Introduction
Cholangiocarcinoma (CCA) is an incurable and lethal
cancer which arises from the epithelial cells of the bile ducts.
Early diagnosis, with potential surgical intervention, has been the
exception rather than the rule with only 30% of patients qualifying
for attempted surgical cure (1).
Despite advances, the median survival is <24 months (2). 5-Fluorouracil (5-FU), an
antimetabolite, is currently the standard treatment for CCA.
Although 5-FU yields great clinical benefit in patients with
advanced CCA, the response rates of ~10–40% and the survival
benefits when 5-FU is used alone are very low (3). The prognosis for the majority of
patients with advanced CCA remains poor due to intrinsic or
acquired chemoresistance. Therefore, identification of the
signaling molecules involved in mediating the response of CCA to
5-FU is required to determine the underlying mechanisms of 5-FU
resistance.
A common cause of treatment failure in CCA is
chemoresistance, which may be related to the redox state of cancer
cells and the tumor microenvironment, where growth factors play
important roles. Hepatocyte growth factor (HGF) was originally
identified as a mitogenic protein for hepatocytes, and it can
promote CCA cell invasiveness through dyslocalization of E-cadherin
and induction of cell motility by distinct signaling pathways
(4,5). Met, which is overexpressed in the
tissues of CCA patients, is known to be the only specific receptor
for HGF. NK4 is a fragment of HGF, consisting of the N-terminal (N)
and four kringle domains (K4) of HGF. It competitively inhibits HGF
binding to Met and inhibits the phosphorylation of Met tyrosine
kinase. It was the first-identified specific inhibitor for the
HGF/Met pathway (5).
The HGF/Met pathway has become a hot target in
anticancer drug development (6,7).
Activation of the HGF/Met pathway in cancer cells contributes to
resistance to chemical and physical treatment of cancer (8,9).
Recent studies revealed that activation of the HGF/Met pathway is a
novel mechanism by which non-small cell lung cancers become
resistant to EGFR inhibitors (10).
It has been reported that combined treatment with an adenoviral
vector expressing NK4 (Ad-NK4) with gemcitabine may be a promising
approach for treating pancreatic cancer, and that this combination
therapy may decrease the risks of side effects (11). In addition, NK4 gene therapy
combined with cisplatin inhibits tumor growth and the metastasis of
squamous cell carcinoma (11).
Murine colon cancer CT26 cells expressing NK4 demonstrated enhanced
5-FU-induced cell apoptosis via downregulation of intracellular
signaling of the HGF/Met pathway (12). In brief, NK4 is a potential
regulator of chemotherapy resistance in many types of malignances.
Therefore, treatment with NK4 may offer a new therapeutic option
for the inhibition of chemoresistance and better outcomes for
cancer patients.
A review summarized the epigenetic mechanisms
triggering resistance to three commonly used agents in colorectal
cancer, including 5-FU (13).
Studies undertaken to determine the molecular mechanisms of
intrinsic and acquired resistance to 5-FU have found various
alterations of drug target(s) and metabolism (14,15),
such as key enzymes of 5-FU anabolism and catabolism including
thymidylate synthase (TS) (16),
thymidine phosphorylase (TP) (17)
and dihydropyrimidine dehydrogenase (DPD). It had been confirmed in
tissue and from mRNA levels that negative DPD (not TS) expression
is significantly associated with the enhanced tumor cell
proliferation and poorer prognosis in patients with intrahepatic
cholangiocarcinoma (18,19). In addition, altered expression of
apoptosis-regulating genes also can lead to resistance to 5-FU
(20). Since research is limited
concerning the combination of NK4 gene therapy and 5-FU
chemotherapy in CCA, the aim of this study was to investigate the
mechanism by which NK4 regulates the 5-FU response in HuCC-T1
cells.
Materials and methods
Reagents
Antibodies against Met, Bcl-2 and Bax were obtained
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit
monoclonal antibodies against phospho-Met (Tyr1003) were obtained
from Cell Signaling Technology (Danvers, MA, USA). Antibodies
against β-actin and anti-goat/rabbit immunoglobulin G
(IgG)-horseradish peroxidase (HRP) were obtained from BioWorld
(Atlanta, GA, USA). 5-FU and cisplatin were obtained from Qilu
Pharmaceutical Co., Ltd. (Shandong, China), and doxorubicin was
from Nanjing KeyGen Biotech., Co., Ltd. (Nanjing, China). All the
other chemicals used were of analytical reagent grade.
Cell cultures
HuCC-T1 human cholangiocarcinoma cells (ATCC,
Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s
medium (DMEM; Gibco Laboratories, Grand Island, NY, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA,
USA) in 5% CO2 at 37°C until reaching a confluence range
of 50–60%. Then the human NK4 expression plasmid pcDNA3/NK4
(provided by Professor Toshikazu Nakamura, Osaka University, Japan)
was transfected into HuCC-T1 using X-tremeGENE HP (Roche Molecular
Biochemicals, Indianapolis, IN, USA). After 48 h, the cells were
cultured in selective medium containing 900 μg/ml G418 (Sigma, St.
Louis, MO, USA) for the selection of resistant colonies. The
expression of NK4 protein was assessed by western blotting. A
transfectant expressing a high level of NK4 was designated as
Hu-NK4. Cells transfected with empty vector pcDNA3 alone were
designated as Hu-Em and used as the control.
Cell growth assay
Hu-Em and Hu-NK4 cells were cultured in 96-well
plates at a density of 5×103 cells/well overnight, and
then treated with 5-FU (0, 0.1, 1, 10 and 100 μM), cisplatin (0,
0.01, 0.1, 1 and 10 μM) or doxorubicin (0, 0.01, 0.1, 1 and 10 μM).
Cells were cultured for 48 and 72 h. During the last 4 h,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma) was added to a final concentration of 1 mmol/l. The
formazan product was dissolved in dimethyl sulfoxide (150 μl), and
the absorbances were read on a microplate reader (Bio-Rad
Laboratories, Hercules, CA, USA) at 490 nm. Each result was
corrected by values from a control study. Experiments were
performed in triplicate and repeated at least three times.
Real-time reverse transcription
(RT)-PCR
Total RNA was isolated from cells using the TRIzol
total RNA isolation kit (Invitrogen) according to the
manufacturer’s protocol. RNA was eluted with RNase-free water.
RT-PCR was performed using the Transcriptor First Strand cDNA
Synthesis kit (Roche Molecular Biochemicals) according to the
manufacturer’s protocol. Briefly, reactions were incubated at 65°C
for 10 min, at 55°C for 30 min, and then at 85°C for 5 min.
Oligonucleotide primers (TYMS forward, 5′-CCTGCTCACGTACATGAT TGC-3′
and reverse 5′-TTT GGGAAAGGTCTGGGTTC-3′; TYMP forward,
5′-GCTGCTGTATCGTGGGTCA-3′ and reverse, 5′-GAGAATGGAGGCTGTGATGAG-3′;
DPYD forward, 5′-CAATGAGATGCCTGAAATGTG-3′ and reverse,
5′-AAGTCAGACCAAGTGGGTTGT-3′; β-actin forward,
5′-TCACCCACACTGTGCCCATCTACGA-3′ and reverse,
5′-CAGCGGAACCGCTCATTGCCAATGG-3′) were designed using Primer 5
software, and synthesized by Invitrogen. Real-time monitoring of
PCR products was carried out using the SYBR-Green Master Mix (Roche
Molecular Biochemicals) and the Prism 7500 Real-Time PCR Detection
system (Applied Biosystems, Foster City, CA, USA). Cycling
conditions were 95°C for 10 min, followed by 40 repeats of 95°C for
15 sec and 60°C for 1 min. Levels of mRNAs were calculated using
the comparative cycle threshold (ΔΔCT) method and normalized to
β-actin, the internal control, to obtain the relative mRNA level of
each target.
Flow cytometry for cell cycle analysis
and measurement of apoptosis
Hu-Em and Hu-NK4 cells were seeded in a 6-well plate
at a density of 1×105 cells/well and incubated
overnight. Cells were treated with 0, 1, 10 and 100 μM 5-FU for 48
h, then harvested by trypsinization, fixed in ice-cold 70% ethanol
at 4°C overnight. Cells were washed, suspended in 1 ml propidium
iodide (PI) staining solution (50 μg/ml PI, 30 U/ml RNase A, 0.1%
Triton X-100, 4 mM sodium citrate) and incubated at 37°C for 10
min. Typically, 10,000 gated events were collected on a FACScan
machine (Beckton-Dickinson, Franklin Lakes, NJ, USA) and analyzed
by CellQuest software (Beckton-Dickinson). Cell cycle analysis was
carried out using FlowJo software (Tree Star, San Carlos, CA, USA).
Apoptotic cells were measured by flow cytometry using the FITC
Annexin V Apoptosis Detection Kit I (BD Biosciences, San Diego, CA,
USA) according to the manufacturer’s instructions.
Assays of caspase-3 and caspase-9
activity
Hu-Em and Hu-NK4 cells were cultured in 6-well
plates overnight, and then treated with 0, 1, 10 and 100 μM 5-FU.
After treatment with 5-FU for 48 h, caspase-3 and -9 activity was
assayed using the Caspase Colorimetric Assay kit (Nanjing KeyGen
Biotech) according to the manufacturer’s protocol. Briefly, the
cells were lysed in a lysis buffer for 30 min in an ice bath. The
lysed cells were centrifuged at 12,000 × g for 10 min, and 200 μg
of the protein was incubated with 50 μl of a reaction buffer and 5
μl of the substrate for caspase-3 and caspase-9, respectively, at
37°C for 4 h. The optical density of the reaction mixture was
quantified spectrophotometrically at a wavelength of 405 nm. The
assay was repeated three times and similar results were
obtained.
Western blot analysis
For Bcl-2 and Bax detection, Hu-Em and Hu-NK4 cells
were serum-starved for 12 h and then incubated with 0, 1, 10 and
100 μM 5-FU for 48 h and total proteins were extracted. For Met
detection, 5-FU and HGF were added to cells, respectively, or their
combination, in which 10 μM 5-FU was incubated with Hu-Em and
Hu-NK4 cells for 48 h, and then 20 ng/ml HGF was added to the cells
1 h before harvested. Then protein extracts were run on a 7%
polyacrylamide gel and were transferred onto polyvinylidene
difluoride (PVDF) membranes (Millipore, Billerica, MA, USA).
Non-specific binding was blocked by incubation of the membranes
with 5% non-fat milk for 2 h, followed by incubation with the
primary antibody at 4°C overnight. The membrane was then washed
with TBST for 15 min. This step was repeated four times. After
being washed, the membrane was incubated with HRP-conjugated
secondary antibodies for 1 h at room temperature, washed with TBST
for 10 min and repeated four times, and visualized by an enhanced
chemiluminescence kit (Amersham, Piscataway, NJ, USA).
Statistical analysis
Data are presented as the means ± SE. Statistical
analysis was carried out using the independent-samples t-test or
one-way ANOVA, with P<0.05 considered to indicate a
statistically significant difference.
Results
NK4 overexpression increases the
sensitivity of HuCC-T1 cells to 5-FU
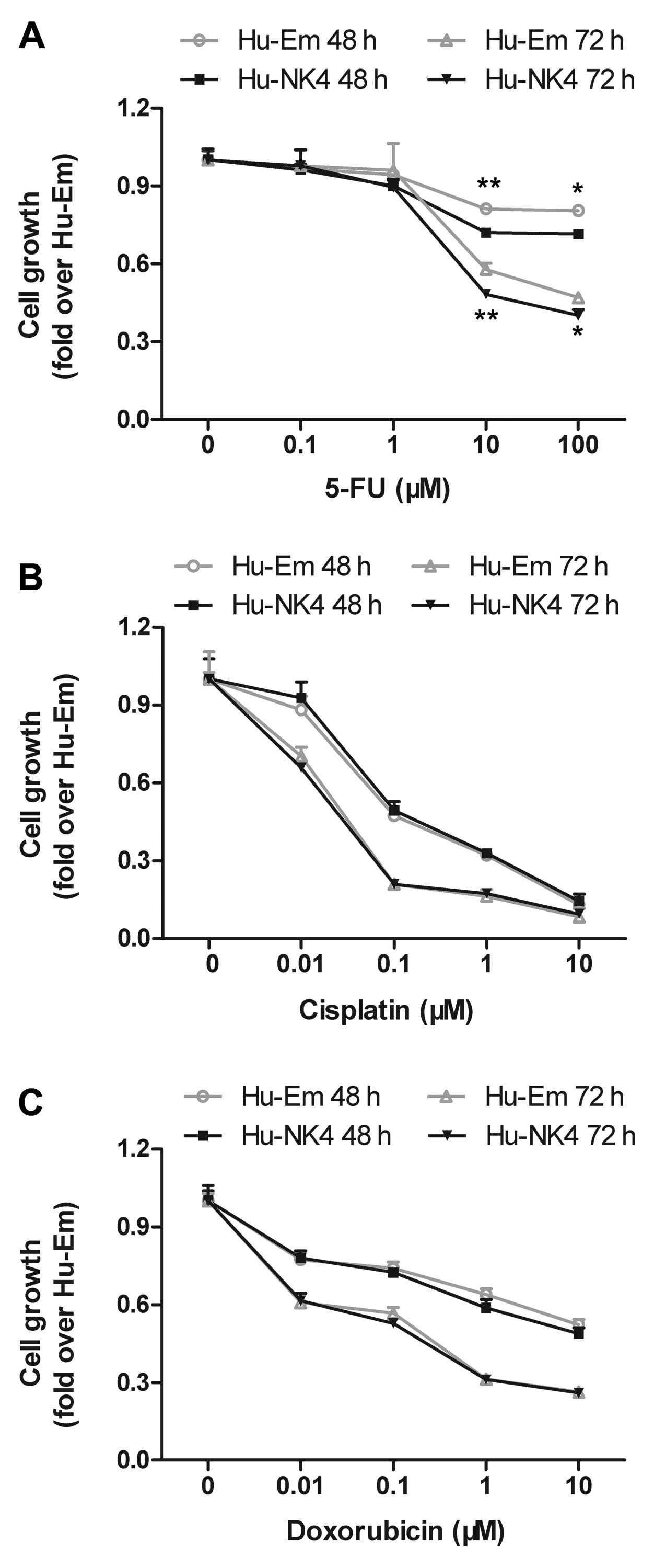
To select the best partner for NK4 gene therapy from
among conventional chemotherapeutic agents, we examined the effect
of 5-FU, cisplatin and doxorubicin on the in vitro
proliferation of NK4-expressing HuCC-T1 cells by MTT assay. There
was little difference in cell proliferation between the Hu-Em and
Hu-NK4 cells under conditions without drugs. All drugs reduced the
cell proliferation of both transfectants in a dose-dependent
manner. 5-FU, in particular, dose-dependently reduced the cell
proliferation of Hu-NK4 cells to a greater extent when compared to
that of Hu-Em cells and to a greater exent than did cisplatin and
doxorubicin (Fig. 1). From the
results of the cell proliferation assay, we selected 5-FU as the
best chemotherapeutic agent for combination with NK4 gene therapy
and examined the assays using 5-FU thereafter. Furthermore, this
method indicated that treatment with 10 μM of 5-FU actually
resulted in a significant, time-dependent growth inhibition of
HuCC-T1 cells.
mRNA expression levels of TYMS, TYMP and
DPYD were slightly altered
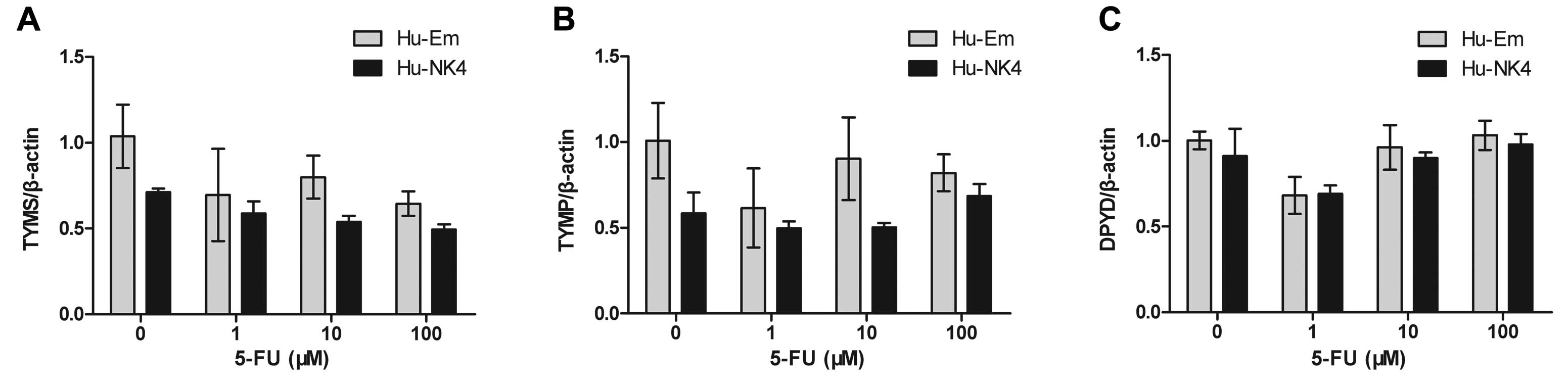
When considering the additive effect of NK4 gene
expression in combination with 5-FU on cell growth, we hypothesized
that NK4 gene expression influences 5-FU metabolism. Therefore, we
assessed the expression of the metabolic enzymes of 5-FU. The mRNA
levels of TYMS (encoding TS), TYMP (encoding TP) and DPYD (encoding
DPD) tended to be slightly decreased in Hu-NK4 cells, but the gene
expression levels were not significantly different from that in the
Hu-Em cell line (Fig. 2).
NK4 overexpression enhances 5-FU-induced
apoptosis of HuCC-T1 cells
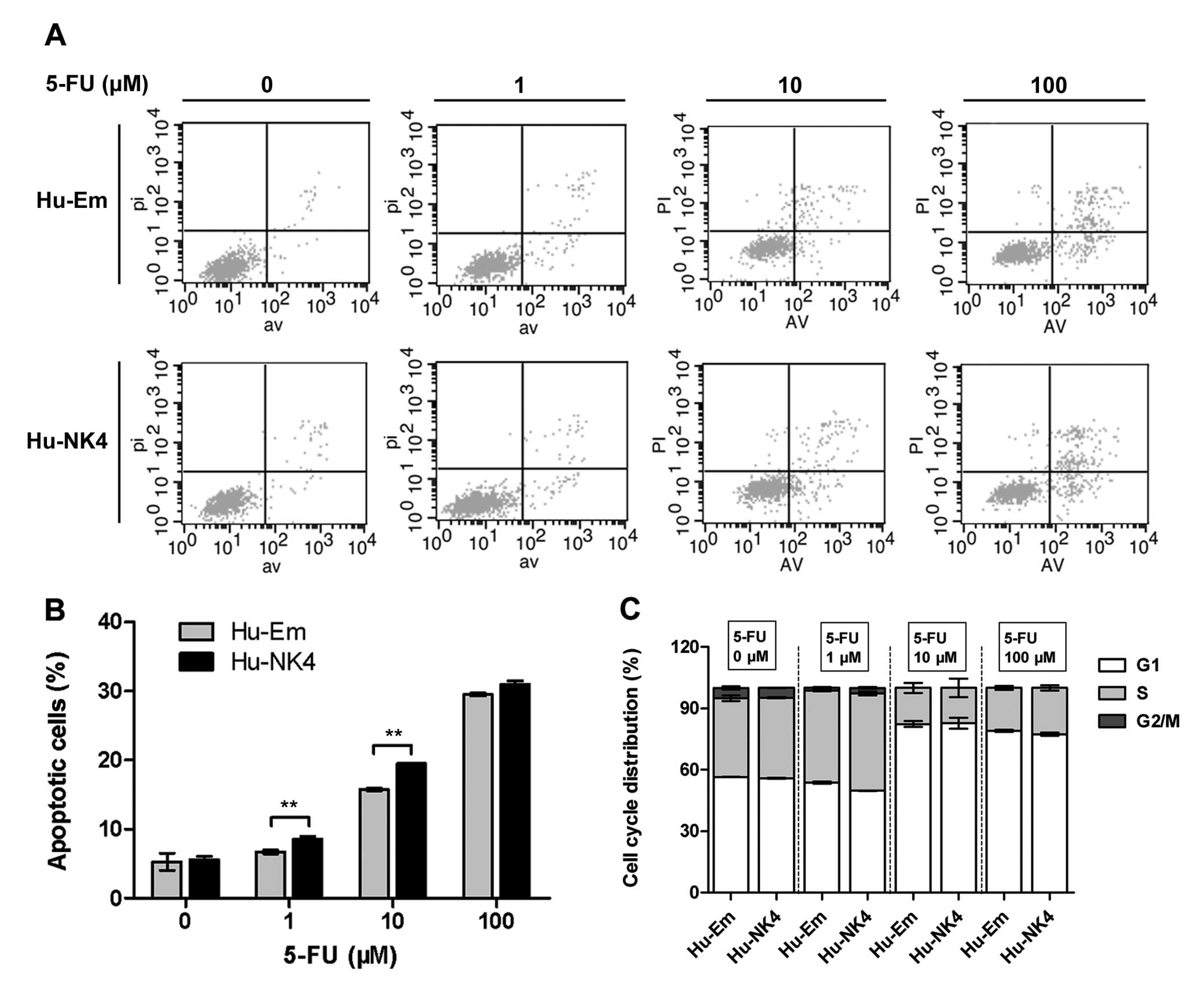
Since NK4 overexpression enhanced cell sensitivity
to 5-FU, we investigated its role in mediating the 5-FU-response.
We examined whether apoptosis is involved in the cytotoxic effect
induced by the combination of NK4 gene expression and 5-FU. Flow
cytometric analysis showed that 5-FU at concentrations of 1 and 10
M increased the proportion of cells undergoing apoptosis at 48 h;
Hu-NK4 cells were affected to a greater extent than Hu-Em cells
(Fig. 3A and B).
5-FU acts as a cytostatic agent by arresting cells
in the G2 phase (21,22). Therefore, we performed cell cycle
analysis following treatment with 5-FU for 24 h to analyze cell
cycle distribution of these transfectants. The proportion of cells
in the G2/M phase of the cell cycle decreased after 1 μM 5-FU
treatment, and even disappeared when the concentration was
increased to 10 and 100 μM. Higher concentrations of 5-FU mainly
caused arrest in the G1 phase and depletion of S phase. There were
no significant differences between Hu-Em and Hu-NK4 cells,
suggesting that NK4 did not affect cell cycle distribution
(Fig. 3C).
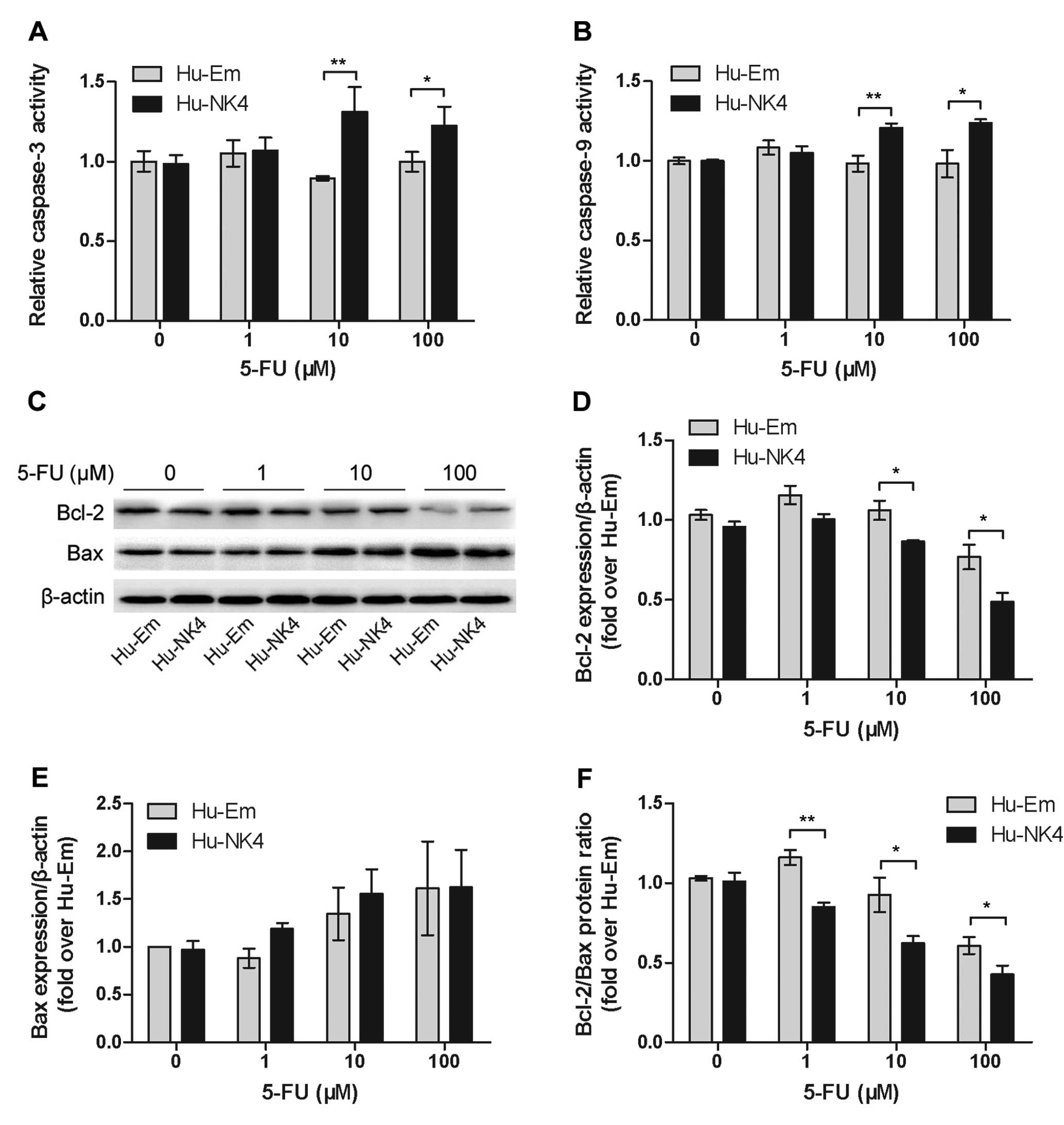
NK4 augments 5-FU-induced apoptosis
through the intrinsic pathway
We measured the activity of caspase-3 and caspase-9,
the critical mediators of apoptosis, by colorimetric assay.
5-FU-induced activation of caspase-3 and caspase-9, except at 1 μM
5-FU, was significantly reduced in the Hu-NK4 cells (Fig. 4A and B). We further investigated the
role of the intrinsic apoptotic pathway in 5-FU resistance. The
pro-apoptotic Bcl-2 family members, Bcl-2 and Bax, are important
initiators of mitochondrial-mediated apoptosis. Western blot
analysis revealed that 5-FU dose-dependently downregulating Bcl-2
and upregulating Bax expression (Fig.
4C-F). Bcl-2 expression following the different treatments
(except at 1 μM) was higher in Hu-Em cells than that in Hu-NK4
cells. Significant difference in the Bcl-2/Bax ratio, but not in
Bax, was observed between the two groups. These results further
suggest a role for NK4 in regulating the 5-FU-induced intrinsic
apoptotic pathway.
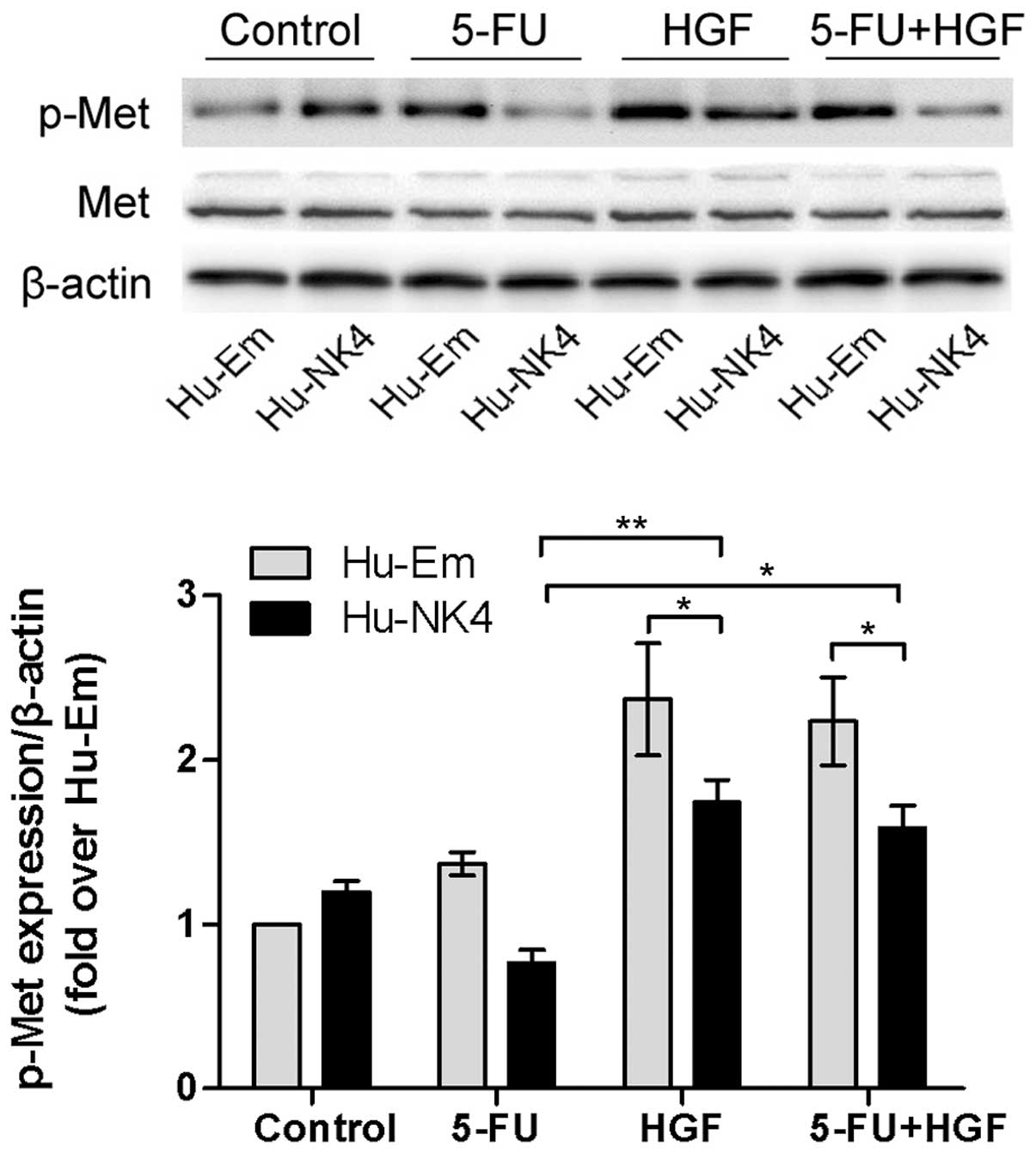
NK4 affects inhibition of HGF/Met
signaling independent of 5-FU
We analyzed 5-FU-induced changes in the expression
levels of phosphorylated Met (p-Met) protein relative to an
untreated, time-matched control. Western blot analysis demonstrated
that phosphorylated Met was downregulated in response to treatment
with 10 μM 5-FU at 48 h in both Hu-Em and Hu-NK4 cells. The level
in the Hu-NK4 cells was decreased similarly when compared to Hu-Em
cells, suggesting that 5-FU had little effect on the HGF/Met
pathway. In our previous study, we demonstrated that the NK4 gene
inhibits HGF-induced activation of Met. To clarify the functional
role of HGF-induced activation of Met and 5-FU-induced
deactivation, we investigated HGF in our study. Western blot
analysis indicated that p-Met expression levels were increased in
the HGF stimulation group, which was inhibited by NK4 transfectant
and 5-FU treatment, and c-Met was strongly inhibited in the Hu-NK4
cell group, while there was no significant difference between the
HGF stimulation group and HGF and 5-FU co-treatment group (Fig. 5). The result revealed that both NK4
and 5-FU were inhibitors of HGF-induced phosphorylation of Met, but
they may be independent factors.
Discussion
In the present study, we demonstrated that the
combination of NK4 gene therapy with chemotherapeutic agents,
particularly 5-FU, enhanced the growth inhibition of HuCC-T1 cells.
When we assessed the mechanism involved, 5-FU was found to exert an
additional effect on apoptosis through the intrinsic pathway. The
association between NK4 and 5-FU prompted us to further investigate
the role of the intrinsic intracellular signaling of the HGF/Met
pathway. NK4 gene expression alone exerts potent antitumor activity
by downregulating the HGF-induced phosphorylation of Met. 5-FU was
another HGF/Met signaling inhibitor independent of NK4. The
influence of NK4 gene expression on enzymes involved in 5-FU
metabolism was not elucidated.
Although 5-FU is the most common chemotherapeutic
drug used in the treatment of CCA, evidence of 5-FU resistance has
been reported both in in vitro studies using human CCA cell
lines (23) and in vivo in
CCA patients (24). Previous
studies have demonstrated that the development of resistance of
cancers to 5-FU may involve mechanisms including alterations in the
exon of several genes including thymidylate synthase (TS) (16), thymidine phosphorylase (TP)
(17) and dihydropyrimidine
dehydrogenase (DPD) (25). In the
present study, when cells were treated with 5-FU, the mRNA levels
of TYMS (encoding TS), TYMP (encoding TP) and DPYD (encoding DPD)
tended to be slightly decreased in the Hu-NK4 cells, while the gene
expression level was not significantly different when compared to
the Hu-Em cell line, consistent with previous reports of
5-FU-resistant CCA cell lines (26).
We next investigated the apoptosis of HuCC-T1 cells
induced by the combination of 5-FU and NK4 gene expression from
various aspects. The increase in the cell apoptosis rate evaluated
by flow cytometry confirmed that NK4 gene expression enhances the
cell apoptosis induced by 5-FU. The results prompted us to further
investigate the apoptotic pathway involved in 5-FU resistance.
Control of the intrinsic pathway is known to be governed by the
Bcl-2 family members, and dysregulation of Bcl-2 family members are
a common occurrence in tumorigenesis. Anti-apoptotic family members
contain 4 Bcl-2 homology (BH) domains (BH1-4) and include Bcl-2,
Bcl-XL, Mcl-1 and A1 (27). The
pro-apoptotic molecule Bax normally resides in the cytoplasm and
translocates to the mitochondria following an apoptotic stimulus.
Loss of Bax expression has been shown to decrease sensitivity to
chemotherapies in vitro(28)
and enhance tumorigenesis in vivo(29). Preclinical models have clearly
demonstrated a role for Bcl-2 family members in regulating the
response to anticancer strategies. Furthermore, the ratio of Bcl-2
to Bax protein appears to determine the susceptibility of cells to
apoptotic stimuli (30,31).
Activation of Bax and Bak results in the formation
of pores in the mitochondrial membrane that allow pro-apoptotic
molecules such as cytochrome c, Smac and Omi to be released
into the cytosol. Cytochrome c together with apoptotic
protease-activating factor-1 (APAF-1), procaspase-9 and ATP form
the apoptosome, a complex that results in the cleavage and
activation of caspase-9, the initiator caspase for the intrinsic
apoptotic pathway (32). Smac
promotes apoptosis by binding to the IAP family of proteins
(33). Binding of Smac to the IAPs
de-represses IAP-mediated inhibition of caspases-3, -7 and -9. When
released from the mitochondria, Smac and Omi bind to members of the
IAP family, alleviating IAP-induced caspase inhibition to promote
apoptosis. In the direct activation model, anti-apoptotic members
of the Bcl-2 family, Bcl-2, Bcl-XL and Mcl-1, inhibit binding of
BH3-only molecules to Bax/Bak, thereby preventing their
activation.
In the present study, 5-FU induced the activation of
caspase-3 and caspase-9. Bcl-2 expression was dose-dependently
downregulated by 5-FU; the level in Hu-NK4 cells decreased to a
greater extend compared to that in the Hu-Em cells. Bax is an
important initiator of mitochondrial-mediated apoptosis. It was
upregulated by 5-FU; a significant difference in the Bcl-2/Bax
ratio, but not in Bax, was observed between the two groups. These
results confirm that the Bcl-2 family and the intrinsic apoptotic
pathway are major effectors of 5-FU-induced apoptosis.
Having identified that 5-FU sensitivity was modified
by NK4, we aimed to ascertain whether it affected the HGF/Met
signaling pathway. The difference in the level of p-Met between
Hu-NK4 and Hu-Em cells was thought to be due to NK4 gene expression
as the difference was observed even at 0 μM of 5-FU. In addition,
5-FU at 10 μM suppressed the phosphorylation of Met in both
transfectants, indicating that 5-FU also has a suppressive effect
on the the phosphorylation of Met, but there was no significant
difference between the HGF stimulation group and HGF and the 5-FU
co-treatment group. Therefore, we demonstrated that NK4 and 5-FU
are two independent inhibitors of the HGF/Met signaling
pathway.
In conclusion, this study identified a novel role
for NK4 as a modulator of 5-FU-induced death in CCA cells through
activation of the intrinsic apoptotic pathway.
Acknowledgements
We are grateful to Professor Toshikazu Nakamura and
Professor Kiyomasa Oka (Osaka University Medical School, Japan) for
their precious gift of the human NK4 expression plasmid pcDNA3/NK4.
We acknowledge the guidance of Professor Zhining Fan, Dr Faming
Zhang and Dr Yun Wang (The Second Affiliated Hospital of Nanjing
Medical University, China) and we also appreciate the excellent
technical assistance of Dr Yanggang Yuan and Dr Chenbo Ji
(Institute of Pediatrics, Nanjing Medical University, China).This
study was supported by grants from the Scientific Research
Foundation for Health of Jiangsu Province (no. H200835) and Science
and Technology Development Foundation of Nanjing Medical University
(2012NJMU228).
References
|
1
|
Marsh Rde W, Alonzo M, Bajaj S, et al:
Comprehensive review of the diagnosis and treatment of biliary
tract cancer 2012. Part I: diagnosis-clinical staging and
pathology. J Surg Oncol. 106:332–338. 2012.PubMed/NCBI
|
|
2
|
Blechacz B, Komuta M, Roskams T and Gores
GJ: Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev
Gastroenterol Hepatol. 8:512–522. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Romiti A, D’Antonio C, Zullo A, et al:
Chemotherapy for the biliary tract cancers: moving toward improved
survival time. J Gastrointest Cancer. 43:396–404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakamura T, Nishizawa T, Hagiya M, et al:
Molecular cloning and expression of human hepatocyte growth factor.
Nature. 342:440–443. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Date K, Matsumoto K, Shimura H, Tanaka M
and Nakamura T: HGF/NK4 is a specific antagonist for pleiotrophic
actions of hepatocyte growth factor. FEBS Lett. 420:1–6. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Comoglio PM, Giordano S and Trusolino L:
Drug development of MET inhibitors: targeting oncogene addiction
and expedience. Nat Rev Drug Discov. 7:504–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sattler M and Salgia R: The MET axis as a
therapeutic target. Update Cancer Ther. 3:109–118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohuchida K, Mizumoto K, Murakami M, et al:
Radiation to stromal fibroblasts increases invasiveness of
pancreatic cancer cells through tumor-stromal interactions. Cancer
Res. 64:3215–3222. 2004. View Article : Google Scholar
|
|
10
|
Yano S, Wang W, Li Q, et al: Hepatocyte
growth factor induces gefitinib resistance of lung adenocarcinoma
with epidermal growth factor receptor-activating mutations. Cancer
Res. 68:9479–9487. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Onimaru M, Ohuchida K, Egami T, et al:
Gemcitabine synergistically enhances the effect of adenovirus gene
therapy through activation of the CMV promoter in pancreatic cancer
cells. Cancer Gene Ther. 17:541–549. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taiyoh H, Kubota T, Fujiwara H, et al: NK4
gene expression enhances 5-fluorouracil-induced apoptosis of murine
colon cancer cells. Anticancer Res. 31:2217–2224. 2011.PubMed/NCBI
|
|
13
|
Crea F, Nobili S, Paolicchi E, et al:
Epigenetics and chemoresistance in colorectal cancer: an
opportunity for treatment tailoring and novel therapeutic
strategies. Drug Resist Updat. 14:280–296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Banerjee D, Mayer-Kuckuk P, Capiaux G,
Budak-Alpdogan T, Gorlick R and Bertino JR: Novel aspects of
resistance to drugs targeted to dihydrofolate reductase and
thymidylate synthase. Biochim Biophys Acta. 1587:164–173. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Longley DB, Allen WL and Johnston PG: Drug
resistance, predictive markers and pharmacogenomics in colorectal
cancer. Biochim Biophys Acta. 1766:184–196. 2006.
|
|
16
|
Leichman CG, Lenz HJ, Leichman L, et al:
Quantitation of intratumoral thymidylate synthase expression
predicts for disseminated colorectal cancer response and resistance
to protracted-infusion fluorouracil and weekly leucovorin. J Clin
Oncol. 15:3223–3229. 1997.
|
|
17
|
Metzger R, Danenberg K, Leichman CG, et
al: High basal level gene expression of thymidine phosphorylase
(platelet-derived endothelial cell growth factor) in colorectal
tumors is associated with nonresponse to 5-fluorouracil. Clin
Cancer Res. 4:2371–2376. 1998.
|
|
18
|
Nishi M, Shimada M, Utsunomiya T, et al:
Role of dihydropyrimidine dehydrogenase and thymidylate synthase
expression in immunohistochemistry of intrahepatic
cholangiocarcinoma. Hepatol Res. 41:64–70. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morine Y, Shimada M, Utsunomiya T, et al:
Role of thymidylate synthase and dihydropyrimidine dehydrogenase
mRNA in intrahepatic cholangiocarcinoma. Surg Today. 42:135–140.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wilson TR, Johnston PG and Longley DB:
Anti-apoptotic mechanisms of drug resistance in cancer. Curr Cancer
Drug Targets. 9:307–319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang L, Wong YP, Cai YJ, Lung I, Leung CS
and Burd A: Low-dose 5-fluorouracil induces cell cycle G2 arrest
and apoptosis in keloid fibroblasts. Br J Dermatol. 163:1181–1185.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Okamoto S, Sakai M, Uchida J and Saito H:
5-Fluorouracil induces apoptotic cell death with G2 phase arrest in
human breast cancer grafted in nude mice. Anticancer Res.
16:2699–2704. 1996.PubMed/NCBI
|
|
23
|
Naus PJ, Henson R, Bleeker G, Wehbe H,
Meng F and Patel T: Tannic acid synergizes the cytotoxicity of
chemotherapeutic drugs in human cholangiocarcinoma by modulating
drug efflux pathways. J Hepatol. 46:222–229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ikeguchi M, Hirooka Y, Makino M and
Kaibara N: Dihydropyrimidine dehydrogenase activity of cancerous
and non-cancerous tissues in liver and large intestine. Oncol Rep.
8:621–625. 2001.PubMed/NCBI
|
|
25
|
Salonga D, Danenberg KD, Johnson M, et al:
Colorectal tumors responding to 5-fluorouracil have low gene
expression levels of dihydropyrimidine dehydrogenase, thymidylate
synthase, and thymidine phosphorylase. Clin Cancer Res.
6:1322–1327. 2000.
|
|
26
|
Namwat N, Amimanan P, Loilome W, et al:
Characterization of 5-fluorouracil-resistant cholangiocarcinoma
cell lines. Chemotherapy. 54:343–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Danial NN: BCL-2 family proteins: critical
checkpoints of apoptotic cell death. Clin Cancer Res. 13:7254–7263.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang L, Yu J, Park BH, Kinzler KW and
Vogelstein B: Role of BAX in the apoptotic response to anticancer
agents. Science. 290:989–992. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heiser D, Labi V, Erlacher M and Villunger
A: The Bcl-2 protein family and its role in the development of
neoplastic disease. Exp Gerontol. 39:1125–1135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yin XM, Oltvai ZN and Korsmeyer SJ: BH1
and BH2 domains of Bcl-2 are required for inhibition of apoptosis
and heterodimerization with Bax. Nature. 369:321–323. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zou H, Li Y, Liu X and Wang X: An APAF-1.
cytochrome c multimeric complex is a functional apoptosome that
activates procaspase-9. J Biol Chem. 274:11549–11556. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nachmias B, Ashhab Y and Ben-Yehuda D: The
inhibitor of apoptosis protein family (IAPs): an emerging
therapeutic target in cancer. Semin Cancer Biol. 14:231–243. 2004.
View Article : Google Scholar : PubMed/NCBI
|