Introduction
Human gastrointestinal stromal tumors (GISTs),
mesenchymal tumors of the gastrointestinal tract (1), originate from the neoplastic
transformation of interstitial cells of Cajal (ICC), which
frequently express mutations in the c-KIT gene and
occasionally in PDGFRA(2).
The resulting mutations of KIT and PDGFRA receptors result in
constitutive activation of receptor kinase activity leading to
downstream effectors that deregulate cell proliferation and
survival, thereby accelerating malignant progression (3). Surgery is currently the first-line
treatment for patients with primary resectable GISTs (4,5).
However, many patients develop recurrent or metastatic disease
despite complete surgical resection (6) and since conventional chemotherapy or
radiotherapy is usually ineffective (7). Thus, there are no ideal methods for
treating such GISTs.
Imatinib mesylate (imatinib) is a tyrosine kinase
(TK) inhibitor that targets BCR-ABL, PDGFR, KIT, DDR and CSFR, and
has been used to treat certain patients with KIT-positive GISTs
having constitutive activating mutations in KIT (8). Although more than 80% of inoperable
KIT-positive GIST patients exhibit clinical benefits from imatinib,
the tumors in most of these patients will eventually progress
(9,10). The activity of imatinib differs
across various types of c-KIT and PDGFRA mutations,
and secondary resistance in imatinib-treated patients often results
from an emerging secondary mutation or amplification of
c-KIT or PDGFRA(11–13).
Approximately 50% of GISTs with secondary resistance to imatinib is
caused by mutations in c-KIT or PDGFRA(14–18).
Several studies have reported that the RTK switch is associated
with imatinib-resistance, but the other mechanisms of secondary
resistance remain to be elucidated.
Although many GISTs are driven by activating KIT
mutations, some tumors only express non-mutated KIT (c-KIT
mutation-negative GISTs) Although c-KIT mutation-positive
GISTs show more frequent liver metastases and higher mortality than
c-KIT mutation-negative GISTs (19,20),
the detailed mechanism of cell migration and metastasis in GIST has
not been characterized. Molecular-based research on GIST has been
hampered by a lack of the availability of suitable animal models
with peritoneal dissemination or liver metastasis using human GIST
tissues.
Our purpose in this study was to establish cell
lines and xenografts models from clinical samples of human GIST
cases and to verify their characteristics in vitro or in
vivo. Tumor tissues were collected and immediately processed
for culture and transplantation into NOG mice. Two cell lines, GK1C
and GK3C, together with three xenografts, GK1X, GK2X and GK3X, were
generated successfully. The established GIST cell lines and
xenografts were characterized using cell morphology, growth
kinetics, immunohistochemistry, drug sensitivity and tumorigenicity
in SCID or nude mice. This new GIST model may be helpful in
improving our understanding of the molecular mechanisms of
c-KIT- or PDGFRA-mediated metastasis and may be
useful for assessments of molecular therapeutics, drug resistance
and in vivo imaging.
Materials and methods
Clinical samples
This study was approved by the Ethics Committee of
the Chamber of Surgeons of the School of Medicine, Keio University
(no. 17–47). Human gastrointestinal stromal tumors (GISTs) were
obtained from patients undergoing surgical resection following
informed patient consent. Between November, 2007 and June, 2010,
GIST tissues from 18 patients were collected and cultured or
implanted into NOG mice. In addition to histological criteria, the
following antibodies were used for immumohistological
classification of all GISTs: CD117, CD34 and Ki-67. The mitotic
count was categorized as follows: <5/50 high power fields (HPF),
5–10/50 HPF, or >10/50 HPF according to the GIST consensus
approach of Fletcher risk classification (21). Metastatic risk was classified as
low, intermediate, or high, respectively, by Miettinen
classification. Patient data including gender, age, tumor size,
clinicopathological and histopathological results were obtained
from the clinical and pathological records. Table I documents the profile of samples
from patients with GISTs in the present study.
 | Table IPatient characteristics. |
Table I
Patient characteristics.
| Clinical sample,
N=18 (%) |
|---|
| Age (years), mean
(range) | 61 (43–75) |
| ≤69 | 16 (77.8) |
| >70 | 5 (22.2) |
| Gender |
| Male | 10 (44.4) |
| Female | 11 (55.6) |
| Tumor size
(cm) |
| ≤5 | 10 (55.6) |
| >5a | 8 (44.4) |
| Mitotic index (/50
HPF) |
| ≤5 | 12 (66.7) |
| >5a | 6 (33.3) |
| Risk of
metastasisb |
| Very low | 2 (11.1) |
| Low | 7 (27.8) |
| Intermediate | 4 (22.2) |
| Higha | 8 (38.9) |
|
Immunohistochemistry |
| KIT | 18 (100) |
| CD34 | 18 (100) |
Primary culture and evaluation of tumor
forming ability
Clinical samples were subjected to mechanical and
enzymatic dissociation. The cells were cultured in RPMI-1640
containing 20% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA,
USA), penicillin-streptomycin mixed solution (penicillin 10,000
units/ml, streptomycin 10,000 μg/ml; Nacalai Tesque Inc., Kyoto,
Japan) and 10 ng/ml SCF (Peprotech Inc., Rocky Hill, NJ, USA). In
all experiments, cells were cultured at 37°C in a humidified 5%
CO2, 95% air atmosphere. After culture, the established
GIST cells were collected and re-suspended in HBSS. Cell
suspensions were then mixed with Matrigel (1:1) (Becton-Dickinson,
San Jose, CA, USA). The cell-Matrigel suspension was then
subcutaneously injected into SCID
(C.B-17/lcr-scid/scidJcl) mice, aged 8 to 10 weeks
(Japan Clea Laboratories, Tokyo, Japan) under anesthesia. Tumor
growth was observed weekly after inoculation. Pieces of
subcutaneous tumors were re-transplanted and embedded in
paraffin.
Implantation of tumor tissues
Tumor tissues from GIST patients were implanted into
NOG (NOD/Shi-scid, IL-2Rrnu) mice. These tumor
tissues were obtained from surgical resection of the primary tumor.
Tumor tissues were collected in serum-free Hanks’ balanced salt
solution (HBSS) medium and immediately processed for
transplantation. The tumor specimens were cut into small fragments
(5 mm3) and kept in a Petri dish containing
physiological saline. The tissue fragments were implanted
bilaterally into 6-week old NOG mice. Tumor growth was monitored
until it reached 1 cm in diameter after three months. These
xenograft lines were maintained by serial passage in NOG mice in
our animal facility. GIST engraftment was assessed by
immunohistochemistry and DNA mutation assay.
Immunohistochemical staining
Specimens were fixed with 4% paraformaldehyde for 24
h at room temperature. Immunohistochemical staining for CD117 (KIT)
was performed on 4-μm sections placed on pre-coated slides with APS
(Matsunami Glass Industries Ltd., Osaka, Japan). Briefly, slides
were incubated with blocking reagent-N101 (Wako Pure Chemicals
Industries, Ltd., Tokyo, Japan) for 20 min. After rinsing in PBST,
avidin and biotin blocking was performed for 15 min each. Slides
were incubated with anti-human CD117 mAb (Dako). A biotinylated
antibody (Vectastain ABC kit) was then used as the secondary
antibody for 30 min, with a 10-min DAB staining reaction. Slides
were counterstained with haematoxylin. Finally slides were
cover-slipped with aqueous mounting medium (Aquatex®,
Merck). Specimens were analyzed under a light microscope, and CD117
positivity was defined as strong membrane and cytoplasmic staining
in at least 50–75% of cells.
Flow cytometric analysis
For the evaluation of CD117 (KIT) and PDGFRA
(CD140a) phosphorylation status, cells were collected and washed
with PBS and fixed with 2% paraformaldehyde (PFA) at 37°C in a
water bath for 10 min. The cells were then washed with PBS and
pelleted by centrifugation (800 × g) for 5 min, and the supernatant
was removed. The tube was mixed to disrupt the pellet and
permeabilized by adding 500 μl of 90% methanol (for
1×106 cells) and incubating on ice for 15 min. After
blocking on ice for 10 min, cells were then washed by PBS and
incubated with primary antibodies against phospho-KIT (Tyr719) and
phospho-PDGFRA (Tyr754) (Cell Signaling Technology, Inc., Danvers,
MA, USA) for 60 min at room temperature. The cells were washed with
PBS before incubation for 30 min with Alexa Fluor 488 donkey
anti-rabbit IgG antibody (Invitrogen, Carlsbad, CA, USA). Each
sample was then analyzed using a FACSCalibur™ (Becton-Dickinson,
Franklin Lakes, NJ, USA). The distribution of cells was analyzed
using FlowJo software (Tomy Digital Biology, Tokyo, Japan).
Assessment of imatinib sensitivity of the
cell lines
Cells were plated in 96-well microplates and
cultured for 12 h before exposure to imatinib (1–100 μM) for 72 h.
The cells were quantified by the WST-8 assay. The optical density
(OD) was determined with Sunrise Rainbow (Wako Pure Chemical
Industries). The rate of inhibition was calculated as follows: % of
inhibition = (OD of treated group - blank)/(OD of control group -
blank) × 100%. The concentration of tested drugs resulting in 50%
growth inhibition (IC50) was calculated.
In vivo drug assay
Tumor tissue was collected in serum-free HBSS medium
and immediately processed for transplantation. The tumor specimen
was cut into small fragments 2 mm3 and kept in a Petri
dish containing physiological saline. The tissue fragments were
implanted bilaterally into 7-week-old nude mice (n=10). Growth
factors, hormones, Matrigel and other supplements were not used.
Drug administration was initiated when tumors in each group
achieved an average volume of 120–350 mm3. Mice were
randomly allocated to control and treatment groups. Treatment
groups consisted of control and imatinib. Each treatment group
included 6–8 mice. Imatinib was administered at a dose of 40
mg/kg/day and given by oral gavage daily for 28 days. Control
animals received saline administration. Tumor volume (TV) was
determined from caliper measurements of tumor length (L) and width
(W) according to the formula LW2/2. TV and body weight
were determined every two to three days and on the day of
evaluation. Relative TV (RTV) on evaluation day was calculated as
the ratio of TV on evaluation day to that on day 1 according to the
following formula: RTV = (TV on evaluation day)/(TV on day 1). The
percentage of tumor growth inhibition (TGI %) was calculated as
follows: TGI (%) = [1 − (tumor volume of treatment group on
evaluation day - tumor volume of treatment group on day 1)/(tumor
volume of control group at evaluation day - tumor volume of control
group on day 1)] × 100%. The percentage of body weight change
(BWC%) was calculated as follows: BWC (%) = [(BW on evaluation day)
- (BW on day 1)]/(BW on day 1) × 100%.
Mutation analysis
DNA was extracted from gastric cancer cell lines
using a QIAmp DNA Mini kit (Qiagen, Düsseldorf, Germany). A
NanoDrop ND-1000 (NanoDrop Technologies) was used to evaluate the
concentration of the samples. c-KIT gene exons 9, 11, 13 and
17 and PDGFRA gene exons 12 and 18 were amplified in PCR
reactions. DNA sequencing was performed by SRL, Inc. Images were
obtained with the SeqScape v2.6 sequence analysis program.
Statistical analysis
Data values are expressed as means ± SD or mean-fold
change. The statistical significances of mean values were
determined by one-way ANOVA first, then by Studen’t t-test. P-value
≤0.05 was considered significant for the ANOVA test. P-value ≤0.01
was considered significant for the Student’s t-test.
Results
Establishment of GIST cell lines and
xenografts from clinical samples
To establish cell lines and xenografts from GIST
tumors, tumor tissue was subcutaneously transplanted to NOG mice or
put into primary culture. GIST cell lines and xenografts were
established from three clinical specimens (GIST1, GIST2 and GIST3).
These specimens were positive for KIT and CD34, which were
classified as high risk GISTs based on tumor size and mitotic rate.
Mutation analyses of c-KIT exons 9, 11, 13, 14 and 17 and
PDGFRA exons 12 and 18 were performed by direct sequencing.
Mutations were detected in c-KIT exon 11: del (550–558), del
(557–558) and del (591–592), respectively, but a PDGFRA
mutation was not detected (Table
II). The two new cell lines (GK1C and GK3C) and three
xenografts (GK1X, GK2X and GK3X) were generated from these clinical
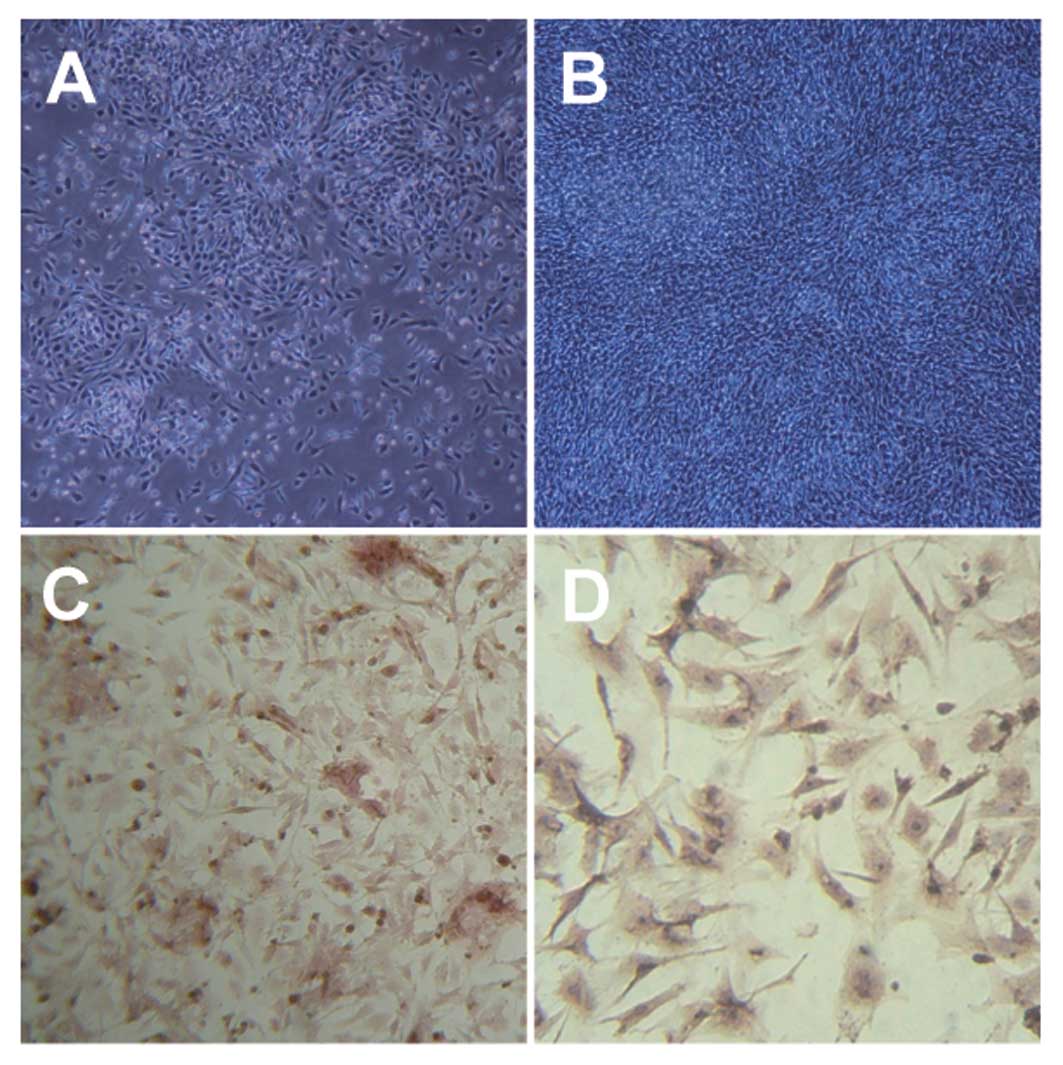
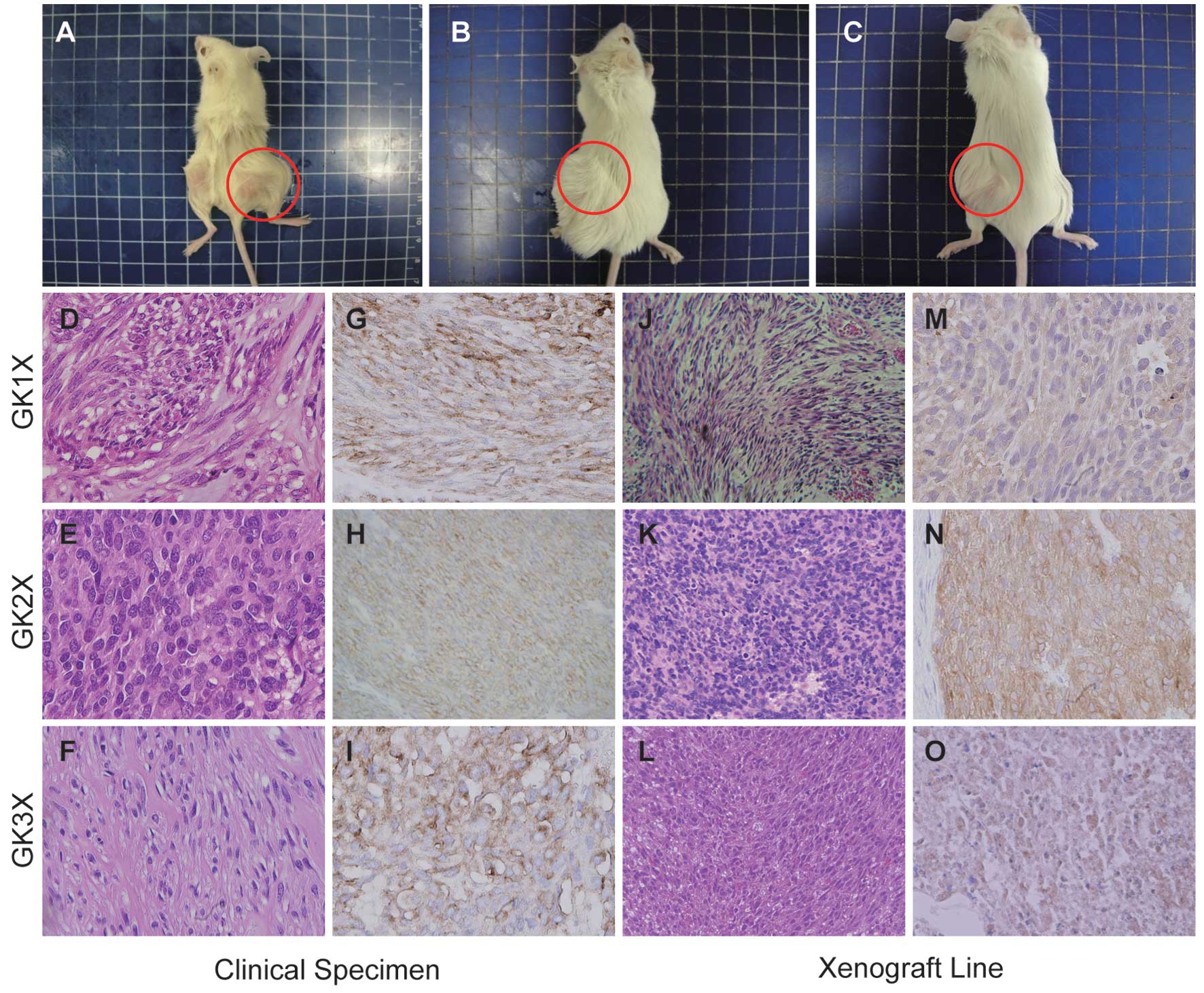
samples (Fig. 1A and B and Fig. 2A-C). The established cells and
xenografts were subject to repeated passages. In xenografts,
tumor-doubling time was found to be ~45 days. Microscopic
examination of the initial human GISTs and the xenografts revealed
similar morphological appearances (Fig.
2D-F and J-L), comprising moderate cellular tissue with
atypical epithelioid and spindle-shaped cells. Immunohistochemical
or immunocytochemical analysis for KIT expression in the 2 cell
lines and 3 xenografts was carried out by DAB staining. These
results indicated that established cells and xenografts were
positive for KIT (Fig. 1C and D and
Fig. 2G-I and M-O).
| Table IIAnalysis of c-KIT and
PDGFRA mutations. |
Table II
Analysis of c-KIT and
PDGFRA mutations.
| ID | Cell line | Xenograft | c-KIT
mutation | PDGFRA
mutation |
|---|
| GIST 1 | GK1C | GK1X | Ex.11: del
(550–558) | WT |
| GIST 2 | - | GK2X | Ex.11: del
(557–558) | WT |
| GIST 3 | GK3C | GK3X | Ex.11: del
(591–592) | WT |
Potential of GIST cell lines for
tumorigenesis
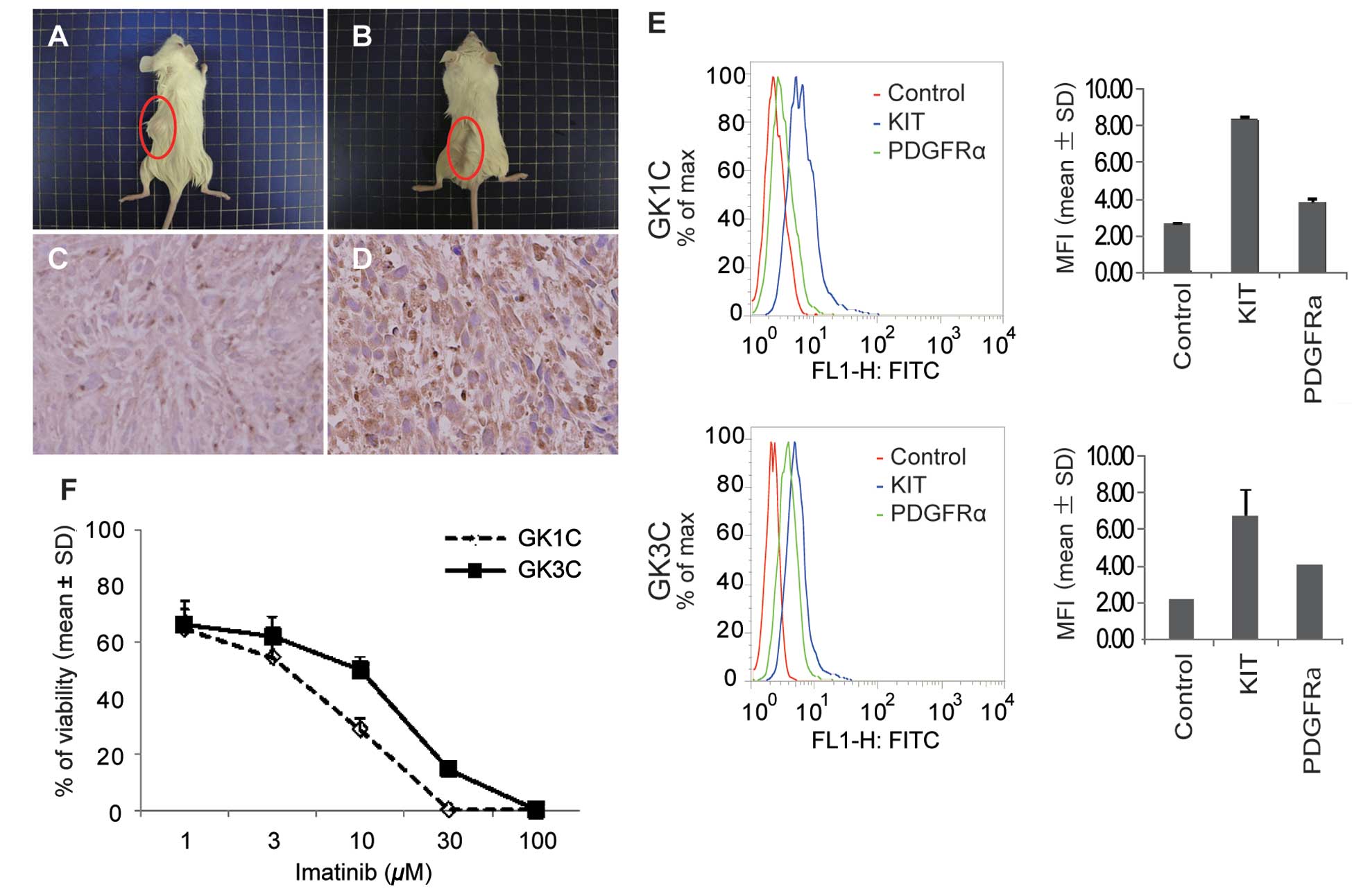
Cells (5×106) from 2 GIST cell lines
(GK1C and GK3C) were injected subcutaneously into SCID mice. Tumor
formation was observed for 12 weeks after injection (Fig. 3A and B). KIT expression was detected
by immunohistochemistry (Fig. 3C and
D). These results indicated that the 2 GIST cell lines
possessed the ability for tumorigenesis.
Intracellular phosphorylation of KIT and
PDGFRA
To investigate the activation of KIT and PDGFRA, we
examined the phosphorylation of the signaling pathway. GK1C and
GK3C cells were treated with anti-phospho-KIT or
anti-phospho-PDGFRA antibody. The degree of phosphorylation was
measured by flow cytometric analysis and expressed as mean
fluorescence intensity (MFI). Phosphorylation of KIT and PDGFRA was
detected in both cell lines. MFI values for phosphorylation of KIT
and PDGFRA in GK1C were 8.40±0.12 and 3.81±0.18, respectively. In
GK3C, MFI values were 6.71±1.38 and 4.05±0.05 (Fig. 3E). In both GIST cell lines, enhanced
phosphorylation of KIT was marked when compared to that of PDGFRA.
These data were consistent with the cell lines having
function-specific proliferation properties of GIST.
In vivo imatinib sensitivity
The effects of imatinib on the growth of GK1C and
GK3C cells were evaluated. Cells were seeded and incubated for 12 h
before treatment, and then exposed to imatinib (0–100 μM) for 72 h.
The percentage of cellular growth was assessed using WST-8. GK1C
and GK3C cells showed sensitivity to imatinib in vitro with
IC50 values of 4.6±0.96 and 11.0±0.17 μM, respectively
(Fig. 3F).
Effect of imatinib on GIST xenograft
models
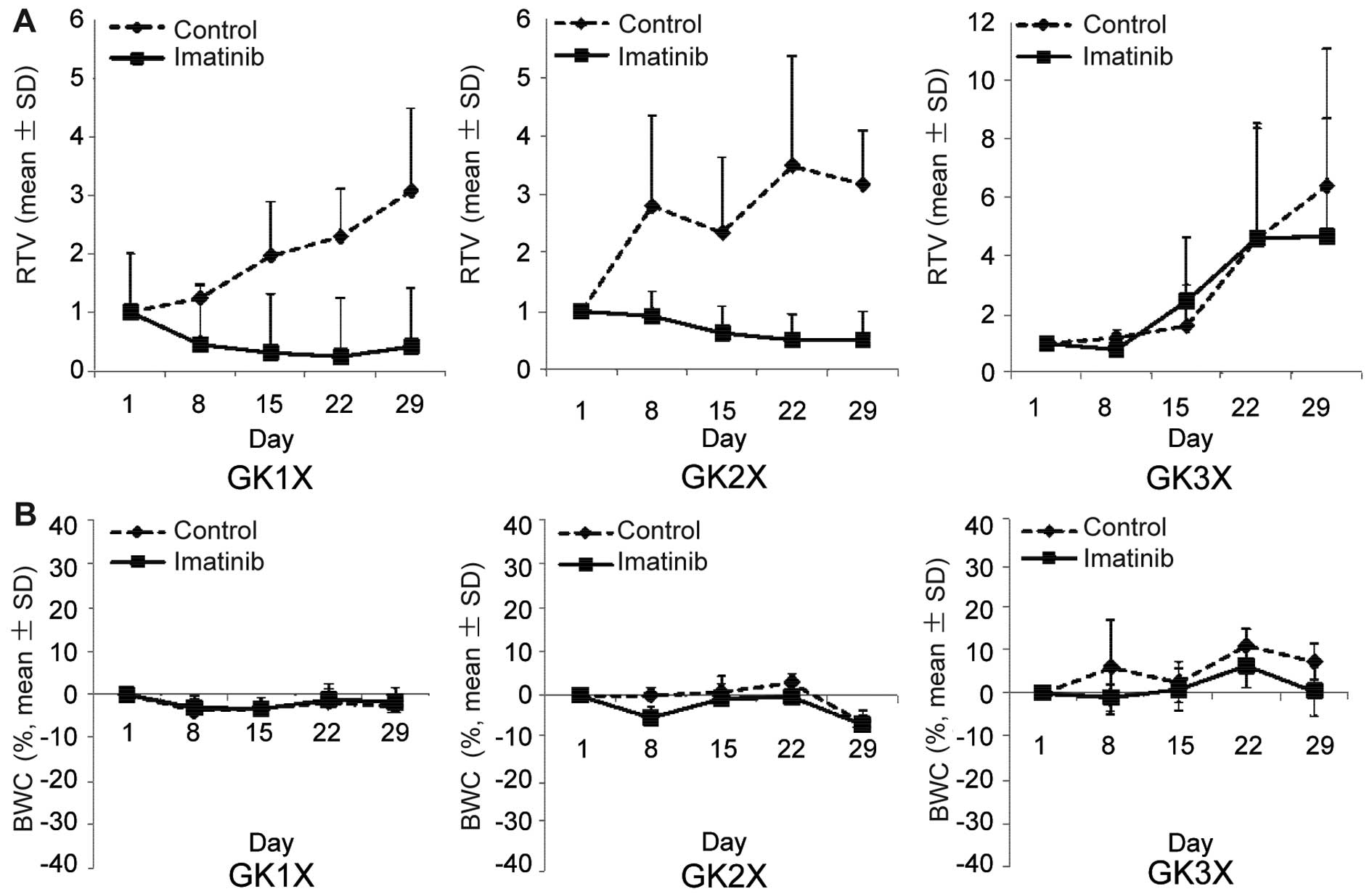
The antitumor activity of imatinib was examined in
the human GIST xenograft model. Mice with tumors derived from GK1X,
GK2X and GK3X were divided into groups for treatment with saline
(control) or imatinib for 28 days. Tumor volume (TV) was evaluated
between groups every three days. Fig.
4A shows the change in TV in each group. The average RTV on the
day of sacrifice for imatinib was 1.86±2.42, whereas for controls
it was 4.21±1.88 (P<0.05). Additionally, the percentage of tumor
growth inhibition for imatinib was 86.7% in GK1X, 84.0% in GK2X and
27.1% in GK3X. The treatment was well tolerated by the mice, with
no signs of toxicity or weight loss during therapy (Fig. 4B). These results indicated that
imatinib inhibited the growth of tumors formed by xenografts
compared to the control.
Discussion
Gastrointestinal stromal tumors (GISTs) are one of
the most common mesenchymal tumors of the gastrointestinal tract
(1–3% of all gastrointestinal malignancies). Until 10 years ago,
these tumors were widely considered variants of smooth muscle
tumors: leiomyomas if benign and leiomyosarcomas if malignant. The
term gastrointestinal autonomic nerve tumor (GANT) also refers to
GIST, based on identical histologic and immunohistochemical
features and c-KIT mutations (22).
The incidence of GIST has been estimated to be 14–20 cases per
million, but minimal incidental GISTs are far more common. Most
GISTs occur on a sporadic basis, but some occur in clinical
syndromes. The most common of these is neurofibromatosis 1, in
which GISTs usually occur in the small intestine, often as
multiple, clinically indolent tumors. Familial GISTs are based on
hereditary c-KIT/PDGFRA-activating mutations, and pediatric GISTs
are linked to loss of succinate dehydrogenase subunit B (SDHB) and
Carney triad and Carney-Stratakis syndromes (CSS), the latter being
an autosomal dominant tumor syndrome combining GIST and
paraganglioma (23).
Two new human GIST cell lines (GK1C and GK3C) and
three xenografts (GK1X, GK2X and GK3X) were established from
surgical tissue samples and characterized. In previous studies, to
improve the cure rate of GIST, researchers have attempted to
establish GIST cell lines and xenografts for basic and clinical
study. However, the success rate of establishing cell lines and
xenografts is very low. There have been reports that several GIST
cell lines and tumors have been generated from clinical specimens
or mice. GIST-T1 was established from a metastatic plural tumor
from a GIST of the stomach in a Japanese woman, and was
characterized by immunohistochemistry, conventional banding
methods, comparative genomic hybridization (CGH), and fluorescence
in situ hybridization (FISH). Immunohistochemically, the
cells were strongly positive for CD34 and c-KIT, but not for
desmin, S-100 protein, or α-smooth muscle actin (24). GIST cell line GIST882 expresses an
activating c-KIT mutation (K642E) in the first part of the
cytoplasmic split tyrosine kinase domain. Notably, the K642E
substitution is encoded by a homozygous exon 13 missense mutation,
and, therefore, GIST882 cells do not express native KIT. GIST882
KIT protein is constitutively tyrosine phosphorylated, but tyrosine
phosphorylation was abolished after incubating the cells with the
selective tyrosine kinase inhibitor imatinib (25). GIST-H1 was established and passaged
more than 60 times over a year. The population doubling time
calculated in log phase of growth was 47.5 h. The cloning
efficiency in soft agar averaged 24.8%. From electron microscopic
evaluation, the cytoplasm was rich in ribosomes and mitochondria.
Immunohistochemical analysis revealed aneuploidy with modal
chromosomal numbers ranging from 60 to 98. The GIST cells
transplanted in nude mice were tumorigenic (26). A culture model of GIST-DR derived
from GIST induced by duodenal reflux was established. GIST-DR
cells, both in vitro and in vivo, were immunopositive
for both KIT and CD34 and imatinib blocked the proliferation of
this cell line (27). Although
GIST-T1 and GIST882, GIST-H1 and GIST-DR (induced by chemical
carcinogenesis), have been established, long-term maintenance in
vitro and in vivo might be difficult.
Our established cell lines and xenografts were
capable of repeated passage for long periods, making them available
for the study of GIST. To investigate their phenotype,
immunohistochemical and cytochemical analyses of KIT expression of
the established cell lines (GK1C and GK3C) and xenografts (GK1X,
GK2X and GK3X) were undertaken. These results showed that the cell
lines and xenografts were positive for KIT. In addition, the
increased phosphorylation of KIT and PDGFRA was detected in both
cell lines, GK1C and GK3C. In both, the intensity enhancement of
phosphorylation of KIT was 2.2- to 4.8-fold higher than that of
PDGFRA.
The discovery of gain-of-function mutations in the
c-KIT proto-oncogene in GIST by Hirota and colleagues
(28) in 1998 was crucial to our
present understanding of the genesis and classification of these
tumors. Sommer et al(29)
reported that constitutive KIT signaling is both critical and
sufficient for induction of GIST and hyperplasia of ICC
(interstitial cells of Cajal). In addition, GIST is known to
represent a discrete neoplastic entity, possibly arising from a
progenitor related to ICC (30,31).
Although, it is well accepted that KIT activity plays an important
role in the tumorigenesis of GIST, little is known regarding the
main cause of c-KIT mutation.
In patients, malignant GISTs often metastasize to
the liver and disseminate within the peritoneal cavity. The
tumorigenicity of GK1C and GK3C cell lines was examined using
immune-deficient SCID mice, into which 5×106 GIST cells
had been transplanted subcutaneously. Both GK1C and GK3C formed
tumors ectopically. Moreover, the tumors were KIT-positive by
immunohistochemical analysis. Recent advances in molecular biology
have highlighted the complicated tumorigenic mechanism of various
tumors. The tumorigenicity of GISTs has been explained by
activating mutations of the c-KIT or PDGFRA gene
(32). Thus, these data indicate
that GK1C and GK3C have the properties of tumorigenic GISTs.
Imatinib inhibits KIT tyrosine kinase activity,
enabling pharmacologic attack on a specific molecular target in
GIST. Early clinical trials with imatinib have resulted in marked
remission of metastatic GIST, a type of tumor that has previously
proven resistant to all other forms of chemotherapy. Imatinib has
also proven highly active in patients with unresectable GISTs
expressing immunohistochemically detectable c-KIT protein (33,34). A
recent report showed that all KIT mutant isoforms, but only a
subset of platelet-derived growth factor receptor α (PDGFRA) mutant
isoforms, were sensitive to imatinib in vitro(35). In this study, imatinib blocked the
proliferation of cell lines (GK1C and GK3C) and xenografts (GK1X,
GK2X and GK3X). These results indicate that the newly established
cell lines and xenografts are useful as models of
imatinib-sensitive GIST.
Within 12 to 36 months, ~50–70% of patients with
GISTs will progress following imatinib therapy (36,37).
The most common cause of secondary resistance (50–80%) is a second
mutation involving the same allele as the initial mutation, a
phenomenon that has not been documented in tumors with primary
resistance (14,38). However, there must be
c-KIT-negative GISTs and the main cause of the c-KIT
mutation has not been fully examined. On the other hand, it has
been reported that imatinib is a substrate for P-glycoprotein, a
multi-drug resistance (MDR) protein, typically involved in
antitumor drug transport (39), and
breast cancer resistance protein (BCRP), a protein implicated in
drug transport in the gut epithelium (40). Increased expression of these
proteins results in decreased drug levels. The mechanisms
underlying the remaining secondary resistance have not yet been
clarified. Our novel GIST cell lines (GK1C and GK3C) are easy to
transplant into SCID mice, and represent a useful model for testing
chemotherapeutic agents in vivo.
In conclusion, our novel models for GIST are
important research tools for investigating relevant cellular
alterations that may be pertinent to the human disease state. In
addition, such models may be beneficial for pre-clinical testing of
new therapeutic strategies.
Abbreviations:
|
GIST
|
gastointestinal stromal tumor
|
|
NOG mice
|
NOD/Shi-scid, IL-2Rrnu
mice
|
|
nude mice
|
BALB/cAJcl-nu/nu mice
|
|
SCID mice
|
FOX CHASE SCID
C.B-17/lcr-scid/scidJcl
|
References
|
1
|
Nowain A, Bhakta H, Pais S, Kanel G and
Verma S: Gastrointestinal stromal tumors: clinical profile,
pathogenesis, treatment strategies and prognosis. J Gastroenterol
Hepatol. 20:818–824. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heinrich MC, Rubin BP, Longley BJ, et al:
Biology and genetic aspects of gastrointestinal stromal tumors: KIT
activation and cytogenetic alterations. Hum Pathol. 33:484–495.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vlahovic G and Crawford J: Activation of
tyrosine kinases in cancer. Oncologist. 8:531–538. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eisenberg BL and Judson I: Surgery and
imatinib in the management of GIST: emerging approaches to adjuvant
and neoadjuvant therapy. Ann Surg Oncol. 11:465–475. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heinrich MC and Corless CL: Gastric GI
stromal tumors (GISTs): the role of surgery in the era of targeted
therapy. J Surg Oncol. 90:195–207. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Samiian L, Weaver M and Velanovich V:
Evaluation of gastrointestinal stromal tumors for recurrence rates
and patterns of long-term follow-up. Am Surg. 70:187–191.
2004.PubMed/NCBI
|
|
7
|
DeMatteo RP, Heinrich MC, El-Rifai WM and
Demetri G: Clinical management of gastrointestinal stromal tumors:
before and after STI-571. Hum Pathol. 33:466–477. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Demetri GD, vonMehren M, Blanke CD, Vanden
Abbeele AD, Eisenberg B, Roberts PJ, et al: Efficacy and safety of
imatinib mesylate in advanced gastrointestinal stromal tumors. N
Engl J Med. 347:472–480. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bauer S, Duensing A, Demetri GD and
Fletcher JA: KIT oncogenic signaling mechanisms in
imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT
is a crucial survival pathway. Oncogene. 26:7560–7568. 2007.
View Article : Google Scholar
|
|
10
|
Sleijfer S, Wiemer E, Seynaeve C and
Verweij J: Improved insight into resistance mechanisms to imatinib
in gastrointestinal stromal tumors: a basis for novel approaches
and individualization of treatment. Oncologist. 12:719–726. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wardelmann E, Buttner R, Merkelbach-Bruse
S and Schildhaus HU: Mutation analysis of gastrointestinal stromal
tumors: increasing significance for risk assessment and effective
targeted therapy. Virchows Arch. 451:743–749. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tornillo L and Terracciano LM: An update
on molecular genetics of gastrointestinal stromal tumours. J Clin
Pathol. 59:557–563. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sleijfer S, Wiemer E and Verweij J: Drug
insight: gastrointestinal stromal tumors (GIST) - the solid tumor
model for cancer-specific treatment. Nat Clin Pract Oncol.
5:102–111. 2008. View Article : Google Scholar
|
|
14
|
Antonescu CR, Besmer P, Guo T, Arkun K,
Hom G, Koryotowski B, et al: Acquired resistance to imatinib in
gastrointestinal stromal tumor occurs through secondary gene
mutation. Clin Cancer Res. 11:4182–4190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Debiec-Rychter M, Dumez H, Judson I, Wasag
B, Verweij J, Brown M, Dimitrijevic S, Sciot R, Stul M, Vranck H,
Scurr M, Hagemeijer A, van Glabbeke M and van Oosterom AT; EORTC
Soft Tissue and Bone Sarcoma Group. Use of c-KIT/PDGFRA mutational
analysis to predict the clinical response to imatinib in patients
with advanced gastrointestinal stromal tumours entered on phase I
and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur
J Cancer. 40:689–695. 2004. View Article : Google Scholar
|
|
16
|
Heinrich MC, Corless CL, Blanke CD,
Demetri GD, Joensuu H, Roberts PJ, et al: Molecular correlates of
imatinib resistance in gastrointestinal stromal tumors. J Clin
Oncol. 24:4764–4774. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mahadevan D, Cooke L, Riley C, Swart R,
Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H,
Nagle R and Bearss D: A novel tyrosine kinase switch is a mechanism
of imatinib resistance in gastrointestinal stromal tumors.
Oncogene. 26:3909–3919. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thao le B, Vu HA, Yasuda K, Taniguchi S,
Yagasaki F, Taguchi T, Watanabe T and Sato Y: Cas-L was
overexpressed in imatinib-resistant gastrointestinal stromal tumor
cells. Cancer Biol Ther. 8:683–688. 2009.PubMed/NCBI
|
|
19
|
Steigen SE and Eide TJ: Trends in
incidence and survival of mesenchymal neoplasm of the digestive
tract within a defined population of northern Norway. APMIS.
114:192–200. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Trent JC and Benjamin RS: New developments
in gastrointestinal stromal tumor. Curr Opin Oncol. 18:386–395.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fletcher CD, Berman JJ, Corless C,
Gorstein F, Lasota J, Longley BJ, Miettinen M, O’Leary TJ, Remotti
H, Rubin BP, Shmookler B, Sobin LH and Weiss SW: Diagnosis of
gastrointestinal stromal tumors: a consensus approach. Int J Surg
Pathol. 10:81–89. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee JR, Joshi V, Griffin JW Jr, et al:
Gastrointestinal autonomic nerve tumor: immunohistochemical and
molecular identity with gastrointestinal stromal tumor. Am J Surg
Pathol. 25:979–987. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: review on morphology, molecular pathology,
prognosis, and differential diagnosis. Arch Pathol Lab Med.
130:1466–1478. 2006.PubMed/NCBI
|
|
24
|
Taguchi T, Sonobe H, Toyonaga S, Yamasaki
I, Shuin T, Takano A, Araki K, Akimaru K and Yuri K: Conventional
and molecular cytogenetic characterization of a new human cell
line, GIST-T1, established from gastrointestinal stromal tumor. Lab
Invest. 82:663–665. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tuveson DA, Willis NA, Jacks T, Griffin
JD, Singer S, Fletcher CD, Fletcher JA and Demetri GD: STI571
inactivation of the gastrointestinal stromal tumor c-KIT
oncoprotein: biological and clinical implications. Oncogene.
20:5054–5058. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu B, Liao G, Liu S, Huang B, Wu S, Zhou
J, Gu H and Zhu H: Characteristics and establishment of cell lines
from human gastrointestinal stromal tumors. Zhong Nan Da Xue Xue
Bao Yi Xue Ban. 35:1138–1144. 2010.PubMed/NCBI
|
|
27
|
Mukaisho K, Miwa K, Totsuka Y, Shimomura
A, Sugihara H, Wakabayashi K and Hattori T: Induction of gastric
GIST in rat and establishment of GIST cell line. Cancer Lett.
231:295–303. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
Muhammad TG, Matsuzawa Y, Kanakura Y, Shinomura Y and Kitamura Y:
Gain-of-function mutations of c-kit in human stromal tumors.
Science. 279:577–580. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sommer G, Agosti V, Ehlers I, Rossi F,
Corbacioglu S, Farkas J, Moore M, Manova K, Antonescu CR and Besmer
P: Gastrointestinal stromal tumors in a mouse model by targeted
mutation of the kit receptor tyrosine kinase. Proc Natl Acad Sci
USA. 100:6706–6711. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kindblom LG, Remotti HE, Aldenborg F and
Meis-Kindblom JM: Gastrointestinal pacemaker cell tumor (GIPACT):
gastrointestinal stromal tumors show phenotypic characteristics of
the interstitial cells of Cajal. Am J Pathol. 152:1259–1269.
1998.
|
|
31
|
Sircar K, Hewlett BR, Huizinga JD,
Chorneyko K, Berezin I and Riddell RH: Interstitial cells of Cajal
as precursors of gastrointestinal stromal tumors. Am J Surg Pathol.
23:377–389. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, et al: PDGFRA activating mutations
in gastrointestinal stromal tumors. Science. 299:708–710. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Joensuu H, Roberts PJ, Sarlomo-Rikala M,
Andersson LC, Tervahartiala P, Tuveson D, Silberman S, Capdeville
R, Dimitrijevic S, Druker B and Demetri GD: Effect of the tyrosine
kinase inhibitor STI571 in a patient with a metastatic
gastrointestinal stromal tumor. N Engl J Med. 344:1052–1056. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
van Oosterom AT, Judson I, Verweij J,
Stroobants S, Donato di Paola E, Dimitrijevic S, Martens M, Webb A,
Sciot R, Van Glabbeke M, Silberman S and Nielsen OS: Safety and
efficacy of imatinib (STI571) in metastatic gastrointestinal
stromal tumours: a phase I study. Lancet. 358:1421–1423.
2001.PubMed/NCBI
|
|
35
|
Heinrich MC, Corless CL, Demetri GD,
Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den
Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S,
Fletcher CD, Silberman S, Dimitrijevic S and Fletcher JA: Kinase
mutations and imatinib response in patients with metastatic
gastrointestinal stromal tumor. J Clin Oncol. 21:4342–4349. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Blanke CD, Rankin C, Demetri GD, Ryan CW,
von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki
RG, Tanaka M, et al: Phase III randomized, intergroup trial
assessing imatinib mesylate at two dose levels in patients with
unresectable or metastatic gastrointestinal stromal tumors
expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol.
26:626–632. 2008. View Article : Google Scholar
|
|
37
|
Verweij J, Casali PG, Zalcberg J, LeCesne
A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC,
Van Glabbeke M, Bertulli R, et al: Progression-free survival in
gastrointestinal stromal tumours with high-dose imatinib:
randomised trial. Lancet. 364:1127–1134. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen LL, Trent JC, Wu EF, Fuller GN,
Ramdas L, Zhang W, Raymond AK, Prieto VG, Oyedeji CO, Hunt KK,
Pollock RE, et al: A missense mutation in KIT kinase domain 1
correlates with imatinib resistance in gastrointestinal stromal
tumors. Cancer Res. 64:5913–5919. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Theou N, Gil S, Devocelle A, Julie C,
Lavergne-Slove A, Beauchet A, Callard P, Farinotti R, Le Cesne A,
Lemoine A, Faivre-Bonhomme L, et al: Multidrug resistance proteins
in gastrointestinal stromal tumors: site-dependent expression and
initial response to imatinib. Clin Cancer Res. 11:7593–7598. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Burger H, Van Tol H, Boersma AW, Brok M,
Wiemer EA, Stoter G and Nooter K: Imatinib mesylate (STI571) is a
substrate for the breast cancer resistance protein (BCRP)/ABCG2
drug pump. Blood. 104:2940–2942. 2004. View Article : Google Scholar : PubMed/NCBI
|