Introduction
Lung cancer is the most common cause of
cancer-related mortality in men and the second most common cause in
women, resulting in 1.61 million new cases and 1.38 million deaths
annually (1). In the treatment of
lung cancer, surgery, chemotherapy, radiotherapy, or their
combination are selected depending on the histological diagnosis,
the stage of the cancer, and the age of the patient. Despite the
significant progress that has been made in developing effective
treatment strategies and the substantial research efforts
undertaken, the prognosis for lung cancer patients remains poor,
and the development of more effective treatments is one of the most
important topics in the field of oncology. Lung cancer is
classified according to histological type: non-small cell lung
cancer (NSCLC) and small cell lung cancer (SCLC). Approximately
85–90% of lung cancers are NSCLC, which is further subdivided into
adenocarcinoma (AC), squamous cell carcinoma (SCC), and large cell
carcinoma (LCC) (2). While SCLC
usually responds better to chemotherapy and radiation, NSCLC is
relatively insensitive to both therapeutic modalities (3).
More than 60% of NSCLC cases overexpress the
epidermal growth factor receptor (EGFR), whereas no overexpression
is detected in SCLC (4). These
receptors play an important role in tumor cell survival, and
activated phosphorylated EGFR results in the phosphorylation of
downstream proteins that cause cell proliferation, invasion,
metastasis and inhibition of apoptosis. Therefore, EGFR tyrosine
kinase inhibitors (TKIs), gefitinib and erlotinib, are currently
used to treat NSCLC (5). Despite
initial responses, patients eventually experience disease
progression due to unknown mechanisms of acquired resistance.
Acquired resistance of NSCLC to TKIs is thought to be associated
with a second mutation in the EGFR kinase domain (6).
Phosphatidylinositol 3-kinase (PI3K) is a key
downstream component of the EGFR pathway and plays significant
roles in cell survival, proliferation, growth and cytoskeleton
rearrangement (7–10). The expression of PI3K has been
related to EGFR TKI resistance in preclinical models (11–13).
Engelman et al(14) found
that expression of the kinase domain mutant H1047R of PI3Kα in
mouse lung induced ACs in vivo. Mutant PIK3CA has been
implicated in the pathogenesis of several types of cancers,
including colon cancer, gliomas, gastric cancer, breast cancer,
endometrial cancer and lung cancer (15). These mutations can maintain
PI3K/Akt/mTOR signaling under conditions of growth factor
deprivation and thus transform cells. Therefore, PI3K is a novel
target for more effective treatment of NSCLC.
We designed and synthesized a new series of
imidazo[1,2-a]pyridine derivatives with the goal of developing a
novel structural class of potent PI3K inhibitors. Among these
compounds, HS-173 strongly inhibited PI3K activity (16). In the present study, we investigated
the anticancer activity of HS-173 in NSCLC cells through disruption
of the PI3K/Akt/mTOR pathway or inhibition of PI3K activity.
Materials and methods
Cell lines and reagents
The human NSCLC cell lines (A549, H1299 and
NCI-H596) were purchased from the Korean Cell Line Bank (KCLB,
Seoul, Republic of Korea). Cells were cultured in Roswell Park
Memorial Institute (RPMI)-1640 supplemented with 10% fetal bovine
serum (FBS) and 1% penicillin/streptomycin. Cell cultures were
maintained at 37ºC in a CO2 incubator. Ethyl
6-(5-(phenylsulfonamido)pyridin-3-yl)imidazo[1,2-a]pyridine-3-carboxylate
(HS-173) was synthesized according to our previous methods
(16). HS-173 was dissolved in
dimethyl sulfoxide (DMSO) at a concentration of 10 mM before use.
DMSO was added to the cells at 0.1% (v/v) as a solvent control.
Cell proliferation assay
The inhibitory effect of HS-173 on NSCLC cell lines
was assessed by measuring
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
dye absorbance. Briefly, cells were plated at a density of
3–5×103 cells/well in 96-well plates for 24 h. The
medium was then removed, and cells were treated with either DMSO as
a control or various concentrations (0.1–10 μM) of HS-173. After
the cells were incubated for 48 h, 100 μl of MTT solution (2 mg/ml)
was added to each well, and the plate was incubated for another 4 h
at 37ºC. The formed formazan crystals were dissolved in DMSO (100
μl/well) with constant shaking for 5 min. The absorbance of the
solution was then measured with a microplate reader at 540 nm. This
assay was conducted in triplicate.
Cell cycle analysis
A549 cells were plated in 100-mm culture dishes. The
next day, the cells were treated with various concentrations of
HS-173 for 12 h. Floating and adherent cells were collected and
fixed with ice-cold 70% ethanol at 4ºC overnight. After washing
with PBS, the cells were subsequently stained with 50 μg/ml
propidium iodide (PI) and 100 μg/ml RNase A for 1 h in the dark,
and then subjected to flow cytometric analysis to determine the
percentage of cells in specific phases of the cell cycle
(sub-G1, G0/G1, S and
G2/M). Flow cytometric analysis was performed using a
FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA) equipped with a 488-nm argon laser. Flow cytometric data
analysis was conducted using FlowJo software (Tree Star, Inc., San
Carlos, CA, USA). All the experiments were performed in
triplicate.
Western blotting
Total cellular proteins were extracted with lysis
buffer containing 1% Triton X-100, 1% Nonidet P-40, and the
following protease and phosphatase inhibitors: aprotinin (10
mg/ml), leupeptin (10 mg/ml; ICN Biomedicals, Asse-Relegem,
Belgium), phenylmethylsulfonyl fluoride (1.72 mM), NaF (100 mM),
NaVO3 (500 mM), and
Na4P2O7 (500 mg/ml; Sigma-Aldrich,
St. Louis, MO, USA). The proteins were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto nitrocellulose membranes. The blots were
immunostained with the appropriate primary antibodies followed by
secondary antibodies conjugated to horseradish peroxidase. Antibody
binding was detected with an enhanced chemiluminescence reagent
(Amersham Biosciences, Piscataway, NJ, USA). The primary antibodies
against p-Akt (Ser473), p-Akt (Thr308), Akt,
p-mTOR (Ser2448), mTOR, p-p70S6K1 (Thr389),
p70S6K1, PARP, cleaved caspase-3 and β-actin were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Bax was
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). The secondary antibodies were purchased from Amersham
Biosciences.
Immunofluorescence and confocal
analysis
A549 cells were plated on 18-mm cover glasses in
RPMI-1640 medium and incubated for 24 h so that ~70% confluence was
reached. The cells were then incubated in the presence or absence
of HS-173 (1 μM), washed twice with PBS, and fixed in acetic
acid:ethanol solution (1:2) for 5 min at room temperature. Cells
were blocked in 1.5% horse serum in PBS for 1 h at room
temperature, and then incubated overnight at 4ºC with primary
antibodies including p-Akt, p-mTOR, p-p70S6K, p-cdc2 (Cell
Signaling Technology, Inc.), cyclin B1, and p-cdc25C (Santa Cruz
Biotechnology, Inc.) in a humidified chamber. After washing twice
with PBS, the cells were incubated with fluorescein-labeled
secondary antibody (1:100; Dianova, Hamburg, Germany) for 1 h at
room temperature. The cells were also stained with
4,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei. The
slides were then washed twice with PBS and covered with DABCO
(Sigma-Aldrich) before being viewed with a confocal laser scanning
microscope (Olympus, Tokyo, Japan).
DNA fragmentation assay
Terminal deoxynucleotidyl transferase (Tdt)
dUTP-mediated nick-end labeling (TUNEL) was performed using the
ApopTag Plus Peroxidase In Situ Apoptosis Detection kit (Chemicon,
Temecula, CA, USA) according to the manufacturer's instructions.
Briefly, A549 cells were plated onto an 18-mm cover glass in
RPMI-1640 medium at ~70% confluence for 24 h at 37ºC. The cells
were then treated with HS-173 (1 μM) for 24 h. They were fixed in
acetic acid:ethanol solution (1:2) for 5 min at room temperature
and then washed with PBS. Cells were incubated with 20 μg/ml
proteinase K (Takara Shuzo Co. Ltd.) for 10–15 min at room
temperature and reacted with Tdt enzyme for 60 min at 37ºC. Cells
were then incubated with anti-digoxigenin conjugate at room
temperature for 30 min, incubated with diaminobenzidine solution
and counterstained with methyl green. Apoptotic cells were observed
in cross-section in randomly selected microscopic fields at a final
magnification of ×400.
In vitro measurement of mitochondrial
membrane potential (MMP)
MMP was assessed with a JC-1 Mitochondrial Membrane
Potential Assay kit (Cayman Chemical Co., Ann Arbor, MI, USA) using
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide (JC-1), which exhibits potential-dependent accumulation in
mitochondria. This dye forms J-aggregates under high MMP, causing a
shift in fluorescence from green to red. A549 cells were plated on
18-mm cover glasses in RPMI-1640 medium and incubated for 24 h.
When cells reached ~70% confluency, cells were then treated with
HS-173 (1 μM) for 6 h; 100 μl of JC-1 solution at a final
concentration of 12.5 μg/ml was then added to each well and the
plate was incubated for another 15 min at 37ºC. After washing twice
with PBS, the cells were also stained with DAPI to visualize the
cell nuclei. The slides were then washed twice with PBS and covered
with DABCO before viewing with a confocal laser scanning microscope
(Olympus, Tokyo, Japan). Data were represented by the level of the
red:green ratio.
Analysis of cytochrome c
localization
A549 cells were plated on 18-mm cover glasses in
RPMI-1640 medium. When cells reached ~70% confluency, they were
treated with HS-173 (1 μM) for 6 h. To label the mitochondria,
cells were incubated with 100 nM mitochondrion-specific dye
(MitoTracker® Red FM; Molecular Probes Inc., Eugene, OR,
USA) for 45 min at 37ºC prior to fixation. After washing twice with
PBS, cells were fixed in acetic acid:ethanol solution (1:2) for 5
min at room temperature. Cells were incubated overnight at 4ºC with
cytochrome c antibody (1:20; Santa Cruz Biotechnology,
Inc.). After washing twice with PBS, the cells were incubated with
mouse fluorescein-labeled secondary antibody (1:50; Dianova). The
cells were also stained with DAPI to visualize the nuclei. The
slides were then washed twice with PBS and covered with DABCO
before viewing with a confocal laser scanning microscope.
Statistical analysis
All data were analyzed using GraphPad Prism
(GraphPad software). Data are expressed as the means ± SD, and
P-values ≤0.05 were considered to indicate a statistically
significant result.
Results
HS-173 inhibits cell proliferation
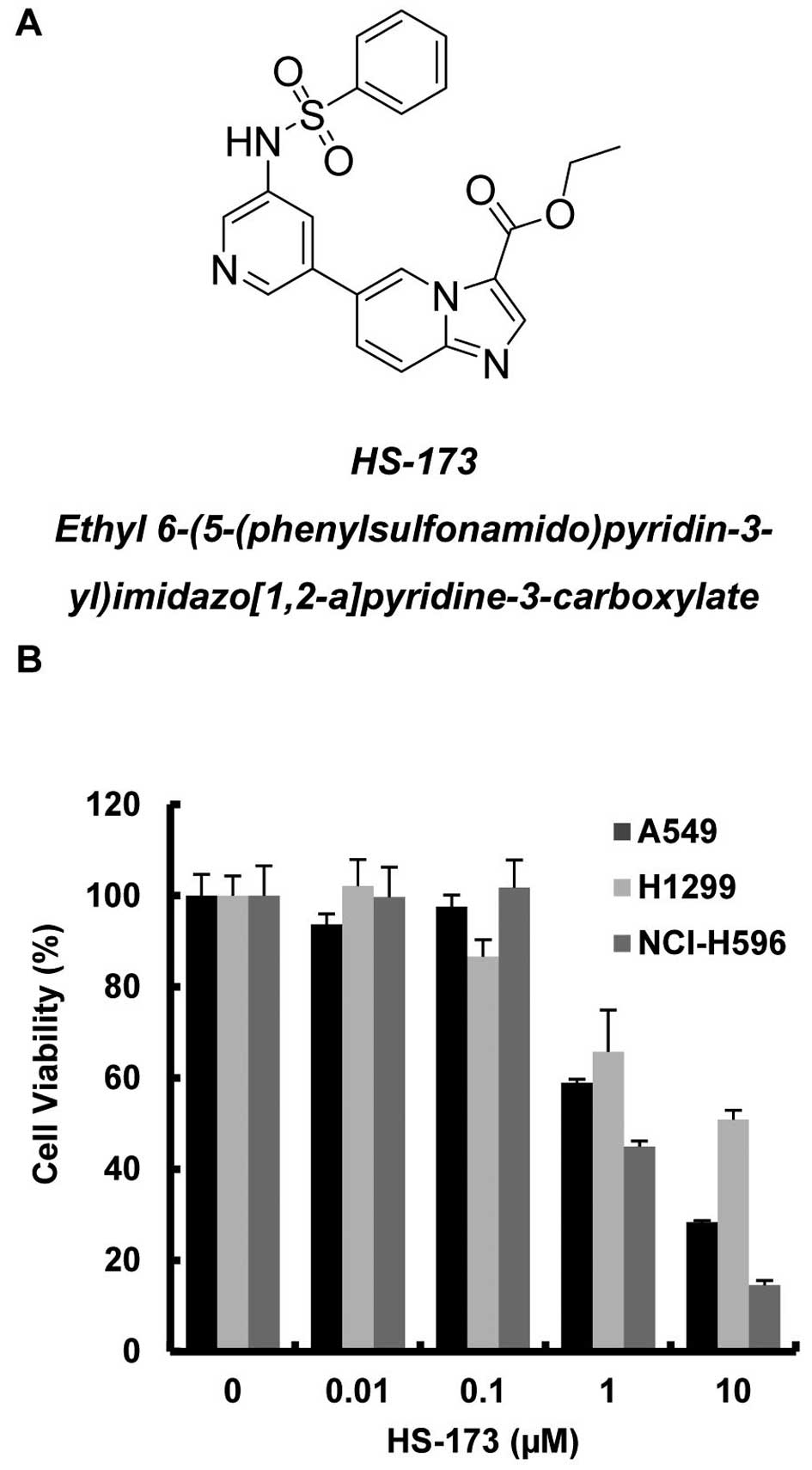
In our previous study, HS-173 demonstrated a high
binding affinity for the ATP-binding site of PI3Kα (Fig. 1A) and antitumor activity against
breast cancer through inhibition of the PI3K/Akt/mTOR pathway
(17). This pathway is closely
associated with the development and progression of NSCLC as well as
breast cancer (18). To assess the
effect of HS-173 on NSCLC proliferation, 3 NSCLC cell lines, A549,
H1299 and NCI-H596, were exposed to various concentrations of
HS-173 for 48 h and then analyzed by MTT assay. As shown in
Fig. 1B, treatment with HS-173
resulted in a dose-dependent reduction in viable cancer cells. The
IC50 of HS-173 was 1, 10 and 0.95 μM in A549, H1299 and
NCI-H596 cells, respectively.
HS-173 inhibits the PI3K-Akt-mTOR
pathway
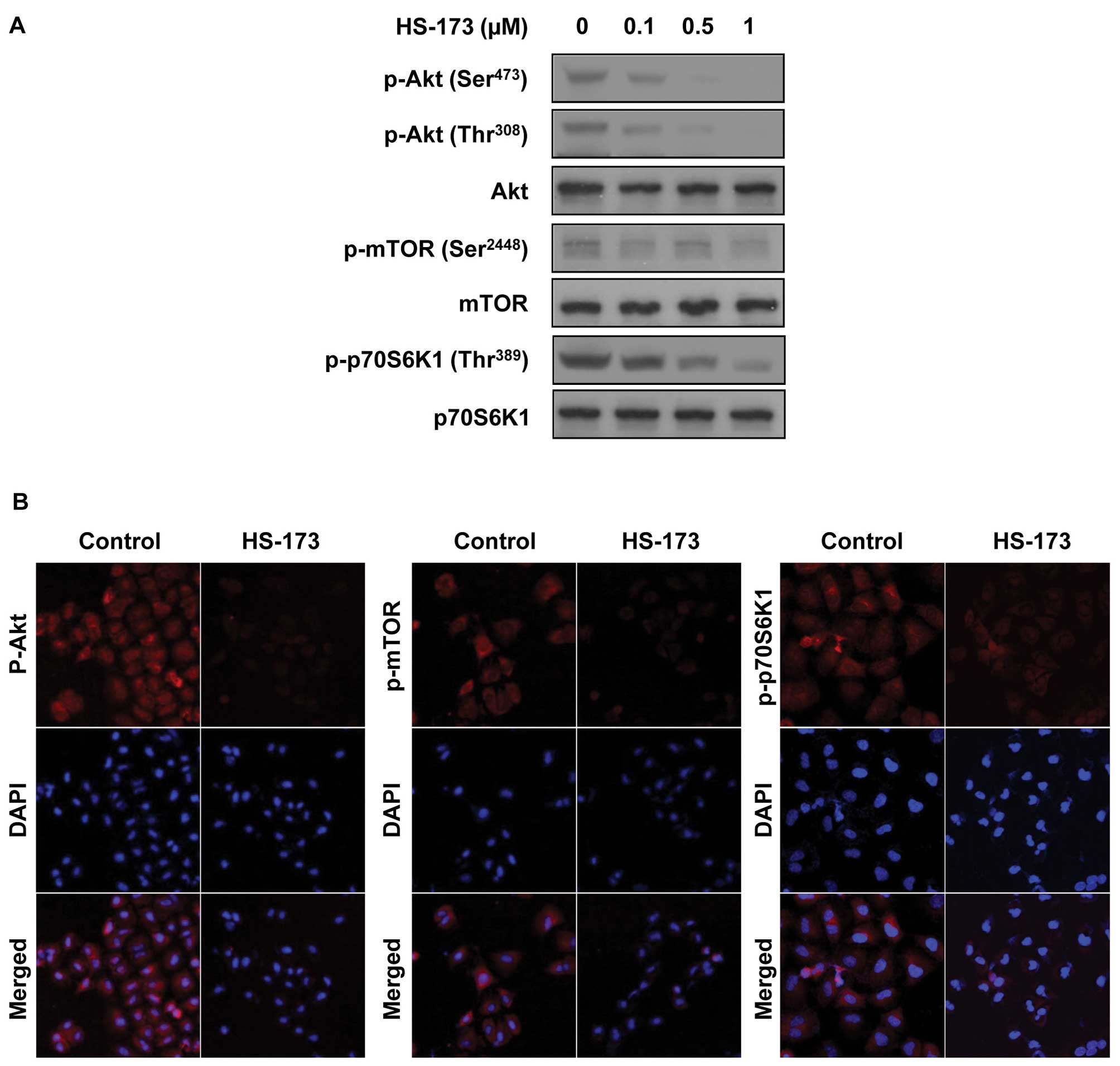
To assess the effect of HS-173 on intracellular
signaling in NSCLC, A549 cells were exposed to various
concentrations of HS-173 in vitro for 2 h, lysed and
analyzed by western blotting. As shown in Fig. 2A, the phosphorylation of Akt, mTOR
and p70S6K1 was effectively suppressed, indicating complete
suppression of the PI3K pathway. Similar to the results of the
western analysis, confocal microscopy data showed that HS-173 also
strongly suppressed phosphorylation of Akt, mTOR and p70S6K1
(Fig. 2B).
HS-173 causes cell cycle arrest
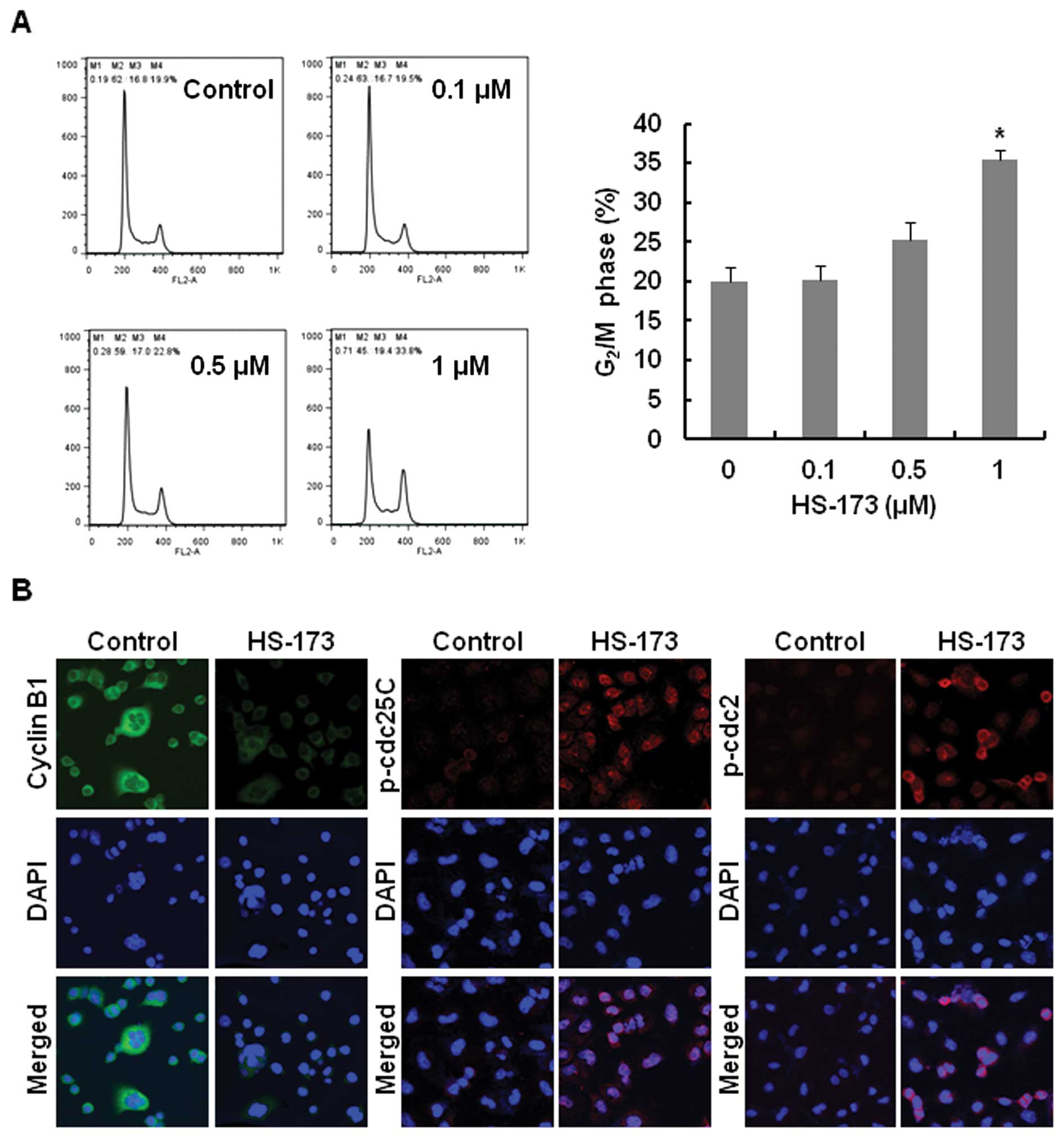
To assess the effect of HS-173 on the cell cycle,
A549 cells were exposed to various concentrations of HS-173 for 12
h, then fixed with ice-ethanol and analyzed using flow cytometry.
As shown in Fig. 3A, HS-173 induced
cell cycle arrest at the G2/M phase in a dose-dependent
manner. To further clarify the mechanism of HS-173-induced cell
cycle arrest at the G2/M phase, we investigated the
expression of proteins related to the G2/M checkpoint
using fluorescence confocal microscopy. HS-173 triggered a decrease
in expression of cyclin B1, as well as an increase in
phosphorylation of cdc2 and cdc25C (Fig. 3B).
HS-173 induces apoptosis
To determine whether the effects of HS-173,
including inhibition of the PI3K/Akt/mTOR pathway and cell cycle
arrest, are due to the induction of apoptosis, we analyzed the
ability of HS-173 to induce apoptosis of A549 lung cancer cells.
First, apoptosis by HS-173 was confirmed by DNA fragmentation assay
(Fig. 4A). As a result, treatment
with 1 μM HS-173 for 24 h increased TUNEL-positive cells. In
addition, to study the effect of HS-173 on MMP, the drug-treated
cells were exposed to the fluorescent cationic dye JC-1. As shown
in Fig. 4B, treatment with HS-173
induced the loss of MMP 5-fold when compared to that of the
controls. This change in MMP can trigger the release of
mitochondrial cytochrome c into the cytosol, a hallmark of
intrinsic pathway-mediated apoptosis. Therefore, we investigated
the release of cytochrome c by HS-173 in A549 lung cancer
cells. As shown in Fig. 4C, we
observed that treatment of HS-173 synergistically increased
cytochrome c release along with a concomitant decrease in
the-co-localization of cytochrome c to the mitochondria. We
also examined the expression of proteins related to apoptosis by
western blotting. As expected, HS-173 caused increased expression
of cleaved caspase-3, PARP and Bax in a dose-dependent manner
(Fig. 4D). Overall, these data
suggest that HS-173 causes apoptosis by directly affecting
mitochondria and activating caspases.
Discussion
Since more than 60% of NSCLC cases express EGFR, the
treatment modalities for NSCLC have involved the development of
EGFR TKIs. Two EGFR TKIs, gefitinib and erlotinib, have been
approved by the FDA for the treatment of locally advanced or
metastatic NSCLC that has failed at least one prior chemotherapy
regimen. However, they are only effective in cancer patients with
mutated and overactive EGFR. It has been reported that patients
with lung cancer usually develop drug-resistance by unknown
mechanisms (19). Therefore,
current treatments of lung cancer are not satisfactory, with a mean
survival of <1 year for advanced lung cancer patients,
regardless of treatment regimen (20).
EGFR plays a pivotal role in lung cancer by
activating several oncogenic signaling pathways. Among these, the
PI3K/Akt/mTOR pathway has been intensely investigated for its
pivotal role in regulating cell proliferation, survival and
metabolism. Therefore, inhibition of EGFR tyrosine kinase by
gefitinib leads to the suppression of the PI3K pathway. Recently,
several inhibitors targeting the PI3K pathway have been developed
and are being evaluated in preclinical studies and early clinical
trials. These include pan-PI3K and isoform-specific PI3K
inhibitors, dual PI3K-mTOR inhibitors, mTOR catalytic site
inhibitors and AKT inhibitors (21–24).
In the development of potent PI3K inhibitors, we screened numerous
compounds and finally developed a novel compound, HS-173 (17). HS-173 demonstrated antitumor
activity by inhibiting the PI3K pathway in liver and breast cancer
(16). In the present study, we
investigated the anticancer effect of HS-173 and its mechanism of
action in NSCLC cell lines.
Notably, although the PI3K pathway is activated in
NSCLC cell lines, Akt activity is selectively reduced in response
to gefitinib in NSCLC cell lines whose growth is inhibited by
gefitinib, while its activity is not reduced in cells resistant to
gefitinib (12). Our study showed
that HS-173 inhibited the growth of NSCLC cell lines which are
resistant to gefitinib as well as the PI3K pathway. This supports
the concept that the inhibition of the PI3K pathway can overcome
the resistance of gefitinib in NSCLC.
In the present study, the inhibition of the
PI3K/Akt/mTOR pathway by HS-173 led to cell cycle arrest during the
G2/M phase. In most mammalian cells, mitosis is
triggered by activation of the cyclin-dependent kinase cdc2.
Activation of this kinase is a multistep process that starts with
the binding of cyclin B1 (25).
Phosphorylated and inhibited cdc2 is dephosphorylated by cdc25C
phosphatase, which is also phosphorylated and inhibited by Chk2
(26). It has been reported that
suppression of AKT enhances the activation of Chk2, resulting in
increased inactive phosphorylation of cdc25C and cdc2 (27). In order to investigate whether the
blockade of G2/M phase transition induced by HS-173 was
due to the modulation of these regulatory proteins, we evaluated
the expression levels of cyclin B1, p-cdc25C and p-cdc2.
Phosphorylation of cdc2 at the Tyr15 residue is involved in the
arrest of dividing cells at the G2/M phase transition
(28). In the present study,
treatment with HS-173 decreased the level of cyclin B1 and
increased p-cdc25C and p-cdc2. The G2/M checkpoint is an
important cell cycle checkpoint in eukaryotic organisms ranging
from yeast to mammals (29). DNA
damage and inhibition of the PI3K pathway by anticancer drugs,
cytotoxic methylating agents and radiation can promote cell cycle
arrest at the G2 phase (30). In addition, inhibition of protein
synthesis during the G2 phase prevents the cell from
undergoing mitosis. PI3K signaling has been reported to contribute
to protein synthesis through the PI3K/Akt/mTOR pathway (31). Therefore, DNA damage and inhibition
of protein synthesis via the inhibition of the PI3K/Akt/mTOR
pathway by HS-173 may lead to G2/M arrest.
The PI3K/Akt/mTOR pathway is involved in cell
survival. Disruption of MMP induces the release of cytochrome
c from mitochondria and is associated with the activation of
caspase-3. In addition, MMP can be regulated by Bcl-2, which is
known to play a role in maintaining MMP by binding to mitochondria.
In contrast, Bax, a pro-apoptotic Bcl-2 family member, translocates
to the mitochondria and perturbs MMP (32). In the present study, HS-173
disrupted the MMP of A549 cells, and increased the release of
cytochrome c, the level of cleaved capase-3 and Bax. These
data suggest that apoptosis induced by HS-173 may be mediated
through activation of the intrinsic apoptotic pathway.
Our present study demonstrated that disruption of
the PI3K/Akt/mROR pathway or inhibition of PI3K activity by the
specific inhibitor, HS-173, significantly sensitized NSCLC cells to
apoptosis induced by chemotherapeutics in vitro. Our
findings suggest that HS-173 may be used to treat NSCLC cases
resistant or sensitive to EGFR TKIs.
Acknowledgements
This study was supported by the Korean Health
Technology R&D Project (A120266 and A110862) and the National
Research Foundation of Korea (NRF) funded by the Ministry of
Education, Science and Technology (NRF-2012-0002988,
2012R1A2A2A01045602 and 2012-0003009) and Inha University
Grant.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Roggli VL, Vollmer RT, Greenberg SD,
McGavran MH, Spjut HJ and Yesner R: Lung cancer heterogeneity: a
blinded and randomized study of 100 consecutive cases. Hum Pathol.
16:569–579. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuwabara K, Matsuda S, Fushimi K, et al:
Differences in practice patterns and costs between small cell and
non-small cell lung cancer patients in Japan. Tohoku J Exp Med.
217:29–35. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gazdar AF: Epidermal growth factor
receptor inhibition in lung cancer: the evolving role of
individualized therapy. Cancer Metastasis Rev. 29:37–48. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shepherd FA, Rodrigues Pereira J, Ciuleanu
T, et al: Erlotinib in previously treated non-small-cell lung
cancer. N Engl J Med. 353:123–132. 2005.PubMed/NCBI
|
|
6
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vanhaesebroeck B, Guillermet-Guibert J,
Graupera M and Bilanges B: The emerging mechanisms of
isoform-specific PI3K signalling. Nat Rev Mol Cell Biol.
11:329–341. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Engelman JA: Targeting PI3K signalling in
cancer: opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Janmaat ML, Kruyt FA, Rodriguez JA and
Giaccone G: Response to epidermal growth factor receptor inhibitors
in non-small cell lung cancer cells: limited antiproliferative
effects and absence of apoptosis associated with persistent
activity of extracellular signal-regulated kinase or Akt kinase
pathways. Clin Cancer Res. 9:2316–2326. 2003.
|
|
12
|
Engelman JA, Jänne PA, Mermel C, et al:
ErbB-3 mediates phosphoinositide 3-kinase activity in
gefitinib-sensitive non-small cell lung cancer cell lines. Proc
Natl Acad Sci USA. 102:3788–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamasaki F, Johansen MJ, Zhang D, et al:
Acquired resistance to erlotinib in A-431 epidermoid cancer cells
requires down-regulation of MMAC1/PTEN and up-regulation of
phosphorylated Akt. Cancer Res. 67:5779–5788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Engelman JA, Chen L, Tan X, et al:
Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D
and PIK3CA H1047R murine lung cancers. Nat Med. 14:1351–1356. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Samuels Y, Wang Z, Bardelli A, et al: High
frequency of mutations of the PIK3CA gene in human cancers.
Science. 304:5542004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee H, Jung KH, Jeong Y, Hong S and Hong
SS: HS-173, a novel phosphatidylinositol 3-kinase (PI3K) inhibitor,
has anti-tumor activity through promoting apoptosis and inhibiting
angiogenesis. Cancer Lett. 328:152–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim O, Jeong Y, Lee H, Hong SS and Hong S:
Design and synthesis of imidazopyridine analogues as inhibitors of
phosphoinositide 3-kinase signaling and angiogenesis. J Med Chem.
54:2455–2466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alvarez M, Roman E, Santos ES and Raez LE:
New targets for non-small-cell lung cancer therapy. Expert Rev
Anticancer Ther. 7:1423–1437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pao W, Miller V, Zakowski M, et al: EGF
receptor gene mutations are common in lung cancers from ‘never
smokers’ and are associated with sensitivity of tumors to gefitinib
and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311. 2004.
|
|
20
|
Bast RC, Kufe DW, Pollock RE, et al:
Cancer of the lung. Holland-Frei Cancer Medicine. 5th edition. BC
Decker Publishing Inc; Hamilton, ON: 2000
|
|
21
|
Yang L, Dan HC, Sun M, et al: Akt/protein
kinase B signaling inhibitor-2, a selective small molecule
inhibitor of Akt signaling with antitumor activity in cancer cells
overexpressing Akt. Cancer Res. 64:4394–4399. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Workman P: Inhibiting the phosphoinositide
3-kinase pathway for cancer treatment. Biochem Soc Trans.
32:393–396. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schultz RM, Merriman RL, Andis SL, et al:
In vitro and in vivo antitumor activity of the
phosphatidylinositol-3-kinase inhibitor, wortmannin. Anticancer
Res. 15:1135–1139. 1995.PubMed/NCBI
|
|
24
|
Rowinsky EK: Targeting the molecular
target of rapamycin (mTOR). Curr Opin Oncol. 16:564–575. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lindqvist A, Rodriguez-Bravo V and Medema
RH: The decision to enter mitosis: feedback and redundancy in the
mitotic entry network. J Cell Biol. 185:193–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tyagi A, Singh RP, Agarwal C, Siriwardana
S, Sclafani RA and Agarwal R: Resveratrol causes Cdc2-tyr15
phosphorylation via ATM/ATR-Chk1/2-Cdc25C pathway as a central
mechanism for S phase arrest in human ovarian carcinoma Ovcar-3
cells. Carcinogenesis. 26:1978–1987. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuo PL, Hsu YL and Cho CY: Plumbagin
induces G2-M arrest and autophagy by inhibiting the AKT/mammalian
target of rapamycin pathway in breast cancer cells. Mol Cancer
Ther. 5:3209–3221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
O'Farrell PH: Triggering the
all-or-nothing switch into mitosis. Trends Cell Biol. 11:512–519.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cuddihy AR and O'Connell MJ: Cell-cycle
responses to DNA damage in G2. Int Rev Cytol. 222:99–140. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hirose Y, Katayama M, Mirzoeva OK, Berger
MS and Pieper RO: Akt activation suppresses Chk2-mediated,
methylating agent-induced G2 arrest and protects from
temozolomide-induced mitotic catastrophe and cellular senescence.
Cancer Res. 65:4861–4869. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang X and Proud CG: The mTOR pathway in
the control of protein synthesis. Physiology (Bethesda).
21:362–369. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ashe PC and Berry MD: Apoptotic signaling
cascades. Prog Neuropsychopharmacol Biol Psychiatry. 27:199–214.
2003. View Article : Google Scholar
|