Introduction
Lung cancer was the most commonly diagnosed cancer
as well as the leading cause of cancer death in males in 2008
globally. It was the fourth most commonly diagnosed cancer and the
second leading cause of cancer death among females. Lung cancer
accounted for 1.61 million new cases and 1.38 million deaths in
2008 alone, representing 12.7% of new cancers and 18.2% of cancer
mortality (1).
Crizotinib (PF-02341066; Pfizer) was identified as
an orally bioavailable, potent, ATP competitive small-molecular
inhibitor of the catalytic activity of MET kinase (2,3), and
belongs to the 3-benzyloxy-2-aminopyridine series of kinase
inhibitors (4). Crizotinib is
highly selective for anaplastic lymphoma kinase (ALK) and c-Met
kinases and acts by binding to the adenosine triphosphate (ATP)
binding site of the ALK enzyme (5).
Crizotinib entered into phase I clinical trial in 2006 as a highly
selective MET inhibitor (2,6). The ongoing phase II trial was designed
for ALK-rearranged NSCLC patients who had more than one previous
chemotherapy regimen (7). In phase
I and II trials, crizotinib was shown to be highly active in
patients with advanced ALK-positive NSCLC (8). Based on the phase I and II trial data,
in August 2011 crizotinib was approved by the US Food and Drug
Administration (FDA) for the treatment of patients with locally
advanced or metastatic ALK-positive NSCLC.
Cancer stem cells (CSCs) may be responsible for the
tumorigenesis and contribute to the resistance of cancer cells to
therapeutic interventions (9,10).
Cancer stem cells have an ability to exclude fluorescent
DNA-binding dye (Hoechst 33342) and resist Hoechst 33342 staining
due to the ABCG2 (BCRP1) transporter (11). Other researchers have also
designated the small subset of cells as ‘side population (SP)’
cells, which are a minor population of cells that has been
identified in a variety of cancers and have many CSC-like
properties (12,13).
However, to the best of our knowledge, there is no
evidence showed the effects of crizotinib on Lewis lung carcinoma.
In our studies, we found the antitumor activities of crizotinib on
LLC cells in vivo and in vitro. Crizotinib inducted
apoptosis and G1 arrest in LLC cells by activating the
Smad signaling pathways.
Materials and methods
Cell culture
LLC cells were obtained from the American Type
Culture Collection (ATCC, Bethesda, MD, USA) and grown in RPMI-1640
medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine
serum and antibiotics (100 U/ml penicillin and 100 μg/ml
streptomycin). Cells were maintained in a humidified cell incubator
with 5% CO2 at 37°C.
Side population (SP) cell analysis
LLC cells were suspended at 1×106
cells/ml and then incubated at 37°C for 60 min with 5 μg/ml Hoechst
33342 (Sigma Chemicals, St. Louis, MO, USA). The control cells were
cultured in the presence of 500 μM verapamil (Sigma). After
incubation, 1 μg/ml propidium iodide (PI; KeyGen, Nanjing, China)
was added to identify dead cells. Analysis and sorting of the SP
cells was performed using a FACSVantage SE cytometer
(Becton-Dickinson, San Jose, CA, USA). Hoechst 33342 was excited
using a UV laser at 350 nm and fluorescence emission was measured
at 402–446 nm for Hoechst blue and 640 nm for Hoechst red.
Fluorescent in situ hybridization
Fluorescent in situ hybridization is
currently the gold standard method used in clinical trials to
detect the ALK fusion gene, and it was the first FDA-approved
method for detecting the ALK fusion (8). FISH was done on LCC SP and MP cells
with the use of a break-apart probe specific to the ALK locus
(Vysis LSI ALK Dual Color, Break Apart Rearrangement Probe; Abbott
Molecular, Abbott Park, IL, USA) according to the manufacturer’s
instructions on LLC MP and SP cells. ALK-positive B cell was used
as a positive control.
Colony formation assay
Cells were seeded at 200 cells/well in 24-well
tissue culture plates. After 24 h, cells were treated with various
concentrations of crizotinib (0, 10, 20, 30 and 40 nM for each).
Three days after treatment, colonies were stained with 0.05%
crystal violet containing 50% methanol and counted. The colonies
were counted in 4 to 5 random fields for each of the duplicate
samples by using a microscope at ×100 magnification. The
IC50 value for crizotinib was determined, and was
applied to cells for 0, 6, 12, 24 and 48 h.
Apoptosis assay
Apoptosis was determined using an apoptosis
detection kit (KeyGen). Briefly, cells were collected, washed twice
in ice-cold PBS, and then resuspended in binding buffer at a
density of 1×106 cells/ml. The cells were simultaneously
incubated with fluorescein-labeled Annexin V and propidium iodide
(PI) for 20 min, and flow cytometric (FCM) analysis performed using
a FACSCalibur machine (Model FACSC 420, Baltimore, MD). Data were
analyzed using CellQuest software (BD Biosciences, Baltimore, MD,
USA), and the number of living cells (Annexin
V−/PI−), early apoptotic cells (Annexin
V+/PI−), damaged cells (Annexin
V−/PI+) and necrotic cells (Annexin
V+/PI+) was determined (14).
Measurement of caspase-3, -8 and -9
activities
Caspase activities were measured by colorimetric
assay kit according to the manufacturer’s instructions. After
harvesting, cells were washed in ice-cold PBS and lysed; proteins
were extracted and stored at −80°C until use. Cell lysate (20 μl)
was added to a buffer containing a p-nitroaniline (pNA)-conjugated
substrate (80 μl) for caspase-3 (Ac-DEVD-pNA) (KeyGen; KGA203), -8
(Ac-IETD-pNA) (KeyGen; KGA302), or -9 (LEHD-pNA) (KeyGen; KGA402).
Incubation was performed at 37°C with shaking (500 rpm for 1 min)
and then at room temperature for 2 h. The released pNA in each well
was measured using a plate-reading luminometer (Thermo Fisher
Scientific, Beijing, China). Data were collected from three
independent experiments.
Cell cycle analysis
Cells were collected, centrifuged at 1,500 × g for 5
min, and the pellet was resuspended in 100 μl PBS at a density of
1×106 cells/ml. Cold ethanol (900 μl of 70%) was added
to the mixture for 1 h on ice. Cells were collected by
centrifugation at 1,500 × g for 5 min. The pellet was then
resuspended in 100 μl PBS containing RNaseA (0.2 mg/ml) (Sigma) and
left at room temperature for 30 min. Cells were recovered by
centrifugation and the pellets were resuspended in 350 μl PBS
containing 50 μg/ml PI (KeyGen) and analyzed by flow cytometry
using a FACSCalibur machine (BD Biosciences).
Xenograft assays
All experiments with animals were performed
according to the guidelines of the China Medical University Ethics
Committee. NU/NU nude mice (Crl: NU-Foxn1nu) 6–8-weeks old were
purchased from Charles River (Wilmington, MA, USA). SP
(1×104 in 200 μl), MP cells (1×106 in 200
μl), or LLC cells (1×105 in 200 μl) were subcutaneously
injected into the axilla of each mouse. After the tumor diameter
reached 3–5 mm, the mice were divided randomly into four groups
(untreated, crizotinib, verapamil and crizotinib combined with
verapamil) and received a 100 μl intratumoral injection of PBS,
crizotinib, verapamil, or crizotinib and verapamil. Two injections
were administered at 10 a.m. and 4 p.m. every 2 days. Tumor growth
was then monitored for 40 days. Every 10 days until the end of the
experiment, one mouse from each group was randomly selected to be
anesthetized, photographed and sacrificed. The tissues recovered
were subjected to further analysis. For each tumor, measurements
were made using calipers, and tumor volume was calculated as
follows: length × width2 × 0.52 (15). Tumors were subsequently fixed in 4%
paraformaldehyde for 24 h, then embedded in paraffin.
Quantification of intratumoral
microvessels
For immunohistochemical staining of CD31, endogenous
peroxidase activity was blocked in 4 μm tumor sections with 3%
hydrogen peroxide for 30 min. Antigen retrieval was performed in
citrate buffer (10 mM, pH 6.0) for 30 min at 95°C in a pressure
cooker. CD31 antibodies (Sigma) were incubated with sections at
1:500 overnight at 4°C. Sections were then incubated with a
biotinylated secondary antibody for 60 min at room temperature,
followed by incubation with a streptavidin horseradish peroxidase
(HRP) complex (Beyotime, Beijing, China) for 60 min at room
temperature. Bound antibody was visualized with
3,3′-diaminobenzidine tetrahydrochloride (DAB, Beyotime). Sections
were also counterstained with hematoxylin (Beyotime). Microvessel
density was detected using the method of Ivkovic-Kapicl et
al(16). Regions of highest
vessel density were located at low magnification (x40), then the
number of vessels were counted at ×200 magnification. Three high
magnification fields were counted for each tumor section and the
mean microvessel density value was recorded for each. Any
individual endothelial cells, or endothelial cell cluster, that was
clearly separated from adjacent microvessels was counted as a
single microvessel.
Model of crizotinib bound to Smad3
The structure of Smad3 (PDB code: 1MK2) (17) with crystallographic resolutions of
less than 3.0 Å, were retrieved from the Protein Data Bank
(http://www.rcsb.org). The molecular structure of
crizotinib (CID_11626560) was downloaded from PubChem Compound
(http://www.ncbi.nlm.nih.gov/pccompound). Data were
imported into the modeling software SYBYL-X 1.3 (Tripos
International, St. Louis, MO, USA). All non-protein components such
as water molecules, metal ions, and lipids were deleted and
hydrogen atoms were added to the protein structures. The
interaction of crizotinib and Smad 3 protein was analyzed by
SYBYL-X 1.3.
RT-PCR
Total RNA was isolated from MP and SP cells using an
RNeasy Mini kit (Biomed, Beijing, China). cDNA was reverse
transcribed with 1 μg of total RNA using a Takara Reverse
Transcription kit (Takara Dalian, Dalian, China) and was amplified
using the following primers. CD133 primers were
5′-ACCGACTGAGACCCAACATC-3′ (sense) and 5′-GG TGCTGTTCAGTTCTCCA-3′
(antisense). ABCG2 primers were 5′-AGCTGCAAGGAAAGATCCAA-3′
(sense) and 5′-TCCAGACACACCACGGATAA-3′ (antisense). GAPDH
primers were 5′-AGAAGGCTGGGGCTCATTTG-3′ and (sense) and
5′-AGGGGCCATCCACAGTCTTC-3′ (antisense) and used as an internal
control. The PCR products were electrophoresed on a 1.5% agarose
gel, and visualized by ethidium bromide staining under a UV imaging
system (UVP, LLC; Upland, CA, USA).
Affymetrix microarray analysis
Microarray experiments were conducted according to
standard protocols for Affymetrix Genome U133 Plus 2.0 arrays
(Affymetrix, Inc., Santa Clara, CA, USA) (18). Briefly, using 1 μg of total RNA,
cDNA and biotinated cRNA synthesis was performed using the GeneChip
expression 3′ amplification reagents (one-cycle cDNA synthesis and
IVT labeling) kits of Affymetrix following the manufacturer’s
protocols. Fragmented cRNA was applied to the hybridization and
scanning of the array was performed following the manufacturer’s
protocols.
Antibodies and western blotting
Crude xenograft lysates were then centrifuged at
14,000 × g for 10 min, and cleared lysates were collected and
separated by 10% SDS-polyacrylamide gel electrophoresis and
transferred to nitrocellulose membranes. Membranes were blocked in
5% BSA-TBST then incubated with primary antibodies. Antibodies used
in our study are summarized in Table
I. The reaction was followed by probing with peroxidase-coupled
secondary antibodies including anti-mouse IgG, anti-rabbit IgG, or
anti-goat IgG antibodies at dilutions ranging from 1:1,000 to
1:2,000 (Amersham Biosciences, Needham, MA, USA), and binding
results were visualized by enhanced chemiluminescence (Amersham
Pharmacia, Piscataway, NJ, USA).
 | Table IThe antibodies used in the western
blot analysis. |
Table I
The antibodies used in the western
blot analysis.
| Protein | Producer | Catalog no. | Dilution |
|---|
| ABCG2 | Abcam (Cambridge,
UK) | ab24114 | 1:200 |
| CD133 | | ab19898 | 1:200 |
| P-Smad3 | Santa Cruz
Biotechnology (Santa Cruz, CA, USA) | sc-130218 | 1:200 |
| Smad3 | | sc-101154 | 1:200 |
| P-Smad2 | | sc-135644 | 1:100 |
| Smad2 | | sc-6200 | 1:100 |
| Smad4 | | sc-7966 | 1:200 |
| β-actin | | sc-47778 | 1:1,000 |
Statistical analysis
Each experiment was performed in triplicate.
Statistical analysis was performed using one-tailed Student’s
t-test (unilateral and unpaired). Differences with a P-value
<0.05 were considered to indicate a statistically significant
result.
Results
Side population (SP) fraction and main
population (MP) fraction in LLC show no ALK-rearrangement
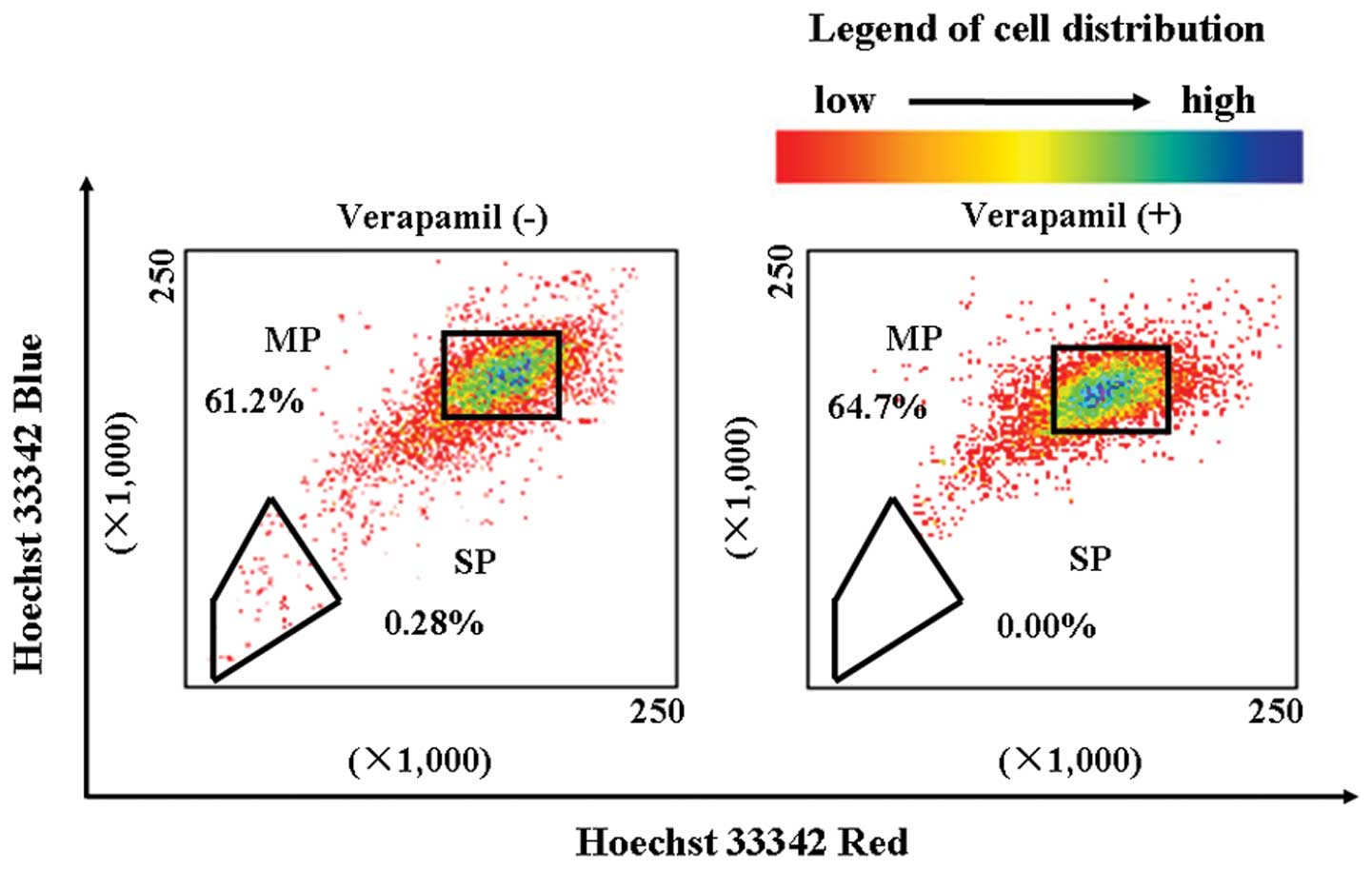
LLC cells were labeled with Hoechst 33342 and
analyzed by flow cytometry. We found that the SP cell fraction
comprised 0.28% of the total cells, and that this population
disappeared following treatment with the selective ABCG2
transporter inhibitor, verapamil (Fig.
1). The SP and MP of LLC cells were tested for ALK
rearrangements by FISH analysis. A break-apart FISH probe has been
used to detect the ALK fusion (Fig.
2A), and the probes are designed for the telomeric and
centromeric sides of the break points. The terminal part of the der
(3) harboring the 3′ end of ALK
showed red signal and loss of the 5′ end of ALK showed green
signal. SP and MP cells showed undetectable ALK rearrangement
(Fig. 2C and D). In contrast, ALK
rearrangement was readily detectable in an ALK-rearranged
anaplastic large B cell lymphoma (Fig.
2B).
Crizotinib exposure inhibits
proliferation and induces apoptosis and G1 arrest in MP
and SP cells
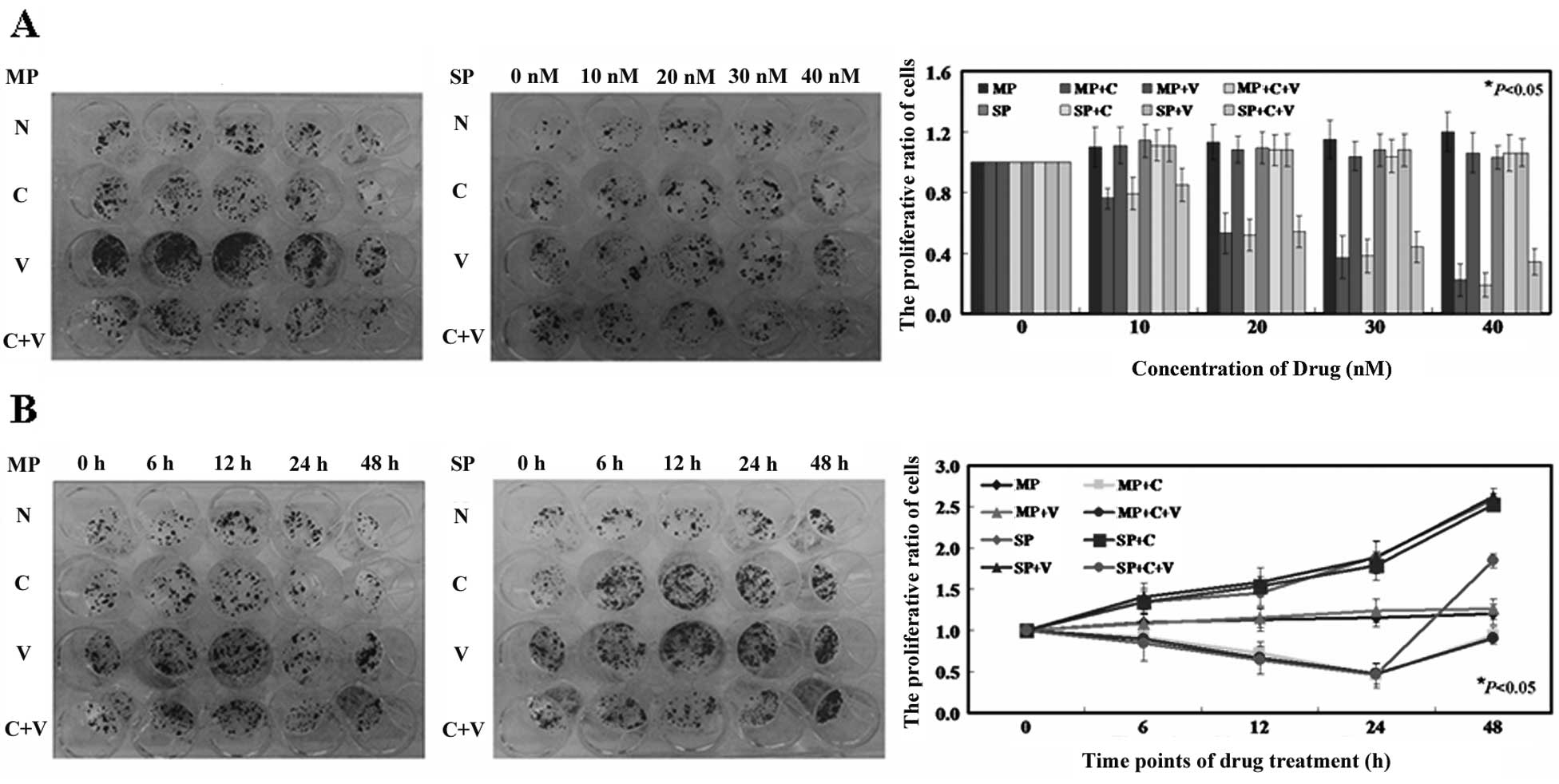
As shown in Fig. 3A,
the inhibitory effects of crizotinib on MP cells and SP cells were
determined by colony formation assay. The IC50 value of
crizotinib for MP cells was 21.3 nM. Of note, the SP cells showed
no significant changes after crizotinib treatment. However, the SP
cells showed a cell survival rate of 50.0±0.6% following a combined
treatment of crizotinib (22.4 nM) and verapamil (500 μM), compared
with 105.3±0.4% survival of SP cells treated with crizotinib (22.4
nM) alone (Fig. 3A, P<0.05). The
growth curves obtained demonstrate that crizotinib inhibited the
growth of SP and MP cells, and this inhibition was dependent on
both concentration and time (Fig. 3A
and B). In addition to the inhibitory effect on proliferation,
crizotinib also induced apoptosis in the MP cells. The apoptotic
ratio of MP cells exposed to 21.3 nM crizotinib was 4.36±0.15% by
using Annexin V and PI double staining, whereas 0.21±0.09% was
observed in the untreated MP cells (Fig. 3C, P<0.05). Similarly, SP cells
after the combined treatment of crizotinib and verapamil showed a
higher apoptotic ratio (4.67±0.14%) compared with untreated SP
cells (0.19±0.05%) and SP cells after crizotinib treatment
(0.63±0.08%) (Fig. 3C, P<0.05).
Next, the effects of crizotinib on cell cycle progression were
examined. As shown in Fig. 3D, MP
cells treated with crizotinib and SP cells treated with crizotinib
and verapamil were observed to arrest in the G1 phase of
the cell cycle. For example, the percentages of MP cells in the
G1 phase following treatment with crizotinib was
67.4±3.5% and 68.4±3.2% of SP cells after the combined treatment of
crizotinib and verapamil, respectively (P<0.05). Furthermore,
the activity of caspase-3, -8 and -9 was significantly increased in
the crizotinib-treated MP cells and crizotinib and
verapamil-treated SP cells (Fig.
3E, P<0.05). Verapamil showed no antitumor effects on MP and
SP cells (Fig. 3, P>0.05).
Crizotinib inhibits tumor growth and
angiogenesis in vivo
Tumorigenicity was examined using immune-deficient
mice, into which SP or MP cells of LLC were subcutaneously
transplanted. Nonsorted LLC cells formed xenografts in mice at
1×105 cells. Transplantation of 1×105 MP
cells consistently failed to form tumors in all mice (n=5), while
1×106 MP cells showed tumor formation in one of five
mice. The tumor volume of MP cell xenografts was smaller than that
of SP cells. In contrast, transplantation of 1×104 SP
cells produced tumors in all of five mice and the tumor volume were
larger than MP group and LLC group (Fig. 4A). We next determined whether
crizotinib displays antitumor properties in established xenograft
tumor models. As shown in Fig. 4B,
the tumor volume of crizotinib-treated LLC mice was 1.1- to
3.2-fold less than untreated LLC mice (P<0.05). Tumor size was
significantly decreased in the crizotinib-treated LLC groups
(225±29 mm3) compared to the untreated group (PBS:
834±41 mm3) by 40 days after treatment. Similarly, tumor
weight for the crizotinib-treated LLC group was also significantly
reduced compared to the untreated LLC group (Fig. 4C, P<0.05). We assessed intratumor
microvessels by immunostaining for CD31 expression. Representative
examples are shown in Fig. 4D. The
blood vessel density of tumors from mice in the crizotinib-treated
LLC group (1.2±0.52 per mm2) was significantly reduced
compared to the untreated LLC group (4.6±0.64 per mm2)
(P<0.05). We also found that crizotinib and verapamil-treated SP
mice displayed similar effects to crizotinib treated LLC mice. The
tumor volume and weight of SP mice after the combined treatment of
crizotinib and verapamil were significantly lower than that of
untreated SP mice (Fig. 4B and C,
P<0.05). Consistent with in vitro results, verapamil
showed no antitumor effects on LLC group and SP group in
vivo (Fig. 4, P>0.05).
Crizotinib binds to Smad3 and activates
Smad4-dependent signaling
To explore the possible proteins that could interact
with crizotinib, we applied the modeling software SYBYL-X 1.3 and
found that crizotinib could dock into Smad3. Fig. 5B showed the binding sites between
crizotinib and Smad3. To investigate whether crizotinib can play an
anti-tumor role by activating Smad signaling pathway, we performed
western blot analysis using antibodies that recognize
phosphorylated, active Smad family members. Western blot analysis
showed that phosphorylated levels of Smad2 and Smad3 significantly
increased in crizotinib treated LLC cell xenografts and crizotinib
and verapamil treated SP cell xenografts (Fig. 5D). Total protein levels of Smad2 and
Smad3 remained unchanged (Fig. 5D).
In contrast, the total protein level of Smad4 significantly
increased in LLC xenografts and SP xenografts after treatment
(Fig. 5D). Interestingly, the level
of Smad3 mRNA showed no changes in LLC xenografts and SP xenografts
after treatment by using Affymetrix microarray analysis (Fig. 5A) (Table II). The results confirmed
crizotinib activates Smad3 protein, but does not affect Smad3
transcription. The traits of LLC SP cells maintained in both in
vitro and in vivo experiments were evidenced by
detecting CD133 and ABCG2 expression (Fig. 5C).
| Table IISummary of Smad RNAs in LLC cell
xenografts and SP cell xenografts |
Table II
Summary of Smad RNAs in LLC cell
xenografts and SP cell xenografts
| LLC cell
xenografts | SP cell
xenografts |
|---|
|
|
|---|
| RNA ID | Ct | RNA ID | Ct |
|---|
| Untreated |
| ABCG2 | 0.014 | ABCG2 | −1.425 |
| CD133 | 0.023 | CD133 | −2.993 |
| Smad3 | 2.054 | Smad3 | 2.047 |
| Smad2 | −1.534 | Smad2 | −1.556 |
| Smad4 | 2.861 | Smad4 | 3.452 |
| GAPDH | −2.245 | GAPDH | −2.131 |
| Crizotinib |
| ABCG2 | a | ABCG2 | −1.367 |
| CD133 | 0.024 | CD133 | −2.975 |
| Smad3 | 2.391 | Smad3 | 2.889 |
| Smad2 | −1.231 | Smad2 | −1.572 |
| Smad4 | −3.884 | Smad4 | 3.425 |
| GAPDH | −2.028 | GAPDH | −2.226 |
| Verapamil |
| ABCG2 | 0.034 | ABCG2 | −1.352 |
| CD133 | 0.045 | CD133 | −2.934 |
| Smad3 | 2.196 | Smad3 | 2.717 |
| Smad2 | −1.264 | Smad2 | −1.595 |
| Smad4 | 2.885 | Smad4 | 3.314 |
| GAPDH | −2.168 | GAPDH | −2.157 |
| Crizotinib and
verapamil |
| ABCG2 | 0.035 | ABCG2 | −1.453 |
| CD133 | a | CD133 | −2.763 |
| Smad3 | 2.201 | Smad3 | 2.624 |
| Smad2 | −1.376 | Smad2 | −1.528 |
| Smad4 | −3.874 | Smad4 | −3.642 |
| GAPDH | −2.264 | GAPDH | −2.142 |
Discussion
As noted in the introduction, crizotinib was
approved by the US Food and Drug Administration (FDA) for the
treatment of patients with locally advanced or metastatic
ALK-positive NSCLC in August, 2011. However, no reports showed
other applications and mechanisms of crizotinib in lung cancer
treatment. In the present study, we confirmed the antitumor effects
of crizotinib on Lewis lung carcinoma in vitro and in
vivo. Crizotinib in LLC MP cells decreased proliferation and
induced G1 arrest and apoptosis. Additionally, crizotinib in LLC
xenografted tumors inhibited tumor growth. We found intratumor
microvessels were significantly reduced after treatment with
crizotinib. However, crizotinib showed inhibitory effects on SP
cells only combined with verapamil. SP cells are refractory to
Hoechst 33342 dye-staining and certain drugs due to the ABCG2
(BCRP1) transporter (11,19). The activity of the transporter is
inhibited by verapamil, which results in the disappearance of the
SP streak from the FACS plot (20,21).
Consistent with previous studies, we confirmed crizotinib was able
to decrease the proliferation of SP cells when the activity of
ABCG2 was inhibited by verapamil.
Several potential oncogenic drivers have been
identified in NSCLC, such as EGFR, BRAF, KRAS, MET, HER2 and ALK
(22–24). Crizotinib is a potent and selective
ATP-competitive inhibitor of ALK receptor tyrosine kinases and
oncogenic variants (2,3,25). A
recent study showed that crizotinib as a potential
radiation-sensitizing agent in multiple established NSCLC cell
lines with varying expression level of EML4-ALK (26). In the present study, we found
another mechanism of crizotinib in LLC cells. To the best of our
know-ledge, no reports showed ALK-rearrangement in LCC cells.
Currently several laboratories rely on FISH analysis of mitotic or
interphase tumor nuclei and identify a ‘split’ hybridization signal
to establish the presence of an ALK-rearrangement (27). We also confirmed no
ALK-rearrangement in MP and SP of LLC cells by FISH analysis. The
result indicated that crizotinib may play its antitumor role in LLC
cells though other signaling pathways. The identification of
candidate binding sites of proteins with crizotinib is the first
step to detect the signaling pathway. In our previous study, we
found crizotinib docked into Smad3 by using iGEMDOCK (version 2.1)
and p-Smad3 was increased in A549 cells after crizotinib treatment
(28). In this study, we confirmed
crizotinib could dock into the active center of Smad3 by SYBYL-X
1.3. Activated Smad3 (P-Smad3) was detected in LLC cells after
crizotinib treatment and SP cells after crizotinib and verapamil
treatment by western blot analysis. The downstream protein of
Smad3, Smad2 was also activated and Smad4 expression was increased.
Phosphorylated Smad2/3 could form complexes with Smad4 and
translocate to the nucleus. The complexes regulated the
transcription of target genes by direct binding to specific DNA
sequences (29,30). Previous studies have demonstrated
that Smad activation could induce apoptosis and cell cycle arrest
(31–33). Consistent with these studies, we
also found activation of Smad signaling could induce G1
arrest and apoptosis in LCC cells.
Collectively, these studies are the first to
determine the effects of crizotinib on Lewis lung carcinoma cells
in vivo and in vitro. We demonstrate that crizotinib
plays a tumor suppressor role in LLC cells through Smad signaling.
These results provide a novel insight into possible therapeutic
application of crizotinib.
Acknowledgements
We thank Miss Jing-Jing Wang for her valuable
comments and excellent technical assistances.
References
|
1
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 6:69–90. 2011. View Article : Google Scholar
|
|
2
|
Christensen JG, Zou HY, Arango ME, et al:
Cytoreductive antitumor activity of PF-2341066, a novel inhibitor
of anaplastic lymphoma kinase and c-Met, in experimental models of
anaplastic large-cell lymphoma. Mol Cancer Ther. 6:3314–3322. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zou HY, Li Q, Lee JH, et al: An orally
available small-molecule inhibitor of c-Met, PF-2341066, exhibits
cytoreductive antitumor efficacy through antiproliferative and
antiangiogenic mechanisms. Cancer Res. 67:4408–4417. 2007.
View Article : Google Scholar
|
|
4
|
Timofeevski SL, McTigue MA, Ryan K, et al:
Enzymatic characterization of c-Met receptor tyrosine kinase
oncogenic mutants and kinetic studies with aminopyridine and
triazolopyrazine inhibitors. Biochemistry. 48:5339–5349. 2009.
View Article : Google Scholar
|
|
5
|
Li Y, Ye X, Liu J, et al: Evaluation of
EML4-ALK fusion proteins in non-small cell lung cancer using small
molecule inhibitors. Neoplasia. 13:1–11. 2011.PubMed/NCBI
|
|
6
|
Ou SH, Bazhenova L, Camidge DR, et al:
Rapid and dramatic radiographic and clinical response to an ALK
inhibitor (crizotinib, PF02341066) in an ALK translocation-positive
patient with non-small cell lung cancer. J Thorac Oncol.
5:2044–2046. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Crinò L, Kim D, Riely GJ, et al: Initial
phase II results with crizotinib in advanced ALK-positive non-small
cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol.
29:75142011.
|
|
8
|
Kwak EL, Bang YJ, Camidge DR, et al:
Anaplastic lymphoma kinase inhibition in non-small-cell lung
cancer. N Engl J Med. 363:1693–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klonisch T, Wiechec E, Hombach-Klonisch S,
et al: Cancer stem cell markers in common cancers - therapeutic
implications. Trends Mol Med. 14:450–460. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dingli D and Michor F: Successful therapy
must eradicate cancer stem cells. Stem Cells. 24:2603–2610. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kondo T, Setoguchi T and Taga T:
Persistence of a small subpopulation of cancer stem-like cells in
the C6 glioma cell line. Proc Natl Acad Sci USA. 101:781–786. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu C, Wei Q, Utomo V, et al: Side
population cells isolated from mesenchymal neoplasms have tumor
initiating potential. Cancer Res. 67:8216–8222. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Szotek PP, Pieretti-Vanmarcke R, Masiakos
PT, et al: Ovarian cancer side population defines cells with stem
cell-like characteristics and Mullerian Inhibiting Substance
responsiveness. Proc Natl Acad Sci USA. 103:11154–11159. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krysko DV, Vanden Berghe T, D’Herde K, et
al: Apoptosis and necrosis: detection, discrimination and
phagocytosis. Methods. 44:205–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alessandri G, Filippeschi S, Sinibaldi P,
et al: Influence of gangliosides on primary and metastatic
neoplastic growth in human and murine cells. Cancer Res.
47:4243–4247. 1987.PubMed/NCBI
|
|
16
|
Ivković-Kapicl T, Knelević-Usaj S,
Panjković M, et al: Immunohistochemical analysis of angiogenesis in
invasive ductal breast carcinoma with correlations to
clinicopathological factor. Vojnosanit Pregl. 63:635–642. 2006.(In
Serbian).
|
|
17
|
Qin BY, Lam SS, Correia JJ, et al: Smad3
allostery links TGF-β receptor kinase activation to transcriptional
control. Genes Dev. 16:1950–1963. 2002.PubMed/NCBI
|
|
18
|
Martínez T and Pascual A: Gene expression
profile in β-amyloid-treated SH-SY5Y neuroblastoma cells. Brain Res
Bull. 72:225–231. 2007.
|
|
19
|
Zillhardt M, Christensen JG and Lengyel E:
An orally available small-molecule inhibitor of c-Met, PF-2341066,
reduces tumor burden and metastasis in a preclinical model of
ovarian cancer metastasis. Neoplasia. 12:1–10. 2010.
|
|
20
|
Tumati V, Kumar S, Yu L, et al: Effect of
PF-02341066 and radiation on non-small cell lung cancer cells.
Oncol Rep. 29:1094–1100. 2012.PubMed/NCBI
|
|
21
|
Hirschmann-Jax C, Foster AE, Wulf GG, et
al: A distinct ‘side population’ of cells with high drug efflux
capacity in human tumor cells. Proc Natl Acad Sci USA.
101:14228–14233. 2004.
|
|
22
|
Bronte G, Rizzo S, La Paglia L, et al:
Driver mutations and differential sensitivity to targeted
therapies: a new approach to the treatment of lung adenocarcinoma.
Cancer Treat Rev. 36(Suppl 3): S21–S29. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garber K: ALK, lung cancer, and
personalized therapy: portent of the future? J Natl Cancer Inst.
102:672–675. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Janku F, Stewart DJ and Kurzrock R:
Targeted therapy in non-small-cell lung cancer - is it becoming a
reality? Nat Rev Clin Oncol. 7:401–414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Challen GA and Little MH: A side order of
stem cells: the SP phenotype. Stem Cells. 24:3–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hadnagy A, Gaboury L, Beaulieu R, et al:
SP analysis may be used to identify cancer stem cell populations.
Exp Cell Res. 312:3701–3710. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shaw AT, Yeap BY, Mino-Kenudson M, et al:
Clinical features and outcome of patients with non-small-cell lung
cancer who harbor EML4-ALK. J Clin Oncol. 27:4247–4253. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xia P, Mou FF and Wang LW: Predictive role
of computer simulation in assessing signaling pathways of
crizotinib-treated A549 lung cancer cells. Asian Pac J Cancer Prev.
13:3119–3121. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jacob D, Davis J, Zhu H, et al:
Suppressing orthotopic pancreatic tumor growth with a
fiber-modified adenovector expressing the TRAIL gene from the human
telomerase reverse transcriptase promoter. Clin Cancer Res.
10:3535–3541. 2004. View Article : Google Scholar
|
|
30
|
Zhu H, Zhang L, Huang X, et al: Overcoming
acquired resistance to TRAIL by chemotherapeutic agents and calpain
inhibitor I through distinct mechanisms. Mol Ther. 9:666–673. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grau AM, Zhang L, Wang W, et al: Induction
of p21waf1 expression and growth inhibition by transforming growth
factor β involve the tumor suppressor gene DPC4 in human pancreatic
adenocarcinoma cells. Cancer Res. 57:3929–3934. 1997.
|
|
32
|
Guo Y and Kyprianou N: Overexpression of
transforming growth factor (TGF) β1 type II receptor restores
TGF-β1 sensitivity and signaling in human prostate cancer cells.
Cell Growth Differ. 9:185–193. 1998.
|
|
33
|
Tang MR, Wang YX, Guo S, et al: CSMD1
exhibits antitumor activity in A375 melanoma cells through
activation of the Smad pathway. Apoptosis. 17:927–937. 2012.
View Article : Google Scholar : PubMed/NCBI
|