Introduction
Transforming growth factor-β (TGF-β) is a
pleiotropic cytokine that has been implicated in wound healing,
angiogenesis and regulation of immune systems. In the field of
tumor biology, TGF-β can act as both a promoter and a suppressor of
tumor progression. In a xenograft model of breast cancer,
endogenous TGF-β reduced the size of the putative cancer stem or
early progenitor cell population, and promoted differentiation of a
progenitor cell population to an intrinsically less proliferative
state (1). Another study in mice
demonstrated that the genetic deletion of TGF-β type II receptor in
mammary epithelium enhanced tumor formation and metastases induced
by polyomavirus middle T-antigen (2). These reports support the function of
TGF-β as a suppressor of tumor progression.
However, many reports underline the tumor-promoting
effect of TGF-β. As tumors grow, tumor cells alter or lose TGF-β
signaling through the genetic deletion of signaling components
including Smad 4 (3), becoming
refractory for TGF-β-mediated growth inhibition of tumors. In
breast and prostate cancer cells, stimulation of the
phosphoinositide-3 kinase/protein kinase B/nuclear factor-κB
pathway with TGF-β promotes the survival of tumor cells (4).
Regarding the contribution of TGF-β to the motility
of tumor cells, TGF-β plays critical roles in their
epithelial-mesenchymal transition (EMT), which is a key step in
metastases to distant organs. During the process of EMT, TGF-β
promotes the expression of transcription factors such as Snail,
Slug, Twist and Zeb1, leading to suppression of E-cadherin
expression in the tumor cells (5).
Given that E-cadherin is a primary epithelial cell-cell adhesion
molecule (6), downregulation of
E-cadherin promotes tumor cell migration and metastasis.
TGF-β also contributes to tumor progression by
suppression of antitumor immune responses. TGF-β dampens the
function of effector immune cell types including helper T cells,
cytotoxic T lymphocytes, natural killer cells and B cells, and
activates regulatory T (Treg) cells, which suppress antitumor
immune responses (7,8). These immunosuppressive functions of
TGF-β decrease the efficacy of cancer immunotherapy.
As TGF-β performs multiple functions in tumor
progression, targeting TGF-β is considered a promising strategy for
the development of anticancer therapy. However, systemic inhibition
of TGF-β functions may also lead to undesirable adverse events.
Transgenic mice expressing a soluble TGF-β type II receptor
exhibited an induced inflammatory response, including lymphocytic
infiltration of the lungs, kidneys and pancreas (9). Other reports demonstrated that a TGF-β
type I receptor inhibitor administered orally induced physeal
dysplasia in rats (10) and
administration of a high dose of a TGF-β-neutralizing antibody
induced epithelial hyperplasia of the tongue in a mouse model of
familial adenomatous polyposis (11). To avoid adverse events induced by a
TGF-β inhibitor, local but not systemic inhibition of TGF-β should
be considered. We previously reported that local inhibition of
TGF-β in tumor-draining lymph nodes contributes to the induction of
potent systemic antitumor immune responses with few adverse effects
(12). In the report, we confirmed
that local inhibition of TGF-β was of value for the development of
TGF-β-targeted strategies against cancers.
Based on the result that the main source of TGF-β in
a tumor-bearing host is tumor cells, we hypothesized that
inhibition of TGF-β release from tumor cells could provide an
avenue for influencing tumor progression via TGF-β. To investigate
this hypothesis, we applied tranilast as an inhibitor of TGF-β
release from tumor cells. Tranilast is an anti-allergic agent that
has been used clinically in Japan, and is reported to inhibit the
release of TGF-β from immune cells (13).
In the present study, we evaluated the effect of
tranilast as an inhibitor of TGF-β release from tumor cells, and
examined whether local inhibition of TGF-β release by tranilast
could suppress EMT-associated motility of tumor cells. Here, we
report that the reduction of TGF-β release from tumor cells by
tranilast suppresses their EMT-associated capacities for metastasis
and invasiveness, and contributes to a decrease in induction of
Treg cells.
Materials and methods
Cells and reagents
The mouse Lewis lung carcinoma cell line LLC1 was
provided by the Cell Resource Center for Biomedical Research,
Tohoku University, Sendai, Japan. The cells were grown in
Dulbecco's modified Eagle's medium (DMEM) (Nacalai Tesque, Inc.,
Kyoto, Japan) supplemented with 10% fetal calf serum, 100 U/ml
penicillin G, and 0.1 mg/ml streptomycin. Tranilast
(N-[3,4-dimethoxycinnamoyl]-anthranilic acid) was kindly provided
by Kissei Pharmaceutical Co., Ltd. (Matsumoto, Japan) and dissolved
in dimethyl sulfoxide.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from LLC1 cells using the
RNeasy Mini kit (Qiagen, Hilden, Germany) and was subjected to the
reverse transcription reaction using the Cloned AMV First-Strand
cDNA Synthesis kit (Life Technologies, Carlsbad, CA, USA). Each
cDNA was amplified with specific paired primers as follows: TGF-β
forward, 5′-CTCCCGTGG CTTCTAGTGC-3′ and reverse, 5′-GCCTTAGTTTGGACA
GGATCTG-3′; Snail forward, 5′-GGAAGCCCAACTATA GCGAGC-3′ and
reverse, 5′-CAGTTGAAGATCTTCCGC GAC-3′; Slug forward,
5′-CTCACCTCGGGAGCATAC AGC-3′ and reverse, 5′-TGAAGTGTCAGAGGAAGGC
GGG-3′; Twist forward, 5′-CGGGTCATGGCTAACGTG-3′ and reverse,
5′-CAGCTTGCCATCTTGGAGTC-3′; vimentin forward, 5′-CACCCTGCAGTC
ATTCAGACA-3′ and reverse, 5′-GATTCCACTTTCCGTTCAAGGT-3′; E-cadherin
forward, 5′-CAGGTCTCCTCATGGCTTTGC-3′ and reverse,
5′-CTTCCGAAAAGAAGGCTGTCC-3′; β-actin forward,
5′-GCCCAGAGCAAGAGAGGTAT-3′ and reverse, 5′-GGC
CATCTCCTGCTCGAAGT-3′.
Quantification of protein levels of
TGF-β1
LLC1 cells were cultured in medium containing
tranilast for 2 days, and the cells and the culture supernatant
were harvested. Cytoplasmic protein of LLC1 cells was extracted
using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). The levels of TGF-β1 in
the cytoplasmic protein of LLC1 and the culture supernatant were
measured using the TGF-β1 Quantikine ELISA kit (R&D Systems,
Inc., Minneapolis, MN, USA), according to the manufacturer's
instructions.
Assays for cell proliferation and
motility
LLC1 cells (1×104 cells/well) were
cultured in 96-well plates with medium containing tranilast for 2
days, and CellTiter 96 AQueous One Solution Reagent (Promega,
Madison, WI, USA) was added to each well. After additional culture
for 4 h, the absorbance was measured at 492 nm.
To evaluate the invasive activity of LLC1, the cells
were cultured in medium containing tranilast for 2 days.
Subsequently, 4×104 cells/well were cultured for 22 h in
5% bovine serum albumin-DMEM containing tranilast in the upper
compartment of a Transwell chamber with a Matrigel-coated membrane
(pore size, 8 μm; BD Biosciences, San Jose, CA, USA) using 24-well
plates. After scraping cells for removal on the surface using
cotton swabs, the membrane was treated with fixation/staining
solution containing 0.5% crystal violet, 12% formaldehyde and 10%
ethanol as described elsewhere (14), and the invaded cells were counted
under a microscope.
To evaluated the adhesive activity of LLC1, the
cells were cultured in medium containing tranilast for 2 days.
Subsequently, 4×104 cells/well were cultured in
fibronectin-coated 96-well plates (BD Biosciences) for 2 h. After
vigorous vortexing, the nonadherent cells were discarded and
treated with a fixation/staining solution. After washing in water
and then drying, 1% SDS solution was added, and the extract
absorbance was measured at 570 nm.
Detection of Treg cells
Spleen cells from normal mice were cultured in
medium in which LLC1 cells had been cultured with tranilast. Two
days later, the cells were stained with anti-CD4, anti-CD25 and
anti-FoxP3 antibodies (eBioscience, Inc., San Diego, CA, USA)
according to the manufacturer's instructions, and analyzed on a
FACScan flow cytometer (Becton Dickinson Immunocytometry System,
Mountain View, CA, USA). Data are presented as dot blots produced
using CellQuest software (Becton Dickinson Immunocytometry
System).
Statistical analysis
The statistical significance of differential
findings between groups was determined by the Student's t-test.
P-values <0.05 were considered to indicate a statistically
significant result. Representative results from three independent
experiments with similar results are shown.
Results
Inhibition of TGF-β1 release from LLC1
cells by tranilast
In the present study, we applied tranilast to
inhibit the release of TGF-β from tumor cells. Tranilast has been
reported to inhibit the release of TGF-β from immune cells;
however, its effects on LLC1 cells remain undefined. First, we
examined whether the release of TGF-β from LLC1 cells would be
inhibited by tranilast in vitro. LLC1 cells were cultured in
medium containing tranilast for 2 days, and subsequently the level
of TGF-β1 in the culture medium was measured by ELISA. The level of
TGF-β1 was significantly decreased by incorporation of tranilast
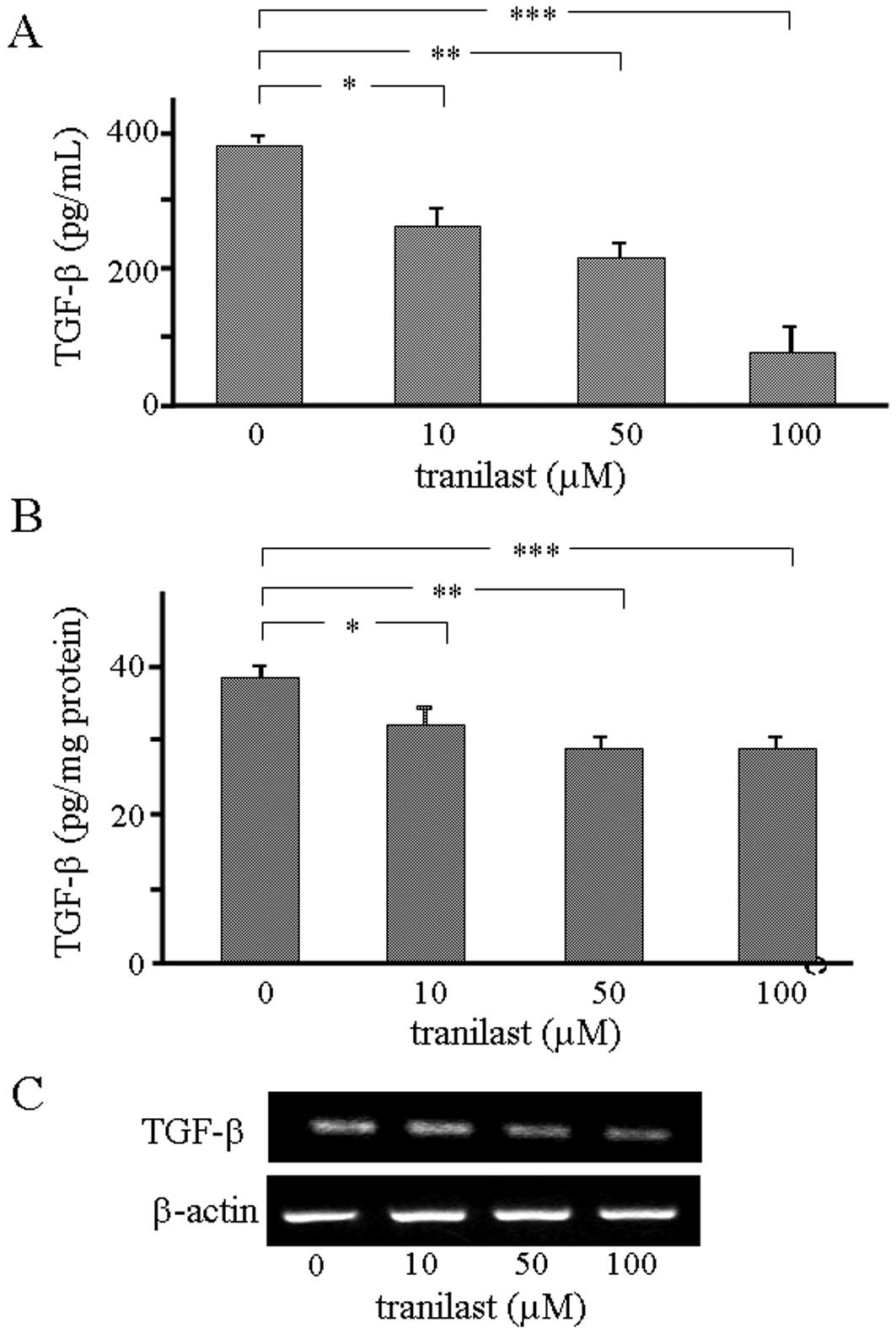
into the culture medium in a dose-dependent manner (Fig. 1A). We confirmed that tranilast
inhibited the release of TGF-β1 from LLC1 cells, even following the
incorporation of a low dose into the culture medium.
Next, we examined whether tranilast affects the
expression of TGF-β1 in LLC1 cells. The cells were cultured in
medium containing tranilast for 5 days, and the level of
cytoplasmic TGF-β1 was measured by ELISA. The data revealed that
the level of cytoplasmic TGF-β1 was significantly decreased by
incorporation of tranilast (Fig.
1B). The level of TGF-β1 mRNA in the cells was also evaluated,
demonstrating that it was downregulated by incorporation of
tranilast into the culture medium at a dosage of 100 μM (Fig. 1C). These data indicate that the
effects of tranilast on TGF-β expression in LLC1 cells are variable
and dose-dependent.
Inhibition of TGF-β1 release has a
minimal effect on the proliferative activity of LLC1 cells
It has been reported that tranilast suppresses the
proliferative activity of tumor cells while having few cytotoxic or
apoptotic effects. We examined the effect of tranilast on the
proliferative activity of LLC1 cells. The data showed that the
proliferation of LLC1 cells was not suppressed markedly by
tranilast at doses ranging from 0 to 100 μM (Fig. 2). At this setting, tranilast
inhibits the release of TGF-β1 from LLC1 cells, without affecting
the proliferative activity of the cells. The findings suggest that
a large dose of tranilast would be required to suppress the
proliferation of LLC1 cells, due to the aggressive nature of their
progression. In this study, we focused on the suppressive effect of
tranilast on TGF-β1 release but not on the proliferation of LLC1
cells, and conducted subsequent experiments using doses of
tranilast ranging from 0 to 100 μM.
EMT-associated motility of LLC1 cells is
influenced by inhibition of TGF-β1 release
We explored the impact of the inhibition of TGF-β1
release from LLC1 cells on cell motility. In order to evaluate the
influence on the invasive activity by inhibition of TGF-β1 release,
we cultured LLC1 cells in a Transwell chamber in medium containing
tranilast. Two days later, the cells that had invaded through the
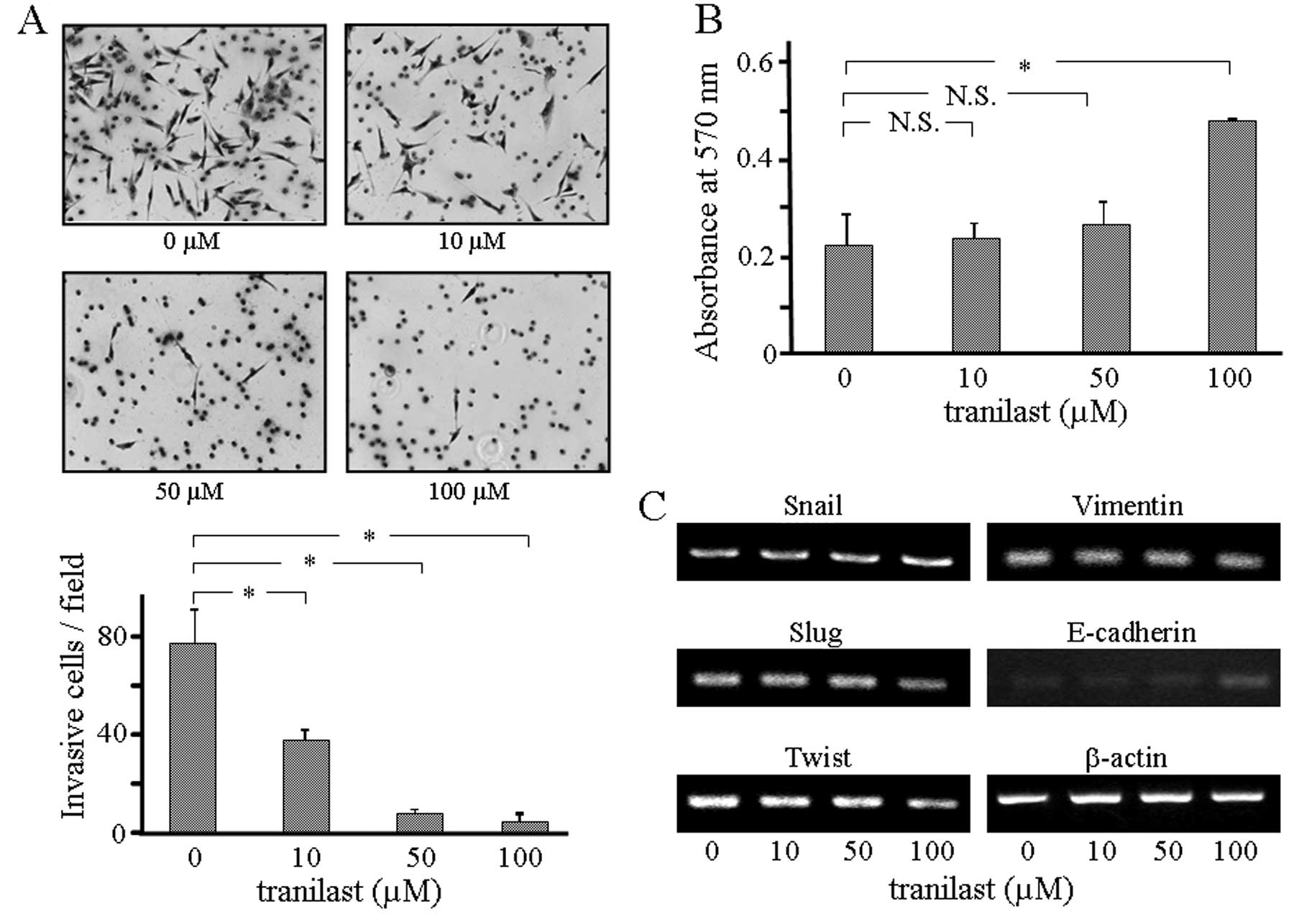
Transwell membrane were counted. As shown in Fig. 3A, invasion of LLC1 cells was
significantly decreased by the incorporation of tranilast into the
culture medium, demonstrating that the invasive activity of LLC1
cells was suppressed by inhibition of TGF-β1 release from the cells
themselves.
Next, we examined whether inhibition of TGF-β1
release from the cells affects the adhesive activity of LLC1 cells.
For this purpose, LLC1 cells were cultured in fibronectin-coated
plates in medium containing tranilast for two days. The number of
adherent LLC1 cells was significantly increased by incorporation of
tranilast at the dose of 100 μM into the culture medium (Fig. 3B), while 10 μM of tranilast in the
culture medium did not affect the adherent LLC1 cell count. These
findings demonstrate that the adhesive activity of LLC1 cells was
decreased by the powerful inhibition of TGF-β1 release from the
cells themselves.
On the basis of our findings concerning LLC1 cell
motility, we aimed to ascertain whether the expression of
EMT-associated markers in LLC1 cells is influenced by inhibition of
TGF-β1 release from LLC1 cells, and we examined mRNA expression of
EMT-associated markers in LLC1 cells cultured in medium containing
tranilast. As shown in Fig. 3C,
among the EMT-associated transcriptional factors examined,
expression of both Slug and Twist was suppressed and that of
E-cadherin was upregulated by incorporation of tranilast. These
results suggest that increased expression of EMT-associated markers
in LLC1 cells can be achieved by powerful inhibition of TGF-β1
release from the cells themselves.
Reduction in Treg cell induction by
inhibition of TGF-β1 release from LLC1 cells
From the viewpoint of tumor immunology, in the tumor
microenvironment, TGF-β plays a crucial role in inducing Treg
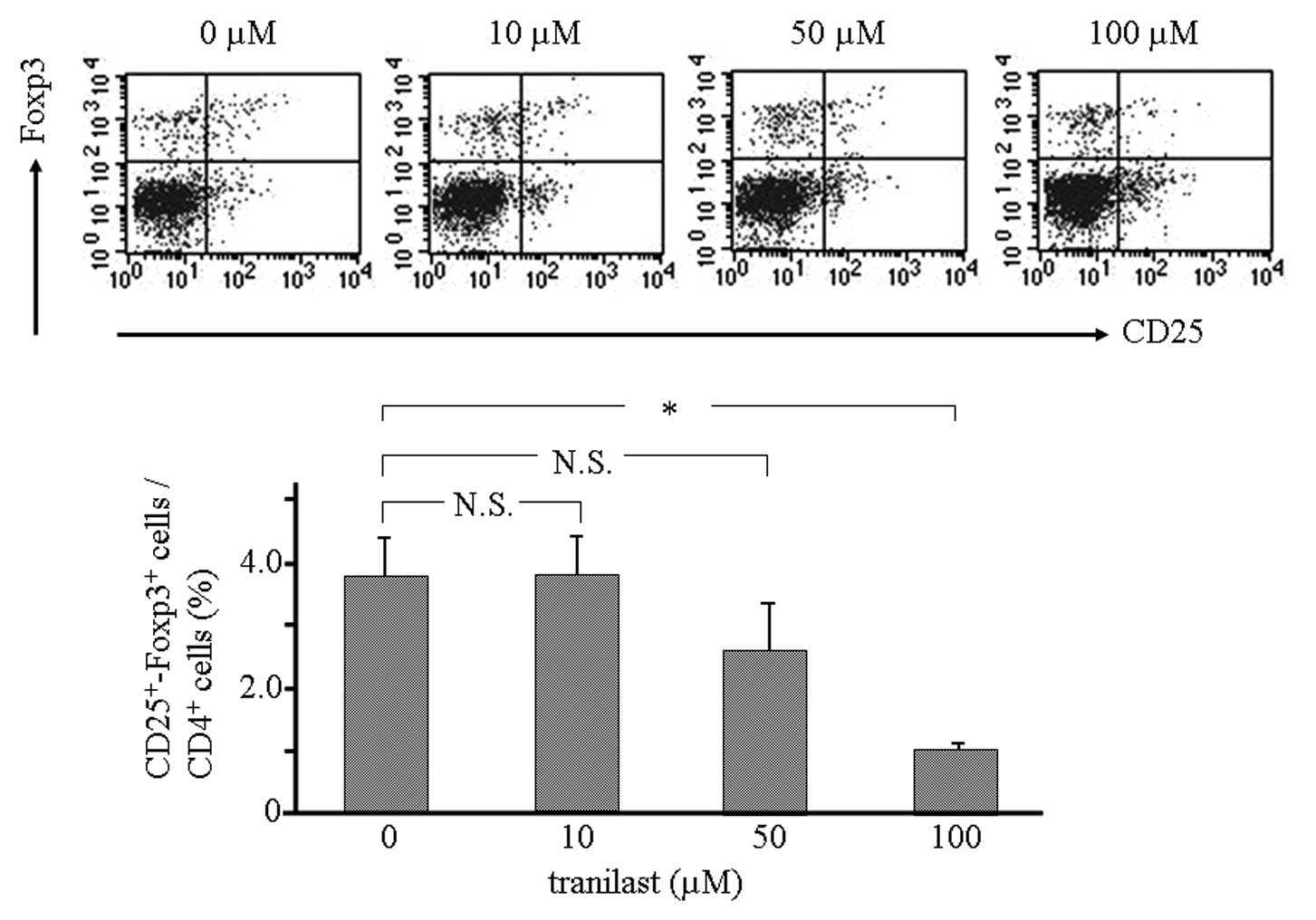
cells. We examined whether inhibition of TGF-β1 release from LLC1
cells suppresses the induction of Treg cells. Spleen cells from
normal mice were cultured in medium in which LLC1 cells had been
cultured with tranilast. Flow cytometry showed that the population
of CD4+CD25+Foxp3+ cells was
significantly decreased by incorporation of tranilast (Fig. 4). These results suggest that local
inhibition of TGF-β1 release from LLC1 contributes to a decrease in
the induction of Treg cells.
Discussion
In the present study, we focused on the release of
TGF-β from tumor cells, and examined the effects of the inhibition
of TGF-β release on their aggressive nature and on the
proliferation of Treg cells. Our data demonstrated that inhibition
of TGF-β1 release from tumor cells by tranilast suppresses their
EMT-associated capacity for motility and contributes to a decrease
in induction of Treg cells in vitro.
Given that tranilast functions to inhibit the
release of TGF-β from immune cells (13), we applied tranilast as a TGF-β
release inhibitor in this study. In clinics in Japan, tranilast is
administered to patient as an anti-allergic agent at the dose of
300 mg/day. At this dose of tranilast, Cmax in peripheral blood may
be estimated to be ~100 μM in humans (15). On the basis of the data, we
conducted an in vitro study in which the culture medium
contained tranilast at concentrating ranging from 0 to 100 μM. In
this setting, the proliferation of LLC1 cells was not affected by
0–50 μM of tranilast in the culture medium, and even a dose of 100
μM did not effectively suppress the proliferation of LLC1
cells.
These findings indicate that in this setting
tranilast did not influence the cell cycle or apoptosis of LLC1
cells, nor did it confer cytotoxic activity against them. Tranilast
has been reported to suppress the proliferation of other types of
tumor cells such as pancreatic, prostate, breast, uterine leiomyoma
and malignant glioma cells (16–20).
The mechanisms underlying this suppressive effect are that
tranilast arrests cell cycle progression through induction of the
cyclin-dependent kinase (CDK) inhibitor. It was also reported that
tranilast induced expression of p21 and p53 as well as G1 arrest,
and decreased CDK2 activity in uterine leiomyoma cells (19) and in human malignant glioma cells
(20). Izumi et al(17) reported that tranilast induced G1
arrest in prostate cancer cells via induction of p53, p21 and p27.
Chakrabarti et al(21)
reported that the proliferative response to tranilast differs
between cell lines. In contrast with our study, higher doses of
tranilast, 300 μM or more, were applied in these previous studies.
However, data from experiments employing a high dose of tranilast
could not translate to cancer treatments in humans. A clinical
study on the effect of tranilast on re-stenosis of the coronary
artery after percutaneous transluminal coronary angioplasty
demonstrated that the re-stenosis rate was significantly decreased
by tranilast at a dose of 600 mg/day. However, adverse events,
including liver dysfunction and gastro-intestinal distress, also
increased ~2-fold as compared with the control cohort (22). Based on this report, although high
doses of tranilast can suppress the proliferation of tumor cells,
they also cause intolerable adverse events in the human clinical
setting.
As described above, tranilast is known to inhibit
the release of TGF-β from immune cells (13). It also has been reported that
tranilast inhibits the release of TGF-β1 and -β2 from human
malignant glioma cells (20). In
the present study, we examined the effect of tranilast on the
release of TGF-β from mouse lung cancer cells, and confirmed that
the level of TGF-β1 in the culture medium was decreased in a
dose-dependent manner by incorporation of tranilast. Even a low
dose (10 μM) of tranilast in the culture medium inhibited the
release of TGF-β1 from tumor cells effectively. Furthermore, we
examined whether tranilast inhibits the expression of TGF-β1 at the
mRNA and cytoplasmic protein levels. Our results demonstrated that
the expression of TGF-β1 mRNA in LLC1 cells was not affected at
lower doses of tranilast, and higher doses of tranilast were
required to suppress the expression of TGF-β1 mRNA. Cytoplasmic
levels of TGF-β1 protein in LLC1 cells were decreased by low doses
of tranilast. These data demonstrate that tranilast affects tumor
cells, causing inhibition of TGF-β1 release and suppression of the
mRNA or protein level of TGF-β1 expression, depending on the dose
incorporated into the culture medium.
Apart from tranilast, other drugs have also been
reported to possess an anti-TGF-β effect. Angiotensin-converting
enzyme (ACE) inhibitors and antagonists of the angiotensin II type
1 receptor (AT1), two commonly used anti-hypertensive drugs, also
have an effect on inhibition of TGF-β. TGF-β production of cardiac
and renal cells was found to be decreased by the AT1 antagonist
losartan through the downregulation of thrombospondin-1, which
activates latent TGF-β (23). Both
ACE and AT1 inhibitors have been reported to have a suppressive
effect on tumor cells; however, captopril, one of the ACE
inhibitors, had no significant effect on expression of TGF-β mRNA
and protein in mouse renal cancer cells (24).
Although various cell types in tumor tissue release
TGF-β, the primary source of TGF-β may be tumor cells. The
aggressiveness of tumor cells is determined by the levels of TGF-β,
which are released by tumor cells themselves. High levels of TGF-β
in tumor cells modulate their metastatic motility. With this in
mind, we focused on downregulation of TGF-β levels in the tumor
environment, and examined the effect of inhibition of TGF-β release
on the motility of tumor cells in vitro. In the setting of
our study, incorporation of tranilast into the culture medium was
able to create an environment where the level of TGF-β1 was
decreased through inhibition of TGF-β1 release. Invasive activity
of LLC1 cells was effectively suppressed by incorporation of even a
low dose (10 μM) of tranilast. However, a higher dose of tranilast
(100 μM) was required to affect the adhesive activity of LLC1
cells. Adhesive activity of tumor cells is associated with the
expression of E-cadherin, which is an epithelial cell-cell adhesion
molecule and a major component of epithelial adherens junction.
TGF-β, as well as other growth factors including hepatocyte growth
factor, insulin-like growth factor and fibroblast growth factor, is
known to provoke the loss of E-cadherin function, leading to
disruption of adhesive activity (25). However, the invasive activity of
tumor cells is associated with integrins, which constitute a family
of transmembrane receptors that bind to the extracellular matrix
extracellularly and to the cytoskeleton intracellularly. TGF-β
induces the de novo expression of several types of
integrins, such as α5β1, αvβ3, αvβ5 and αvβ6, in epithelial cells
(26). These integrins enhance the
migratory and invasive activity of tumor cells. Although both
invasive and adhesive activities are regulated by TGF-β, our data
indicate that the invasive activity of LLC1 cells is more
susceptible than its adhesive activity to the inhibition of the
release of TGF-β. In turn, adhesive activity may be more dependent
on TGF-β than invasive activity.
The modulation of cell motility, called EMT, is the
primary step when tumor cells metastasize to distant organs. The
process of EMT, in which tumor cells modulate their invasive and
adhesive activities, has been reported to be regulated by TGF-β
(6). The alteration of the cell
motility observed in this study suggests that EMT of tumor cells
may be suppressed by inhibition of TGF-β release. When we examined
the expression of transcription factors associated with EMT,
expression of both Slug and Twist was suppressed, and that of
E-cadherin was upregulated. These findings were observed at a
higher (100 μM) but not at a lower (10 μM) dose of tranilast. These
results can be explained by several factors. First, transcriptional
changes in EMT of tumor cells may depend on the level of TGF-β in
the environment. Reduction in the TGF-β1 level by a low dose (10
μM) of tranilast may be insufficient to induce changes of
EMT-associated transcription factors, and a much greater reduction
in the TGF-β level may be needed to do so. Second, on the basis of
the result that a higher dose (100 μM) of tranilast suppresses the
mRNA expression of TGF-β1 in LLC1 cells, downregulation of TGF-β
mRNA may be required to induce the alteration of EMT-associated
transcription factors. It has been reported that EMT-associated
transcription factors, such as Snail, Slug, Twist and Zeb1, that
are upregulated by TGF-β signaling, can affect the expression of
E-cadherin directly or indirectly (5). Both Snail and Slug are direct
inhibitors of E-cadherin, while Twist suppresses the expression of
E-cadherin indirectly (6). Given
that EMT-associated factors are under complex regulation, the
effect of inhibition of TGF-β release from tumor cells on their
expression may depend on the degree of decrease in the level of
TGF-β.
In the tumor microenvironment, the level of TGF-β
can induce Treg cells, contributing to suppression of antitumor
immune responses in tumor-bearing hosts. Controlling the activity
of Treg cells should be one of the strategies employed in cancer
therapy. If the level of TGF-β in the tumor tissue can be
attenuated, the population of Treg cells induced will be decreased,
contributing to improvement of antitumor immune responses. In this
study, we examined whether inhibition of TGF-β release from tumor
cells by tranilast suppresses the induction of Treg cells. Spleen
cells from normal mice were cultured in medium in which LLC1 cells
had been cultured with tranilast, where the level of TGF-β1 had
been reduced. In this setting, the population of Treg cells
(CD4+CD25+Foxp3+ cells) induced
was significantly decreased, depending on the level of TGF-β1 in
the culture medium. These data are consistent with previous reports
that addition of a neutralizing antibody against TGF-β into the
culture supernatant of prostate tumor cells prevented
CD4+CD25− T cells from expressing Foxp3
(27). Another report showed that
the number of Treg cells infiltrating the tumor was decreased by
TGF-β siRNA in mice with colon cancer (28). Attenuation of TGF-β levels in tumor
tissue can decrease the distribution of Treg cells, and inhibition
of TGF-β release from tumor cells contributes to creating an
environment where induction of Treg cells is suppressed.
In conclusion, inhibition of TGF-β release from
tumor cells by tranilast suppresses the EMT-associated metastatic
and invasive capacities of tumor cells and contributes to a
decrease in induction of Treg cells in vitro. These findings
indicate that inhibition of TGF-β release from tumor cells has the
potential to suppress metastasis of tumor cells and improve
systemic antitumor immune responses, providing a new rationale for
the development of TGF-β-targeted molecular immunotherapy against
cancer.
References
|
1
|
Tang B, Yoo N, Vu M, Mamura M, Nam JS,
Ooshima A, Du Z, Desprez PY, Anver MR, Michalowska AM, Shih J,
Parks WT and Wakefield LM: Transforming growth factor-β can
suppress tumorigenesis through effects on the putative cancer stem
or early progenitor cell and committed progeny in a breast cancer
xenograft model. Cancer Res. 67:8643–8652. 2007.
|
|
2
|
Forrester E, Chytil A, Bierie B, Aakre M,
Gorska AE, Sharif-Afshar AR, Muller WJ and Moses HL: Effect of
conditional knockout of the type II TGF-β receptor gene in mammary
epithelia on mammary gland development and polyomavirus middle T
antigen induced tumor formation and metastasis. Cancer Res.
65:2296–2302. 2005.
|
|
3
|
Jakowlew SB: Transforming growth factor-β
in cancer and metastasis. Cancer Metastasis Rev. 25:435–457.
2006.
|
|
4
|
Galliher AJ, Neil JR and Schiemann WP:
Role of transforming growth factor-β in cancer progression. Future
Oncol. 2:743–763. 2006.
|
|
5
|
Talbot LJ, Bhattacharya SD and Kuo PC:
Epithelial-mesenchymal transition, the tumor microenvironment, and
metastatic behavior of epithelial malignancies. Int J Biochem Mol
Biol. 3:117–136. 2012.PubMed/NCBI
|
|
6
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Letterio JJ and Roberts AB: Regulation of
immune responses by TGF-β. Annu Rev Immunol. 16:137–161. 1998.
|
|
8
|
Wrzesinski SH, Wan YY and Flavell RA:
Transforming growth factor-beta and the immune response:
implications for anticancer therapy. Clin Cancer Res. 13:5262–5270.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang YA, Dukhanina O, Tang B, Mamura M,
Letterio JJ, MacGregor J, Patel SC, Khozin S, Liu ZY, Green J,
Anver MR, Merlino G and Wakefield LM: Lifetime exposure to a
soluble TGF-β antagonist protects mice against metastasis without
adverse side effects. J Clin Invest. 109:1607–1615. 2002.PubMed/NCBI
|
|
10
|
Frazier K, Thomas R, Scicchitano M,
Mirabile R, Boyce R, Zimmerman D, Grygielko E, Nold J, DeGouville
AC, Huet S, Laping N and Gellibert F: Inhibition of ALK5 signaling
induces physeal dysplasia in rats. Toxicol Pathol. 35:284–295.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Prud'homme GJ: Pathobiology of
transforming growth factor β in cancer, fibrosis and immunologic
disease, and therapeutic considerations. Lab Invest. 87:1077–1091.
2007.
|
|
12
|
Fujita T, Teramoto K, Ozaki Y, Hanaoka J,
Tezuka N, Itoh Y, Asai T, Fujino S, Kontani K and Ogasawara K:
Inhibition of transforming growth factor-β-mediated
immunosuppression in tumor-draining lymph nodes augments antitumor
responses by various immunologic cell types. Cancer Res.
69:5142–5150. 2009.
|
|
13
|
Suzawa H, Kikuchi S, Ichikawa K and Koda
A: Inhibitory action of tranilast, an anti-allergic drug, on the
release of cytokines and PGE2 from human
monocytes-macrophages. Jpn J Pharmacol. 60:85–90. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanaka K, Honda M, Kuramochi T and Morioka
S: Prominent inhibitory effects of tranilast on migration and
proliferation and collagen synthesis by vascular smooth muscle
cells. Atherosclerosis. 107:179–185. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hiroi M, Onda M, Uchida E and Aimoto T:
Anti-tumor effect of N-[3,4-dimethoxycinnamoyl]-anthranilic acid
(tranilast) on experimental pancreatic cancer. J Nippon Med Sch.
69:224–234. 2002.
|
|
17
|
Izumi K, Mizokami A, Li YQ, Narimoto K,
Sugimoto K, Kadono Y, Kitagawa Y, Konaka H, Koh E, Keller ET and
Namiki M: Tranilast inhibits hormone refractory prostate cancer
cell proliferation and suppresses transforming growth factor
β1-associated osteoblastic changes. Prostate. 69:1222–1234.
2009.PubMed/NCBI
|
|
18
|
Subramaniam V, Chakrabarti R, Prud'homme
GJ and Jothy S: Tranilast inhibits cell proliferation and migration
and promotes apoptosis in murine breast cancer. Anticancer Drugs.
21:351–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shime H, Kariya M, Orii A, Momma C,
Kanamori T, Fukuhara K, Kusakari T, Tsuruta Y, Takakura K, Nikaido
T and Fujii S: Tranilast inhibits the proliferation of uterine
leiomyoma cells in vitro through G1 arrest associated with the
induction of p21waf1 and p53. J Clin Endocrinol Metab.
87:5610–5617. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Platten M, Wild-Bode C, Wick W, Leitlein
J, Dichgans J and Weller M: N-[3,4-dimethoxycinnamoyl]-anthranilic
acid (tranilast) inhibits transforming growth factor-β relesase and
reduces migration and invasiveness of human malignant glioma cells.
Int J Cancer. 93:53–61. 2001.
|
|
21
|
Chakrabarti R, Subramaniam V, Abdalla S,
Jothy S and Prud'homme GJ: Tranilast inhibits the growth and
metastasis of mammary carcinoma. Anticancer Drugs. 20:334–345.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kato K, Tamai H, Hayakawa H, Yamaguchi T,
Kanmatsuse K, Haze K, Aizawa T, Suzuki S, Takase S, Suzuki T,
Nishikawa H, Nakanishi S, Kato O and Nakashima M: Clinical
evaluation of tranilast on restenosis after percutaneous
transluminal coronary angioplasty (PTCA) - a double blind
placebo-controlled comparative study. J Clin Therapy Med. 12:65–85.
1996.
|
|
23
|
Zhou Y, Poczatek MH, Berecek KH and
Murphy-Ullrich JE: Thrombospondin 1 mediates angiotensin II
induction of TGF-β activation by cardiac and renal cells under both
high and low glucose conditions. Biochem Biophys Res Commun.
339:633–641. 2006.PubMed/NCBI
|
|
24
|
Miyajima A, Asano T and Hayakawa M:
Captopril restores transforming growth factor-β type II receptor
and sensitivity to transforming growth factor-β in murine renal
cell cancer cells. J Urol. 165:616–620. 2001.PubMed/NCBI
|
|
25
|
Yilmaz M and Christofori G: Mechanisms of
motility in metastasizing cells. Mol Cancer Res. 8:629–642. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Margadant C and Sonnenberg A:
Integrin-TGF-β crosstalk in fibrosis, cancer and wound healing.
EMBO Rep. 11:97–105. 2010.
|
|
27
|
Liu VC, Wong LY, Jang T, Shah AH, Park I,
Yang X, Zhang Q, Lonning S, Teicher BA and Lee C: Tumor evasion of
the immune system by converting CD4+CD25− T
cells into CD4+CD25+ T regulatory cells: role
of tumor-derived TGF-β. J Immunol. 178:2883–2892. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Conroy H, Galvin KC, Higgins SC and Mills
KH: Gene silencing of TGF-β1 enhances antitumor immunity induced
with a dendritic cell vaccine by reducing tumor-associated
regulatory T cells. Cancer Immunol Immunother. 61:425–431.
2012.
|