Results showed that
HSPA9 expression in HepG2 cells was suppressed nearly 60% by siRNA
(Fig. 3).
HSPA9 overexpression results in partial
cytoplasmic distribution of apoptin
Since HSPA9 and apoptin were found to be interaction
partner proteins in HepG2 cells, we performed immunofluorescence to
study the effect of HSPA9 overexpression on the subcellular
localization of apoptin. Transient co-transfection of plasmid or
siRNA is shown in Fig. 4. Results
showed that HSPA9 overexpression in HepG2 cells resulted in partial
cytoplasmic distribution of apoptin (Fig. 4C).
HSPA9 overexpression in HepG2 cells
significantly affects apoptosis induced by apoptin
To investigate the effect of overexpression or
suppression of HSPA9 on the apoptosis induced by apoptin in HepG2
cells, HepG2 cells were transiently co-transfected with pcDNA4-VP3
and HSPA9 plasmid or siRNA. Twenty-four hours after transfection,
the apoptosis of HepG2 cells was assessed by cytofluorometric
analysis. Annexin V and PI-positive cells were considered as
apoptotic cells. Our data indicated that overexpression of HSPA9 in
HepG2 cells led to a significant reduction in the cell apoptotic
rate from 41.2% (control) to 31.7% (co-transfected group)
(P<0.01). Notably, the apoptotic rate was increased from 43.6%
(co-transfected group) to 52.2% (siRNA group) when HSPA9 was
knocked down by siRNA (P<0.05), whereas overexpression or
suppression of HSPA9 had no effect on the apoptosis of L-02 cells
(Fig. 5C). These results suggest
that HSPA9 inhibits apoptosis induced by apoptin in tumor cells
rather than in normal cells.
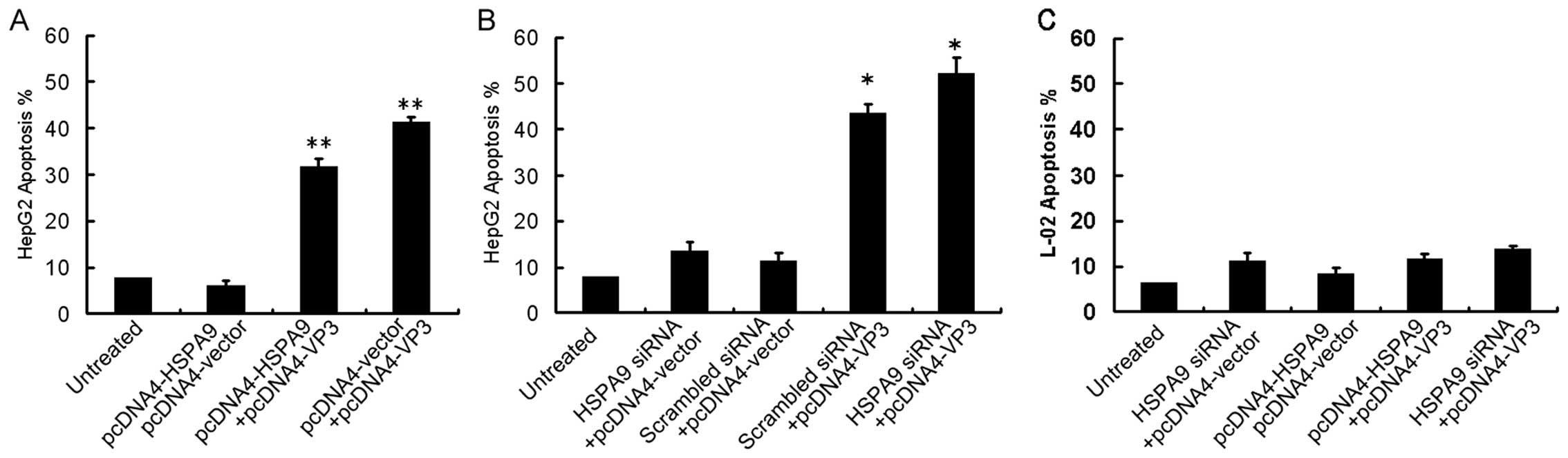 | Figure 5Overexpression or suppression of HSPA9
significantly affects apoptosis induced by apoptin in HepG2 cells,
but not in L-02 cells. (A) HepG2 cells were co-transfected with
pcDNA4-HSPA9 and pcDNA4 vector, or pcDNA4-HSPA9 and pcDNA4-VP3 or
pcDNA4 vector and pcDNA4-VP3, respectively. Apoptosis of control
cells and the transfected cells were tested by cytofluorometric
analysis. (B) HepG2 cells were co-transfected with HSPA9 siRNA and
pcDNA4 vector, or scrambled siRNA and pcDNA4 vector, or scrambled
siRNA and pcDNA4-VP3, or HSPA9 siRNA and pcDNA4-VP3, respectively.
(C) L-02 cells were transfected with HSPA9 siRNA and pcDNA4 vector,
or pcDNA4-HSPA9 and pcDNA4 vector, or pcDNA4-HSPA9 and pcDNA4-VP3
or HSPA9 siRNA and pcDNA4-VP3, respectively. *P<0.05;
**P<0.01, compared with the control. |
Discussion
HSPA9 is a member of the heat shock protein 70
family of chaperones (16) which is
localized in mitochondria, in endoplasmic reticulum, on the plasma
membrane and in cytoplasmic vesicles (17,18).
HSPA9 is a major protein in the mitochondria (28) and it plays a central role in the
elaborate protein translocation system responsible for efficient
import and export of proteins (29–31).
Its role in cell viability and mitochondrial biogenesis was
demonstrated by experimental data. A study of the HSPA9 homologue
(Sscl) knocked out from yeast cells revealed that this was lethal
to these cells (32), and loss of
functional mutations of HSPA9 led to aggregation of yeast
mitochondria (33). Previous
studies focusing on cancer biology suggest that HSPA9 is a protein
which plays a crucial role in anti-apoptosis. It functions by means
of counteracting the ‘stress’ of senescence and apoptosis and as a
‘microevolutionary buffer’ that neutralizes conformational
consequences of mutant proteins. It also aids the acquisition of
functions in chaperone-stabilized rogue proteins and promotes
cellular invasiveness and motility. Furthermore, HSPA9 plays a
pivotal role in the cellular coordination of hyperactivation of
proliferation signals and resistance to radiation, heat, hormones
and chemotherapeutic agents (34).
To date, the underlying mechanism of the specific
toxic effect of apoptin on tumors remains to be elucidated. Maddika
et al(35) reported that the
specific toxic effect of apoptin is independent of death receptors
but is related to loss of mitochondrial membrane potential and the
release of mitochondrial cell-death mediators via a Nur77-dependent
pathway. It has been demonstrated that the nuclear localization of
apoptin and the cytoplasmic translocation of Nur77 are the main
causes for the toxic effect on cancer cells by apoptin (35). Our results revealed that HSPA9
overexpression led to the partial distribution of apoptin in the
cytoplasm. Meanwhile, the apoptin-induced apoptosis rate of HepG2
cells was markedly decreased in response to HSPA9 overexpression.
In contrast, when downregulation of HSPA9 expression occurred in
HepG2 cells due to siRNA treatment, apoptin’s toxicity to HepG2
cells was significantly enhanced in that the apoptosis rate of
HepG2 cells was found to be markedly reduced. These findings
suggest that HSPA9 may inhibit the cancer-specific toxicity
produced by apoptin by means of retaining apoptin’s location in the
cytoplasm. Thus, we deduced that the overexpression of HSPA9 in
HepG2 cells may inhibit the cancer-specific toxicity of apoptin
through two ways. On the one hand, the more HSPA9 that was present
in the cytoplasm, the more apoptin that was retained in the
cytoplasm. At this time, relatively less apoptin was translocated
to the nucleus so that less toxicity was produced by apoptin
accordingly. On the other hand, HSPA9 overexpression may protect
mitochondria from the impairment of pro-apoptosis factor
stimulation, thereby contributing to the inhibition of
cancer-specific toxicity of apoptin. In contrast, downregulation of
HSPA9 levels caused by siRNA treatment attenuated mitochondrial
stability (33), which may
indirectly enhance the activity of apoptin, therefore leading to
the high cancer-specific toxicity of apoptin in HepG2 cells.
Although the overexpression of HSPA9 plays a crucial role in the
underlying mechanism in attenuating the anti-apoptosis effect in
HepG2 cells, still a high number of HepG2 cells died under the same
experimental conditions, suggesting that other factors playing
different roles in cancer-specific toxicity also participated in
this process and were involved in the relationship between apoptin
and HSPA9. However, the precise mechanism still needs extensive
investigation.
Taken together, our study showed that HSPA9 is an
important apoptin-interacting protein. HSPA9 overexpression
prevented apoptin from entering the nucleus to carry out its
apoptosis-promoting activity, while downregulation of HSPA9
expression significantly elevated the cancer-specific toxicity of
apoptin. Strategies to precisely manipulate the downregulation of
HSPA9, so as to strengthen the anticancer effect of apoptin and
shed light on novel effective therapeutic strategies for cancer,
such as liver cancer are needed and deserve to be intensively
studied.
Acknowledgements
This study was supported by grants from the Yunnan
Provincial Science and the Technology Department of P.R. China (no.
2011FB241).
Abbreviations:
|
NMI
|
N-myc interacting protein
|
|
PML
|
promyelocytic leukemia protein
|
|
APC/C
|
anaphase-promoting complex/cyclosome
subunit 1
|
|
DEDAF
|
death effector domain-associated
factor
|
|
Ppil3
|
peptidylprolyl isomerase-like 3
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
HA
|
haemagglutinin
|
References
|
1
|
Noteborn MH, de Boer GF, van Roozelaar DJ,
Karreman C, Kranenburg O, Vos JG, Jeurissen SH, Hoeben RC, Zantema
A and Koch G: Characterization of cloned chicken anemia virus DNA
that contains all elements for the infectious replication cycle. J
Virol. 65:3131–3139. 1991.PubMed/NCBI
|
|
2
|
Danen-Van Oorschot AA, Fischer DF,
Grimbergen JM, Klein B, Zhuang S, Falkenburg JH, Backendorf C, Quax
PH, Van der Eb AJ and Noteborn MH: Apoptin induces apoptosis in
human transformed and malignant cells but not in normal cells. Proc
Natl Acad Sci USA. 94:5843–5847. 1997.
|
|
3
|
Rohn JL, Zhang YH, Aalbers RI, Otto N, Den
Hertog J, Henriquez NV, Van De Velde CJ, Kuppen PJ, Mumberg D,
Donner P and Noteborn MH: A tumor-specific kinase activity
regulates the viral death protein apoptin. J Biol Chem.
277:50820–50827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Teodoro JG, Heilman DW, Parker AE and
Green MR: The viral protein apoptin associates with the
anaphase-promoting complex to induce G2/M arrest and apoptosis in
the absence of p53. Gene Dev. 18:1952–1957. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Danen-Van Oorschot AA, Zhang YH, Leliveld
SR, Rohn JL, Seelen MC, Bolk MW, Van Zon A, Erkeland SJ, Abrahams
JP, Mumberg D and Noteborn MH: Importance of nuclear localization
of apoptin for tumor-specific induction of apoptosis. J Biol Chem.
278:27729–27736. 2003.PubMed/NCBI
|
|
6
|
Sun GJ, Tong X, Dong Y, Mei ZZ and Sun ZX:
Identification of a protein interacting with apoptin from human
leucocyte cDNA library by using yeast two-hybrid screening. Sheng
Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao. 34:369–372. 2002.(In
Chinese).
|
|
7
|
Janssen K, Hofmann TG, Jans DA, Hay RT,
Schulze-Osthoff K and Fischer U: Apoptin is modified by SUMO
conjugation and targeted to promyelocytic leukemia protein nuclear
bodies. Oncogene. 26:1557–1566. 2007.PubMed/NCBI
|
|
8
|
Teodoro JG, Heilman DW, Parker AE and
Green MR: The viral protein apoptin associates with the
anaphase-promoting complex to induce G2/M arrest and apoptosis in
the absence of p53. Gene Dev. 18:1952–1957. 2004.PubMed/NCBI
|
|
9
|
Danen-van Oorschot AA, Voskamp P, Seelen
MC, van Miltenburg MH, Bolk MW, Tait SW, Boesen-de Cock JG, Rohn
JL, Borst J and Noteborn MH: Human death effector domain-associated
factor interacts with the viral apoptosis agonist apoptin and
exerts tumor-preferential cell killing. Cell Death Differ.
11:564–573. 2004.
|
|
10
|
Liu X, Elojeimy S, El-Zawahry AM, Holman
DH, Bielawska A, Bielawski J, Rubinchik S, Guo GW, Dong JY, Keane
T, Hannun YA, Tavassoli M and Norris JS: Modulation of ceramide
metabolism enhances viral protein apoptin’s cytotoxicity in
prostate cancer. Mol Ther. 14:637–646. 2006.PubMed/NCBI
|
|
11
|
Poon IK, Oro C, Dias MM, Zhang J and Jans
DA: Apoptin nuclear accumulation is modulated by a CRM1-recognized
nuclear export signal that is active in normal but not in tumor
cells. Cancer Res. 65:7059–7064. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huo DH, Yi LN and Yang J: Interaction with
Ppil3 leads to the cytoplasmic localization of apoptin in tumor
cells. Biochem Biophys Res Commun. 372:14–18. 2008. View Article : Google Scholar
|
|
13
|
Maddika S, Wiechec E, Ande SR, Poon IK,
Fischer U, Wesselborg S, Jans DA, Schulze-Osthoff K and Los M:
Interaction with PI3-kinase contributes to the cytotoxic activity
of apoptin. Oncogene. 27:3060–3065. 2008. View Article : Google Scholar
|
|
14
|
Maddika S, Bay GH, Kroczak TJ, Ande SR,
Maddika S, Wiechec E, Gibson SB and Los M: Akt is transferred to
the nucleus of cells treated with apoptin, and it participates in
apoptin-induced cell death. Cell Prolif. 40:835–848. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maddika S, Panigrahi S, Wiechec E,
Wesselborg S, Fischer U, Schulze-Osthoff K and Los M: Unscheduled
Akt-triggered activation of CDK2 as a key effector mechanism of
apoptin’s anticancer toxicity. Mol Cell Biol. 29:1235–1248.
2009.PubMed/NCBI
|
|
16
|
Wadhwa R, Kaul SC, Ikawa Y and Sugimoto Y:
Identification of a novel member of mouse hsp70 family. Its
association with cellular mortal phenotype. J Biol Chem.
268:6615–6621. 1993.PubMed/NCBI
|
|
17
|
Singh B, Soltys BJ, Wu ZC, Patel HV,
Freeman KB and Gupta RS: Cloning and some novel characteristics of
mitochondrial Hsp70 from Chinese hamster cells. Exp Cell Res.
234:205–216. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ran Q, Wadhwa R, Kawai R, Kaul SC, Sifers
RN, Bick RJ, Smith JR and Pereira-Smith OM: Extramitochondrial
localization of mortalin/mthsp70/PBP74/GRP75. Biochem Biophys Res
Commun. 275:174–179. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Craig EA, Kramer J, Shilling J,
Werner-Washburne M, Holmes S, Kosic-Smithers J and Nicolet CM:
SSC1, an essential member of the yeast HSP70 multigene family,
encodes a mitochondrial protein. Mol Cell Biol. 9:3000–3008.
1989.PubMed/NCBI
|
|
20
|
Kaul SC, Taira K, Pereira-Smith OM and
Wadhwa R: Mortalin: present and prospective. Exp Gerontol.
37:1157–1164. 2002.PubMed/NCBI
|
|
21
|
Merrick BA, Walker VR, He C, Patterson RM
and Selkirk JK: Induction of novel Grp75 isoforms by 2-deoxyglucose
in human and murine fibroblasts. Cancer Lett. 119:185–190. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wadhwa R, Taira K and Kaul SC: Mortalin: a
potential candidate for biotechnology and biomedicine. Histol
Histopathol. 17:1173–1177. 2002.PubMed/NCBI
|
|
23
|
Wadhwa R, Takano S, Kaur K, Deocaris CC,
Pereira-Smith OM, Reddel RR and Kaul SC: Upregulation of
mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J
Cancer. 118:2973–2980. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peng DJ, Sun J, Wang YZ, Tian J, Zhang YH,
Noteborn MH and Qu S: Inhibition of hepatocarcinoma by systemic
delivery of apoptin gene via the hepatic asialoglycoprotein
receptor. Cancer Gene Ther. 14:66–73. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Drewes G, Ebneth A, Preuss U, Mandelkow EM
and Mandelkow E: MARK, a novel family of protein kinases that
phosphorylate microtubule-associated proteins and trigger
microtubule disruption. Cell. 89:297–308. 1997. View Article : Google Scholar
|
|
26
|
Rohn JL, Zhang YH, Aalbers RI, Otto N, Den
Hertog J, Henriquez NV, Van De Velde CJ, Kuppen PJ, Mumberg D,
Donner P and Noteborn MH: A tumor-specific kinase activity
regulates the viral death protein apoptin. J Biol Chem.
227:50820–50827. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ohtsuka R, Abe Y, Fujii T, Yamamoto M,
Nishimura J, Takayanagi R and Muta K: Mortalin is a novel mediator
of erythropoietin signaling. Eur J Haematol. 79:114–125. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bhattacharyya T, Karnezis AN, Murphy SP,
Hoang T, Freeman BC, Phillips B and Morimoto RI: Cloning and
subcellular localization of human mitochondrial hsp70. J Biol Chem.
270:1705–1710. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koehler CM: New developments in
mitochondrial assembly. Annu Rev Cell Dev Biol. 20:309–335. 2004.
View Article : Google Scholar
|
|
30
|
Rehling P, Brandner and Pfanner N:
Mitochondrial import and the twin-pore translocase. Nat Rev Mol
Cell Biol. 5:519–530. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wiedemann N, Frazier AE and Pfanner N: The
protein import machinery of mitochondria. J Biol Chem.
279:14473–14476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Craig EA, Kramer J and Kosic-Smithers J:
SSC1, a member of the 70-kDa heat shock protein multigene family of
Saccharomyces cerevisiae, is essential for growth. Proc Natl
Acad Sci USA. 84:4156–4160. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kawai A, Nishikawa S, Hirata A and Endo T:
Loss of the mitochondrial Hsp70 functions causes aggregation of
mitochondria in yeast cells. J Cell Sci. 114:3565–3574.
2001.PubMed/NCBI
|
|
34
|
Kaul SC, Deocaris CC and Wadhwa R: Three
faces of mortalin: a housekeeper, guardian and killer. Exp
Gerontol. 42:263–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maddika S, Booy EP, Johar D, Gibson SB,
Ghavami S and Los M: Cancer-specific toxicity of apoptin is
independent of death receptors but involves the loss of
mitochondrial membrane potential and the release of mitochondrial
cell-death mediators by a Nur77-dependent pathway. J Cell Sci.
118:4485–4493. 2005. View Article : Google Scholar
|