Introduction
Histone molecules are octamers composed of dimmers
of the basic histone proteins H2A, H2B, H3 and H4. Acetylation and
deacetylation of histones occur at lysine residues in the
N-terminal tail of all four histone proteins and are mediated by
different families of histone acetylases (HATs) and histone
deacetylases (HDACs). While the activity of HATs leads to a
hyperacetylated and ‘open’ chromatin conformation allowing
transcriptional activity, HDAC activity represses transcriptional
activity by condensing the chromatin package. The activity of HATs
and HDACs is not restricted to histones, and a number of
non-histone proteins have been identified as HAT/HDAC targets
(1,2). Currently, 18 HDACs are known; they are
either zinc-dependent or nicotinamide adenine dinucleotide
(NAD)-dependent and are generally divided into four classes, based
on sequence homology to yeast counterparts. These HDAC substrates
are directly or indirectly involved in numerous important cell
pathways, including cell proliferation, differentiation, and
apoptosis (3).
Overexpression of HDAC enzymes is observed in
various primary human cancer tissues, which include the stomach,
colon, breast, and prostate. Aberrant recruitment of HADCs
correlates with the initiation and progression of various types of
cancer (4,5). As a result, HDACs have become
attractive targets for cancer therapy and HDAC inhibitors (HDACi)
have been discovered with different structural characteristics,
including hydroximates, cyclic peptides, aliphatic acids and
benzamides. Several HDACi have advanced into clinical trials
(6,7). A few of these compounds have shown
potent antitumor activities in various types of hematologic and
solid cancer. Currently, two HDACi - vorinostat (suberoylanilide
hydroxamic acid; SAHA) and romidepsin (FK228; depsipeptide)- have
been approved by the Food and Drug Administration for the treatment
of patients with relapsed cutaneous T-cell lymphoma (7).
Several compounds in clinical developments seem to
have limitations that include low oral bioavailability, poor in
vivo stability, and undesirable safety profiles (1). Hence, there remains a significant
clinical opportunity for efficacious HDACi that are safe and well
tolerated. Previously, we designed and synthesized a series of new
class of hydroxamate histone deacetylase inhibitors. Among them,
S-(E)-3-(1-(1-(benzo[d]oxazol-2-yl)-2-methylpropyl)-1H-1,2,3-triazol-4-yl)-N-hydroxyacrylamide
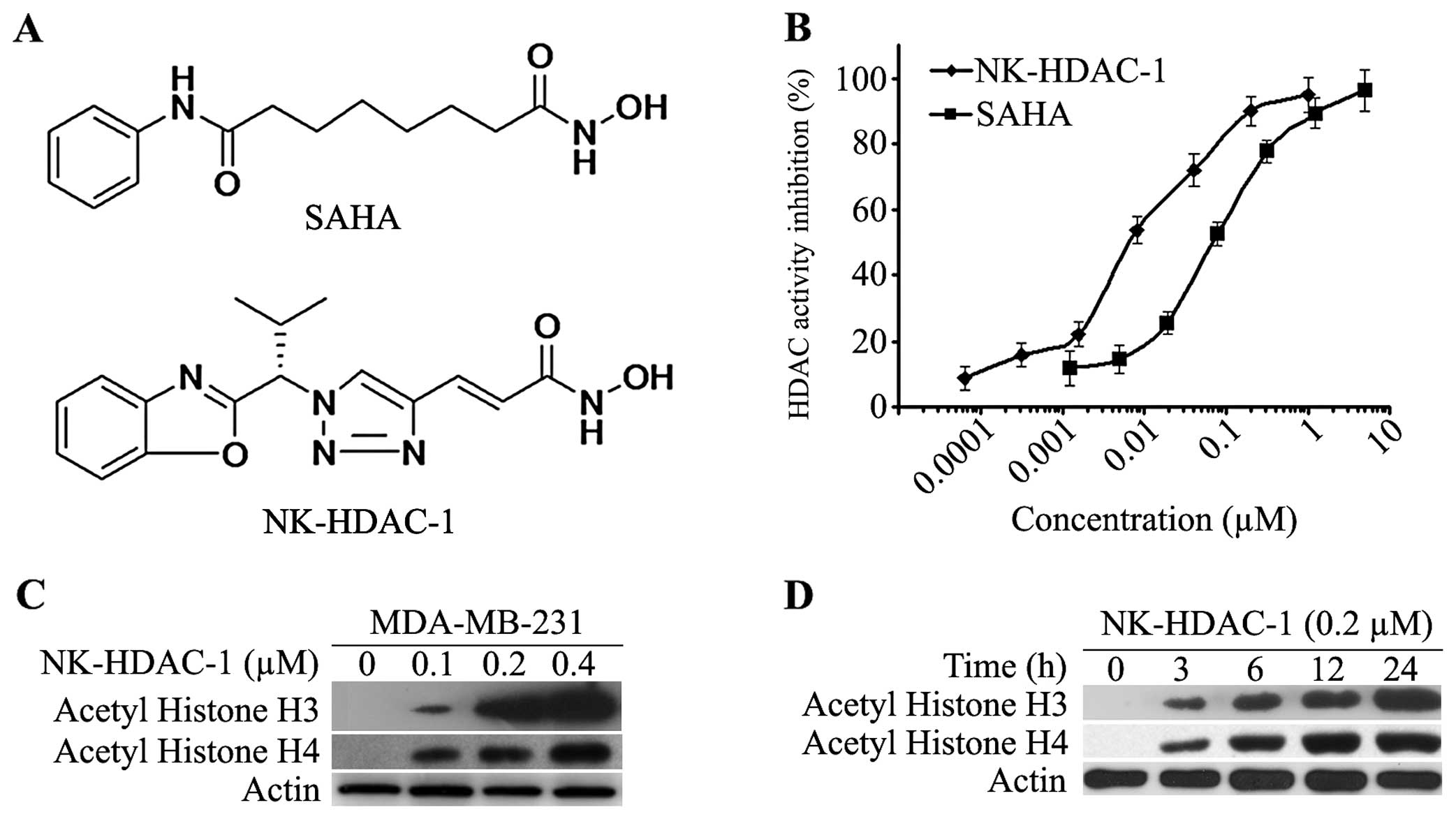
(NK-HDAC-1) (Fig. 1A) has more
potent antitumor activity than SAHA (8,9). In
this study, we examined the functional effects of the novel HDACi
NK-HDAC-1 on the activity of HADC enzyme and on the growth of
breast cancer cells in vitro and in vivo. The
possible molecular mechanisms underlying these effects were
explored. These findings suggested that NK-HDAC-1 could be a potent
HADC inhibitor against breast cancer.
Materials and methods
Cell lines and cell culture
All human breast cancer cell lines used in this
study were purchased from Shanghai Cell Bank, the Institute of Cell
Biology, China Academy of Sciences (Shanghai, China). They were
maintained in a humidified atmosphere containing 5% CO2
at 37 °C in a different medium supplemented with 10% FBS, 100 U/ml
penicillin, and 100 μg/ml streptomycin. Breast cancer cell lines
MDA-MB-231, MCF-7, SKBR3, MDA-MB-453, BT474 and T47D were cultured
in RPMI-1640 or in DMEM medium. An immortalized human mammary
epithelial cell line MCF10A was purchased from the American Type
Culture Collection and cultured in DMEM medium supplemented with 5%
horse serum, 20 ng/ml epidermal growth factor, 0.5 μg/ml
hydrocortisone, 100 ng/ml cholera toxin, 10 μg/ml insulin, and
penicillin/streptomycin.
Inhibition of cell growth in vitro
Cells were seeded in 96-well plates and incubated
overnight. They were then treated for 72 h with varying
concentrations of SAHA (Sigma-Aldrich, St. Louis, MO, USA) or
NK-HDAC-1. Next, 20 μl
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT;
Sigma-Aldrich) solution (5 mg/ml in PBS) was added to each well and
incubated for 4 h at 37°C. After removing the medium, MTT formazan
was dissolved in 150 μl dimethyl sulfoxide (DMSO) and monitored by
a microplate reader at a wavelength of 570 nM. The IC50
value was obtained using CalcuSyn software (Biosoft, Cambridge,
UK).
HDAC enzyme activity assay in vitro
The deacetylase enzyme assay was based on a
homogeneous fluorescence release assay. Purified HeLa cell nuclear
extracts were incubated with NK-HDAC-1 diluted in various
concentrations for 20 min in an assay buffer [25 mM HEPES (pH 8.0),
137 mM NaCl, 1 mM MgCl2, 2.7 mM KCl] at room
temperature. The substrate Ac-Arg-Gly-Lys(Ac)-AMC was added to the
reaction for further incubation at 37°C. The reaction was
discontinued by incubating it with trypsin at room temperature for
20 min, allowing the release of fluorophore from the deacetylated
substrate. The fluorescent signal was detected by a fluorometer at
an excitation of 360 nm, emission of 470 nm, and cutoff at 435 nm.
HeLa cells were harvested by trypsinization, resuspended in two
volumes of buffer (60 mM Tris-HCl, pH 7.4, 30% glycerol, 450 mM
NaCl), and lysed by three freeze and thaw cycles (dry ice and
30°C). Cell debris was removed by centrifugation at 20,000 × g and
the supernatant was aliquoted and stored at −80°C.
Cell cycle analysis by flow
cytometry
Cells were trypsinized, fixed in ice-cold 70%
ethanol, and then stored at −20°C. Prior to analysis, samples were
washed twice in PBS and resuspended in a solution of propidium
iodide (50 mg/ml) and RNase A (0.5 mg/ml) in PBS for 30 min in the
dark. Data collected from 10,000 cells of each sample were analyzed
by a flow cytometer (Becton-Dickinson Co., USA).
Annexin V-FITC apoptosis assay
Annexin V-FITC apoptosis assay was employed to
determine viable, early apoptotic cells. According to the
recommended protocols of the Annexin V-FITC kit (BD Pharmingen,
USA), the cells were seeded at 4×105 cells/ml/well in
6-well plates. Following treatment with SAHA or NK-HDAC-1,
MDA-MB-231 cells were harvested and washed twice with ice-cold
phosphate-buffered saline (PBS) and resuspended in 100 μl binding
buffer. Next, 5 μl Annexin V-FITC and 10 μl propidium iodide were
added and the mixture was incubated for 30 min in the dark.
Finally, 400 μl binding buffer was added to the cells and the
mixture was analyzed using a flow cytometer (Becton-Dickinson
Co.).
Western blotting
Cells were harvested after treatment and whole-cell
lysate was prepared as described below. Cells were trypsinized,
washed with PBS, and then lysed with a buffer containing 50 mM
Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1 mM
dithiothreitol, 1% Nonidet P-40, 0.1% SDS, protease inhibitors (1
mM PMSF, 5 mg/ml aprotinin, 5 mg/ml leupeptin and 5 mg/ml
pepstatin), and phosphatase inhibitors (20 mM β-glycerophosphate,
50 mM NaF and 1 mM Na3VO4). Lysates were
incubated at 4°C for 20 min and centrifuged at 12,000 × g for 15
min. Equal amounts of lysate (40 μg) were resolved by SDS-PAGE and
transferred to the polyvinylidene difluoride membrane (Millipore,
USA). Membranes were blocked in 5% non-fat skim milk/TBST [20 mM
Tris-HCl (pH 7.4), 150 mM NaCl, and 0.1% Tween-20] at room
temperature for 1 h and detected with primary antibodies at room
temperature for 2 h. The membranes were then blotted for 1 h at
room temperature with an appropriate horseradish peroxidase-linked
secondary antibody followed by enhanced chemiluminescence western
blotting detection reagents (Amersham Pharmacia Biotech, USA).
Cytosolic fractions were prepared by using mitochondria/cytosol
fraction kit (BioVision). Primary antibody cyclin D1, p21,
caspases-3, -9, -8, retinoblastoma (Rb), phosphorylated Rb, actin,
and secondary antibody were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-acetyl-histone H3
and H4 were purchased from Millipore. Poly(ADP-ribose) polymerase
(PARP) and cytochrome c were purchased from Cell Signaling
Technology.
RT-PCR
Total RNA was extracted using TRIzol®
reagent (Invitrogen). p21 mRNA expression was detected by RT-PCR
according to the instructions (Qiagen). PCR primers for p21 were:
5′-GCCTGCCGCCGCCTCTTC-3′ and 5′-GCCGCCT GCCTCCTCCCAACTC-3′. GAPDH
was used as an internal control with forward primer,
5′-GGCAAATTCCATGG CACCGTCAAG-3′ and reverse primer, 5′-GCAATGCCAG
CCCCAGCGTCAAA-3′.
Chromatin immunoprecipitation
Analysis was performed by means of a chromatin
immunoprecipitation assay kit protocol (Millipore). Soluble
chromatin from MDA-MB-231 cells was immunoprecipitated with
anti-acetylated histone H3 and H4 antibodies, respectively. PCR
primer for the promoter region of the p21 gene was used to amplify
the DNA isolated from the immunoprecipitated chromatin. p21 primers
were 5′-CAGAGGAGGCGCCATGTCAG-3′ and 5′-CCTGTGGGCGGATTAGGG-3′.
Acute toxicity study in BALB/c mice
BALB/c mice aged 7–8 weeks and weighing 20–22 g were
purchased from the Animal Center of Shandong University. All animal
experiments were performed in compliance with the local Ethics
Committee. Seventy mice were equally divided into seven groups of
five males and five females each. NK-HDAC-1 was administered on the
1st day by intraperitoneal injection to groups of mice at doses of
300, 195, 126.8, 82.4, 53.6 and 34.8 mg/kg body weight at a ratio
of 1/0.65. The control group received 100 μl DMSO only. The
mortality and general conditions, i.e., energy, activity, hair,
feces, behavior pattern, and other clinical signs, of all animals
were observed continuously for 14 days after administration. The
body weights of the mice were measured before dosing and on day 14.
All animals were sacrificed on the 14th day and the major organs of
each mouse were examined after dissection.
Inhibition of tumor growth in vivo
The research protocol was in accordance with the
institutional guidelines of the Animal Care and Use Committee at
Shandong University. The animals were housed under pathogen-free
conditions. Female BALB/c (nu/nu) mice (20±2 g, 4–6 weeks
old) were purchased from the Animal Center of the China Academy of
Medical Sciences (Beijing, China). Breast cancer MDA-MB-231 cells
of 5.0×106 suspended in 100 μl PBS were subcutaneously
inoculated into the lower right flank of the nude mice. The mice
were divided randomly into four groups of six mice each when the
tumors were 100–150 mm3 in size. The control group
received solvent (DMSO) only while the positive control group
received SAHA at 50 mg/kg. The treatment groups received NK-HDAC-1
at the following doses: 100, 50, 30, 10 and 3 mg/kg body weight.
The mice were injected intraperitoneally with 100 μl DMSO, SAHA
(diluted in 100 μl DMSO), and NK-HDAC-1 (diluted in 100 μl DMSO) on
days 2, 3, 4, 6 and 7 of each week. All mice were then treated for
3 weeks. The diameter of the tumor was measured twice a week with a
caliper. The tumor volume was calculated using the following
formula: v=ab2/2, where a and b are
the long diameter and perpendicular short diameter of the tumor,
respectively. At the end of the experiment, tumors were harvested
and tumor weights were determined.
Statistical analysis
All quantitative data were subjected to ANOVA to
determine the existence of significant differences between groups.
For data groups satisfying the ANOVA criteria (P<0.05),
individual comparisons were performed using the Student’s t-test.
P≤0.05 was considered to indicate a statistically significant
difference.
Results
NK-HDAC-1 inhibits total HDAC enzyme
activity and increases acetylation of histone H3 and H4
NK-HDAC-1 was submitted to a deacetylation assay
using a synthetic acetylated substrate to evaluate the inhibitory
effect of NK-HDAC-1 on HDAC activity. As shown in Fig. 1B, NK-HDAC-1 significantly inhibited
total HDAC activity in a dose-dependent manner with an
IC50 value of 0.032±0.009 μM, which was much lower than
that of SAHA (IC50=0.218±0.051 μM).
To confirm the ability of NK-HDAC-1 to inhibit
deacetylation of HDACs in cells, western blotting was used to
analyze acetylation of histone H3 and H4 in MDA-MB-231 breast
cancer cells. NK-HDAC-1 dose-dependently increased the acetylation
of histone H3 and histone H4 after the cells were treated for 24 h
(Fig. 1C). Even 0.1 μM NK-HDAC-1
markedly induced the acetylation of histone H3 and H4. The
acetylation of histone H3 and H4 was detectable after MDA-MB-231
cells were treated with 0.5 μM NK-HDAC-1 for 3 h and they increased
in a time-dependent manner for up to 24 h (Fig. 1D).
NK-HDAC-1 inhibits cell growth in breast
cancer cells
NK-HDAC-1 was also entered into a screen at the
National Cancer Institute (NCI60) where it was tested for growth
inhibitory effects on a panel of cell lines representing various
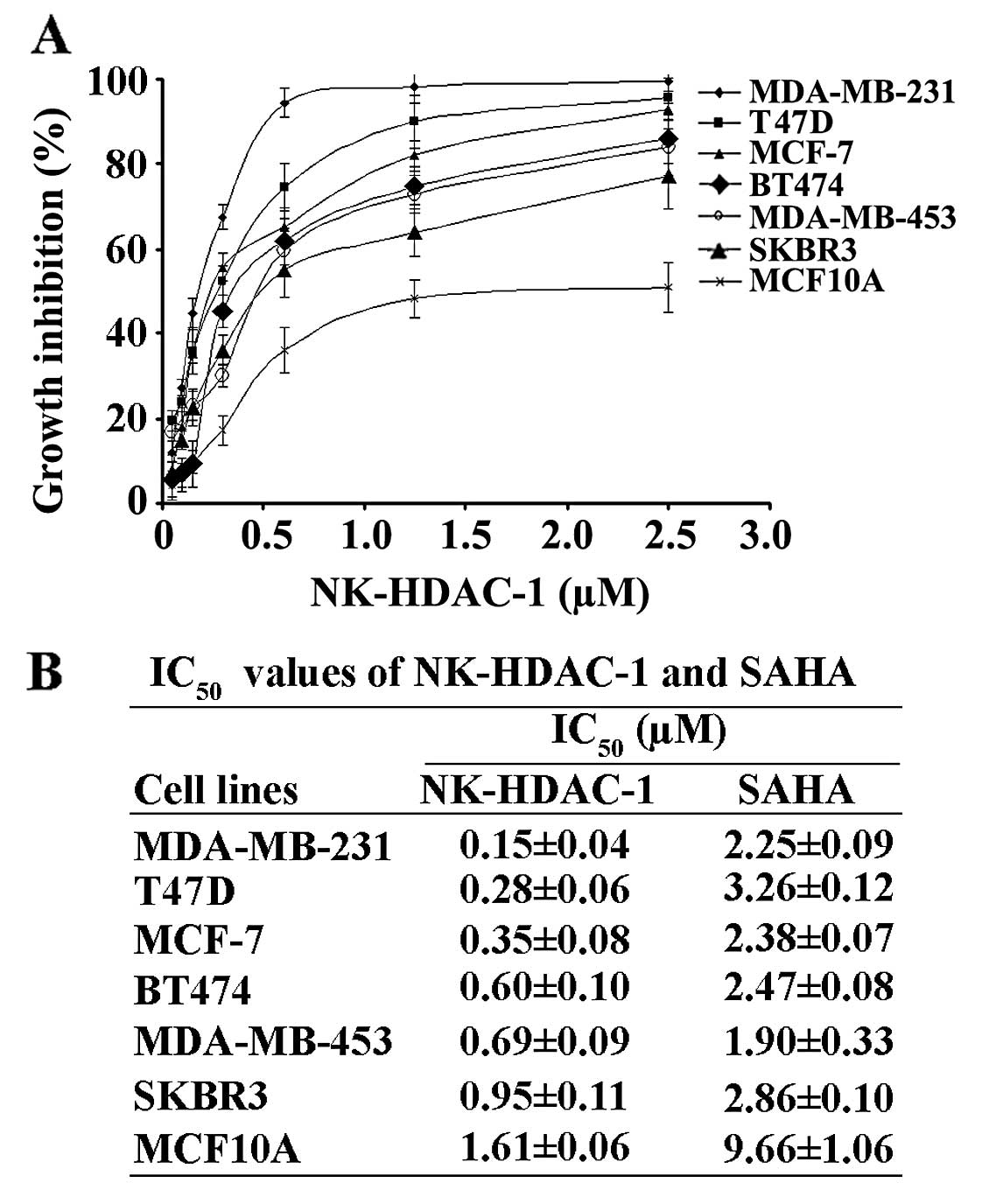
types of cancer. The results showed that NK-HDAC-1 was
approximately 1 order of magnitude more potent than SAHA against
cancer cells growth (9). In order
to test the effects of NK-HDAC-1 on the proliferation of breast
cancer cells, we performed MTT assay in six human cancer cell lines
as well as in an immortalized human breast epithelial cell MCF10A.
After 72 h of treatment of the cells with NK-HDAC-1 and SAHA
separately, NK-HDAC-1 markedly inhibited cell proliferation in all
breast cancer cell lines in a dose-dependent manner (Fig. 2A), but it moderately affected the
proliferation of MCF10A. The IC50 values of NK-HDAC-1
were significantly lower than those of SAHA on all cancer cells
(Fig. 2B). MDA-MB-231 cells were
most sensitive to NK-HDAC-1 among all the breast cancer cells and
were selected for the subsequent experiments to explore the
mechanisms by which NK-HDAC-1 inhibited breast cancer cell
proliferation.
NK-HDAC-1 induces cell cycle arrest in
MDA-MB-231 cells
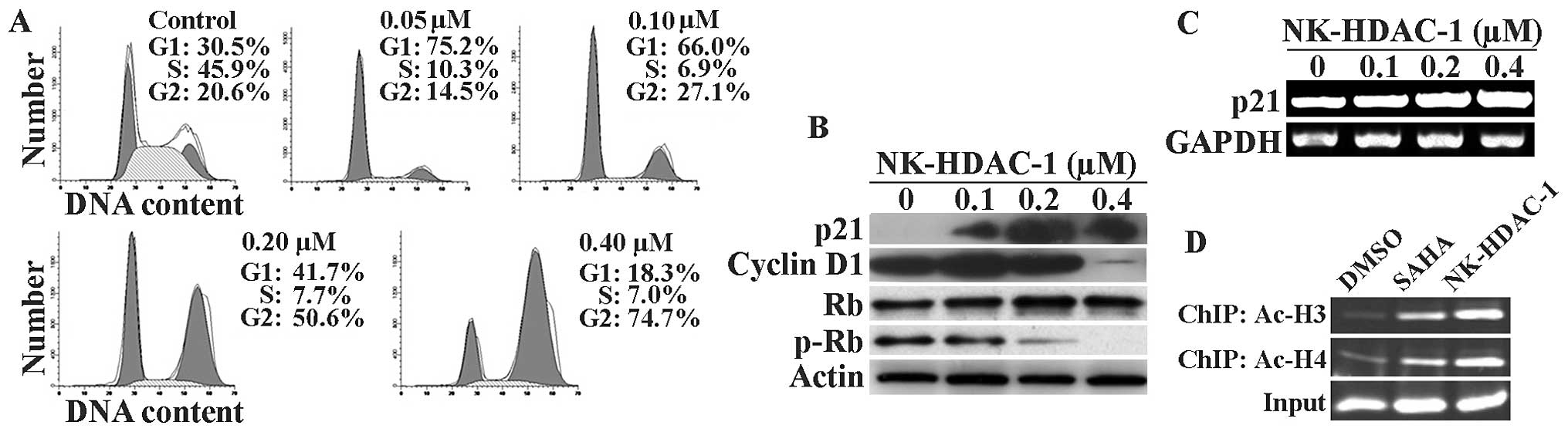
HADCi generally inhibit the growth of tumor cells by
causing them to arrest at a specific phase of the cell cycle. Cell
cycle analysis was performed after 24 h of incubation with
NK-HDAC-1 in order to detect the shifts in cell cycle distribution
before a significant amount of cells underwent apoptosis. NK-HDAC-1
induced G1 arrest at concentrations below 0.20 μM and G2/M arrest
at concentrations above 0.40 μM after 24-h treatments in MDA-MB-231
cancer cells (Fig. 3A). As control,
SAHA was found to cause G1 arrest at concentrations below 1.5 μM
and G2/M arrest at concentrations above 3.0 μM, which were
consistent with previous studies on the effects of SAHA on cell
cycle distribution in breast cancer cells (10) (data not shown).
P21 is generally considered one of the targets of
HDACi and an effector for HDACi-induced anticancer action.
Following treatment with NK-HADC-1, p21 gene expression markedly
increased and cyclin D1 expression markedly decreased. The
phosphorylation levels of Rb decreased while Rb protein was
unchanged after MDA-MB-231 cells were treated with NK-HADC-1 for 24
h (Fig. 3B). As measured by RT-PCR
detection shown in Fig. 3C, p21
mRNA was induced by NK-HADC-1 in a dose-dependent manner in
MDA-MB-231 cells.
To further elucidate whether NK-HADC-1 induced
hyperacetylation of histone H3 and H4 around the promoter region of
p21, chromatin immunoprecipitation was performed using primers
corresponding to the region -597 to -342 bp upstream of the
translation initiation site (ATG) of the p21 gene. Chromatin
fragments from MDA-MB-231 cells cultured with or without NK-HADC-1
for 24 h were immunoprecipitated with antibody against acetylated
histone H3 and H4, respectively. The chromatin fragments were then
isolated and amplified using PCR with p21 primers. Similar to SAHA,
NK-HADC-1 induced hyperacetylation around the promoter region of
p21 in MDA-MB-231 cells (Fig. 3D).
This result strongly suggested that p21 upregulation is mainly due
to enhancement of acetylation of histones H3 and H4 around the
promoter region of p21.
NK-HDAC-1 induces apoptosis in MDA-MB-231
cells by activating both the intrinsic and the extrinsic
pathway
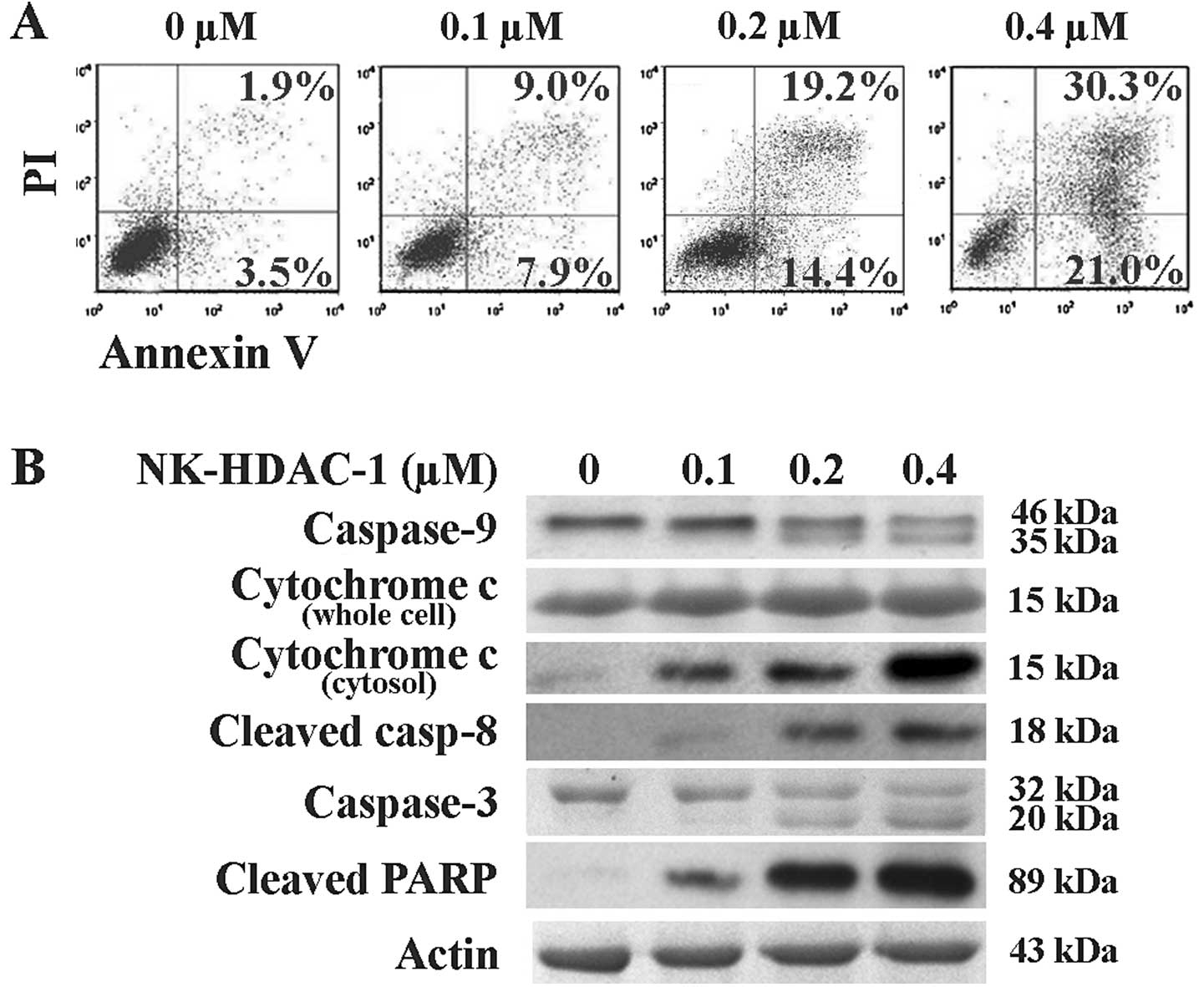
Apoptosis is believed to be the common cause of cell
killing by chemotherapy treatment as well as by HDACi. The rate of
apoptotic cells was then detected through Annexin V assay.
NK-HDAC-1 induced apoptosis in a dose-dependent manner in
MDA-MB-231 cells (Fig. 4A). Early
apoptotic cells increased from 7.9 to 21.0% and late apoptotic
cells increased from 9.0 to 30.3% following NK-HDAC-1 treatment of
0.1 to 0.4 μM for 48 h. These drug concentrations were selected as
they were near the IC50 concentration of NK-HDAC-1 on
MDA-MB-231 cells. Western blot analysis showed that the release of
cytochrome c from mitochondria to the cytosol was
dose-dependently increased while cytochrome c in whole cells
was unchanged (Fig. 4B). Both
caspase-9 and -8 activation was also confirmed by the appearance of
cleaved caspase-9 and cleaved caspase-8. The bands of cleaved
caspase-3 and cleaved PARP, as a result of apoptosis, were clearly
observed (Fig. 4B). Collectively,
these results suggested that the major mechanism of apoptosis
induced by NK-HDAC-1 was activation of the intrinsic and the
extrinsic pathway.
Acute toxicity of NK-HDAC-1 in BALB/c
mice
To investigate the maximal tolerance dose of
NK-HDAC-1 in vivo, acute toxicity analysis in BALB/c
mice was performed. NK-HDAC-1 was injected intraperitoneally into
six groups of ten mice at doses of 300, 195, 126.8, 82.4, 53.6 and
34.8 mg/kg body weight. The control group was injected with solvent
DMSO. Following exposure, mortality and changes in the body weight
of the mice were recorded for 14 days. All ten mice treated with
300 mg/kg died within 2 days. Nine of the ten mice treated with 195
mg/kg died within 2 days. Two mice in the group treated with 126.8
mg/kg and one mouse in the group treated with 82.4 mg/kg died. All
deaths occurred in the first two days and no mice died during the
last 12 days of the observation period. No mice died in the groups
treated with NK-HDAC-1 at doses of 53.6 and 34.8 mg/kg. None of the
surviving mice showed any abnormal signs and the body weights of
the mice were significantly unchanged during the period of the test
(data not shown). The results indicated that the maximal tolerance
dose of NK-HDAC-1 in BALB/c mice was >53.6 mg/kg.
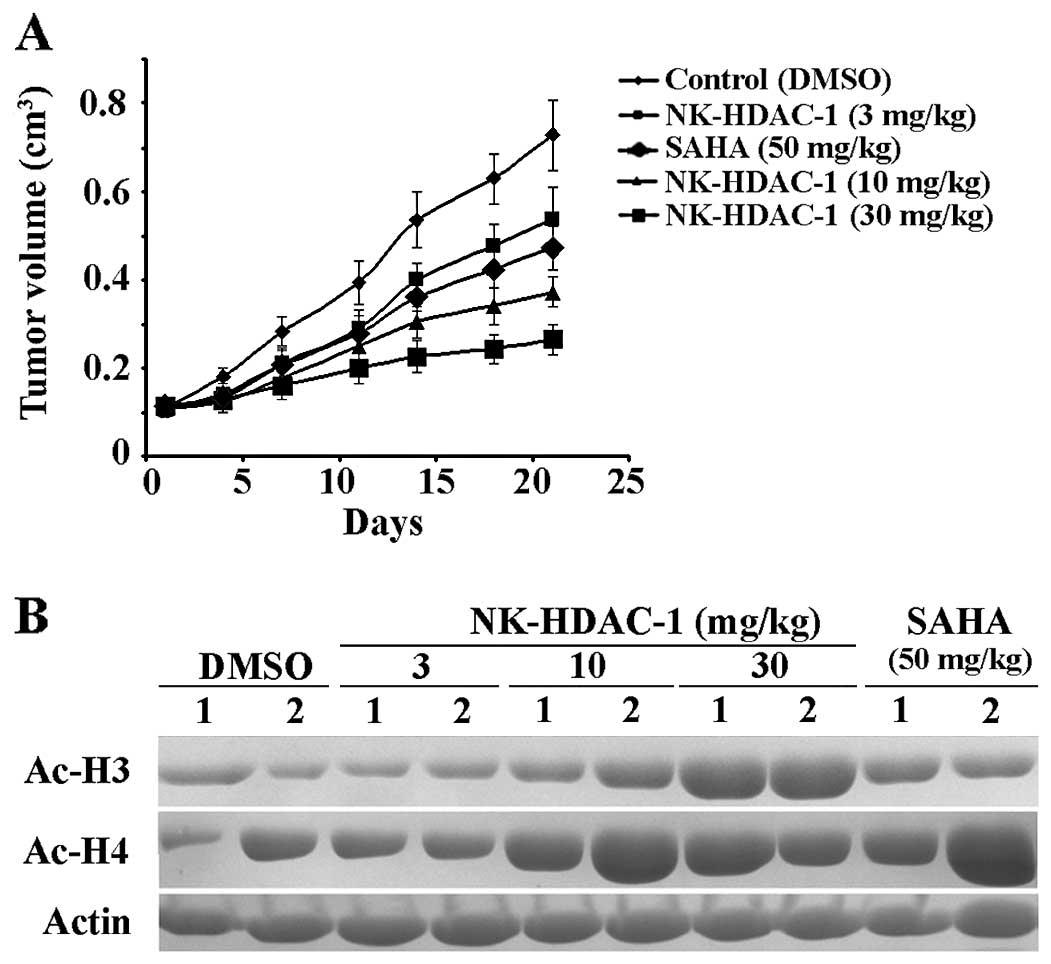
NK-HDAC-1 inhibits breast tumor
growth
To further assess the antitumor effects of NK-HDAC-1
in vivo, athymic mice bearing MDA-MB-231 xenograft were
treated 5 days a week for 21 days with vehicle or SAHA at 50 mg/kg
or with NK-HDAC-1 at doses of 100, 50, 30, 10 and 3 mg/kg (n=6).
Four of the six mice in the group treated with 100 mg/kg NK-HDAC-1
died at the end of the experiment. One of the six mice in the group
treated with 50 mg/kg NK-HDAC-1 died and the mean body weight of
the five surviving mice decreased significantly from 19.8 g on day
1 to 16.8 g on day 21. Compared with the control group, NK-HDAC-1
at doses of 30, 10 and 3 mg/kg did not cause overt signs of
toxicity or significant weight loss (data not shown). Tumor volumes
in MDA-MB-231 xenografts decreased by 63.6, 48.8 and 25.9%,
respectively (Fig. 5A). In the same
experiment, 50 mg/kg SAHA, a positive control, inhibited tumor
growth by 35.0%.
The expression of acetylation of both histone H3 and
H4 in tumor was determined by western blot analysis to investigate
whether NK-HDAC-1 affects HDAC enzyme activity in vivo. Two
tumors from each group were selected for this analysis. As shown in
Fig. 5B, NK-HDAC-1 enhanced the
acetylation of histone H3 and H4 in tumor sample, thus confirming
NK-HDAC-1 inhibited HDAC activity in vivo.
Discussion
Since HDACs regulate a variety of cell functions
that are involved in cell survival, cell cycle progression,
angiogenesis, and immunity, they are considered promising targets
for cancer therapy. In the current study, we provided the antitumor
effect of a novel hydroxamate histone deacetylase inhibitor,
NK-HDAC-1, in breast cancer in vitro and in vivo.
NK-HDAC-1 exhibited more potent antiproliferative
effects than SAHA. In contrast to SAHA with IC50 at
0.218±0.051 μM, NK-HDAC-1 had higher inhibitory effects on total
HDAC enzyme activity with IC50 at 0.032±0.009 μM. The
expression of acetylated histone H3 and H4 was markedly enhanced by
NK-HDAC-1 even at very low concentrations.
NK-HDAC-1 showed a potent growth inhibitory effect
against breast cancer cells in a dose-dependent manner. Moreover,
it was quite resistant to the immortalized human epithelial breast
cell line MCF10A, which was consistent with previous findings that
normal cells appear to be relatively resistant to HDACi (11,12).
Increasing evidence has revealed that HDACi suppress
cancer cell growth by inducing cell cycle arrest at the G1 and/or
G2/M phase (13,14). SAHA caused G1 and/or G2/M arrest in
various cancer cells (15,16). Similarly, NK-HDAC-1 induced G1
arrest at lower concentrations and G2/M arrest at higher
concentrations.
Several studies demonstrated that cyclin-dependent
kinases (CDKs) inhibitor p21 and cyclin D1 are important regulators
of the cell cycle in breast cancer. p21 inhibits CDK2-cyclin E,
with the consequent inhibition of CDK2-dependent phosphorylation of
Rb, thus inhibiting E2F1-dependent gene transcription and
progression into and through the S phase. p21 also inhibits the
kinase activity of CDK2-cyclin A and CDK1-cyclin A, which are
required for progression through the S phase and into G2
respectively. Additionally, p21 inhibits the kinase activity of
CDK1-cyclin B1, thus inhibiting progression through G2 and G2/M
(17). The primary function of
cyclin D is to activate the CDKs CDK4 and CDK6 as a regulatory
partner and then CDKs promote Rb phosphorylation and activation of
E2F-responsive gene (18). In this
study, NK-HDAC-1 markedly increased both p21 protein expression and
mRNA level. Cyclin D1 and the phosphorylation of Rb were
dose-dependently decreased while Rb protein level was unaltered. In
this study, the increase of p21 and the decrease of cyclin D1 may
act together to account for the reduced CDK activity, causing
dephosphorylation of Rb, which blocks E2F activities in the
transcription of genes for G1 progression and G1/S transition. This
study revealed that NK-HDAC-1 differentially regulated cell cycle
progression depending on its concentration, and that its inhibition
of cancer cell growth may be mediated, at least in part, by
blocking cell cycle progression.
HDACi can affect transcription by inducing
acetylation of histones, transcription factors and other proteins
regulating transcription (19). In
this report, we identified that NK-HDAC-1 induced hyperacetylation
of histone H3 and H4 around the promoter region of p21, which may
enhance p21 mRNA level and protein expression.
All HDACi have been reported to activate either an
extrinsic or intrinsic pathway or both of these cell death pathways
in several cancer models (20,21).
The death-receptor pathway is activated when ligands, such as Fas
or TRAIL, bind to their death receptors, causing the activation of
caspase-8. The mitochondrial pathway is activated by stress stimuli
(chemotherapeutic agents) that disrupt the mitochondrial membrane,
causing the release of cytochrome c. Cytochrome c
release leads to apoptosome formation and activation of caspase-9.
Caspases-8 and -9 can then cleave caspases-3, -6, and -7,
culminating in apoptosis (22).
Annexin V assay in this study showed that NK-HDAC-1 promoted
apoptosis in a dose-dependent manner. Western blotting showed that
cytochrome c translated from mitochondria to cytosol with
activation of caspase-9. The activation of caspases-9 and -8
resulted in enhancement of cleaved caspase-3 and PARP. These
results suggested that NK-HDAC-1 induced apoptosis via both the
intrinsic and the extrinsic pathway and that the induction of
apoptosis may be another mechanism responsible for growth
inhibition by NK-HDAC-1.
The acute toxicity experiment indicated that the
maximal tolerance dose of NK-HDAC-1 in BALB/c mice was
higher than 53.6 mg/kg. In the nude mice xenograft model treated
five times a week for three weeks, one in six mice died in the
group treated with 50 mg/kg NK-HDAC-1. However, treatment with
NK-HDAC-1 at doses of 30, 10 and 3 mg/kg inhibited tumor growth
without any signs of toxicity by 63.6, 48.8 and 25.9%,
respectively. These results showed that NK-HDAC-1 had potent
antitumor activity in vivo.
In conclusion, results of the present study
demonstrated that the novel hydroxamate HDAC inhibitor NK-HDAC-1
inhibited the growth of cancer cells in vitro and in
vivo and that the growth inhibitory effect of NK-HDAC-1 may be
mediated by cell cycle arrest and apoptosis. The results suggest
that NK-HDAC-1 is a potential therapeutic candidate for cancer
therapy.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 21072105) and the
Independent Innovation Foundation of Shandong University (no.
2009DX002).
References
|
1
|
Glaser KB: HDAC inhibitors: clinical
update and mechanism-based potential. Biochem Pharmacol.
74:659–671. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou Q, Chaerkady R, Shaw PG, Kensler TW,
Pandey A and Davidson NE: Screening for therapeutic targets of
vorinostat by SILAC-based proteomic analysis in human breast cancer
cells. Proteomics. 10:1029–1039. 2010.PubMed/NCBI
|
|
3
|
Struhl K: Histone acetylation and
transcriptional regulatory mechanisms. Genes Dev. 12:599–606. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marks PA and Xu WS: Histone deacetylase
inhibitors: potential in cancer therapy. J Cell Biochem.
107:600–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hagelkruys A, Sawicka A, Rennmayr M and
Seiser C: The biology of HDAC in cancer: the nuclear and epigenetic
components. Handb Exp Pharmacol. 206:13–37. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma X, Ezzeldin HH and Diasio RB: Histone
deacetylase inhibitors: current status and overview of recent
clinical trials. Drugs. 69:1911–1934. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gryder BE, Sodji QH and Oyelere AK:
Targeted cancer therapy: giving histone deacetylase inhibitors all
they need to succeed. Future Med Chem. 4:505–524. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hou J, Feng C, Li Z, et al:
Structure-based optimization of click-based histone deacetylase
inhibitors. Eur J Med Chem. 46:3190–3200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hou J, Li Z, Fang Q, et al: Discovery and
extensive in vitro evaluations of NK-HDAC-1: a chiral histone
deacetylase inhibitor as a promising lead. J Med Chem.
55:3066–3075. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang L and Pardee AB: Suberoylanilide
hydroxamic acid as a potential therapeutic agent for human breast
cancer treatment. Mol Med. 6:849–866. 2000.PubMed/NCBI
|
|
11
|
Armeanu S, Pathil A, Venturelli S,
Mascagni P, et al: Apoptosis on hepatoma cells but not on primary
hepatocytes by histone deacetylase inhibitors valproate and
ITF2357. J Hepatol. 42:210–217. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garber K: HDAC inhibitors overcome first
hurdle. Nat Biotechnol. 25:17–19. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marks PA and Dokmanovic M: Histone
deacetylase inhibitors: discovery and development as anticancer
agents. Expert Opin Investig Drugs. 14:1497–1511. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ungerstedt JS, Sowa Y, Xu WS, et al: Role
of thioredoxin in the response of normal and transformed cells to
histone deacetylase inhibitors. Proc Natl Acad Sci USA.
102:673–678. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumagai T, Wakimoto N, Yin D, et al:
Histone deacetylase inhibitor, suberoylanilide hydroxamic acid
(Vorinostat, SAHA) profoundly inhibits the growth of human
pancreatic cancer cells. Int J Cancer. 121:656–665. 2007.
View Article : Google Scholar
|
|
16
|
Yin D, Ong JM, Hu J, et al:
Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor:
effects on gene expression and growth of glioma cells in vitro and
in vivo. Clin Cancer Res. 13:1045–1052. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gartel AL: p21(WAF1/CIP1) and cancer: a
shifting paradigm? Biofactors. 35:161–164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JK and Diehl JA: Nuclear cyclin D1: an
oncogenic driver in human cancer. J Cell Physiol. 220:292–296.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gui CY, Ngo L, Xu WS, Richon VM and Marks
PA: Histone deacetylase (HDAC) inhibitor activation of
p21WAF1 involves changes in promoter-associated
proteins, including HDAC1. Proc Natl Acad Sci USA. 101:1241–1246.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang L, Sowa Y, Sakai T and Pardee AB:
Activation of the p21WAF1/CIP1 promoter independent of
p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic
acid (SAHA) through the Sp1 sites. Oncogene. 19:5712–5719.
2000.
|
|
21
|
Rikiishi H: Autophagic and apoptotic
effects of HDAC inhibitors on cancer cells. J Biomed Biotechnol.
2011:8302602011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Emanuele S, Lauricella M and Tesoriere G:
Histone deacetylase inhibitors: apoptotic effects and clinical
implications (Review). Int J Oncol. 33:637–646. 2008.PubMed/NCBI
|