Introduction
A number of studies have shown high levels of the
cyclooxygenase-2 (COX-2) protein in solid tumors (1–6). In
breast cancer, COX-2 expression is a predictor of poor disease-free
and overall survival (4–9). In a retrospective study of 1,576
invasive breast tumors, Ristimaki et al(4) found that elevated COX-2 expression was
associated with a lower survival rate in patients with estrogen
receptor α (ERα)-positive breast tumors. Women whose invasive
breast tumors were ERα-positive but had low levels of COX-2 had an
86% chance of 5-year distant disease-free survival, whereas women
whose tumors were ERα-positive but had high levels of COX-2 had a
76% chance of 5-year distant disease-free survival (4).
Breast cancer patients who have ERα-positive breast
tumors are typically treated with selective estrogen receptor
modulators (SERMs). We previously demonstrated that transfection of
the COX-2 gene into the tamoxifen-sensitive, ERα-positive MCF-7
breast cancer cell line (MCF-7/COX-2) reduced the sensitivity of
MCF-7 cells to tamoxifen by ~5-fold (10). These data suggest that breast cancer
patients who have ERα-positive and COX-2-overexpressing tumors may
not benefit from tamoxifen as much as patients who have low levels
of COX-2 in their ERα-positive breast tumors.
Elevated levels of COX-2 have also been associated
with lymph node and distant metastasis (11,12).
COX-2 has been shown to increase breast cancer cell invasion in
vitro(13–15) and in vivo(16–18).
We and others demonstrated that MCF-7/COX-2 cells are ~3-fold more
invasive than parental MCF-7 cells (13,14).
The decrease in tamoxifen sensitivity and increase in invasive
activity by COX-2 may contribute to the reduced survival rate noted
in patients with ERα-positive, COX-2-overexpressing breast
tumors.
COX-2 utilizes its product prostaglandin
E2 (PGE2) to stimulate protein kinase C (PKC)
activity. We demonstrated that activation of PKC reduced the
anti-proliferative effects of tamoxifen (10), and increased the invasiveness of
MCF-7 cells across a Matrigel basement membrane (13). Although high levels of COX-2 have
been associated with activation of the mitogen activated protein
kinase (MAPK) family (19–21) and the Akt kinase (22), it is not known whether these kinases
mediate COX-2-induced tamoxifen resistance and invasive activity.
In the present study, we report that COX-2 utilizes PKC to increase
the activity of Jun N-terminal kinases (JNKs) to mediate invasion,
but not tamoxifen resistance, in MCF-7 breast cancer cells.
Materials and methods
Cell lines and culture conditions
The MCF-7 human breast cancer cell line was obtained
from the American Type Cell Culture (ATCC, Manassas, VA, USA).
MCF-7/COX-2 cells were generated by stably transfecting plasmids
encoding the COX-2 gene into ERα-positive MCF-7 cells (10,13).
MCF-7/COX-2 cells were obtained from individual colonies, and
continuously cultured in DMEM/F-12 medium containing 5% FBS and 500
μg/ml G418. We selected clone 12, which expressed higher levels of
COX-2 than the parental MCF-7 cells (10,13)
for our studies.
Chemical reagents
Tamoxifen citrate, Gö6976, SP600125 and PD98059 were
purchased from EMD Chemicals (San Diego, CA, USA). Stock solutions
(10 mM) of tamoxifen, Gö6976, SP600125 and PD98059 were prepared in
DMSO and stored at −20°C. All reagents were diluted in culture
medium to the indicated final concentration. Matrigel was purchased
from BD Biosciences (Bedford, MA, USA). Antibodies specific for
phosphorylated ERK (T202/Y204),
phosphorylated p38MAPK (T183/Y185),
phosphorylated Akt (S473), phosphorylated c-Jun
N-terminal kinase (JNK) (T183/Y185), ERK,
p38MAPK, Akt, JNK, and c-Jun were obtained from Cell Signaling
Technology (Danvers, MA, USA). Antibodies specific for β-actin and
Histone H3 were purchased from Sigma-Aldrich Chemical Co. (St.
Louis, MO, USA). Anti-mouse and anti-rabbit secondary antibodies
conjugated with horseradish peroxidase were purchased from Amersham
Life Sciences (Cell Signaling Technology).
Western blotting
Western blotting was performed as previously
described (10,23). MCF-7 parental and MCF-7/COX-2 cells
were plated at 4×105 cells/well in 6-well plates in
DMEM/F-12 medium containing 5% FBS. Two days later, cells were
harvested and cell pellets were lysed. The protein concentration
was determined using the DC protein assay (Bio-Rad Laboratories,
Hercules, CA, USA). Samples were electrophoresed on 12%
polyacrylamide gels (Bio-Rad Laboratories), then transferred to
nitrocellulose membranes (Bio-Rad Laboratories) for western blot
analysis. Membranes were blocked in Tris-buffered saline (20 mM
Tris pH 7.6, 150 mM NaCl) with 0.1% Tween-20 containing 5% non-fat
dry milk (Bio-Rad Laboratories) at room temperature for 30 min.
After washing, the membranes were incubated with primary antibodies
(1:1,000 dilution) overnight at 4°C. The next day, membranes were
washed and incubated with anti-rabbit secondary antibody conjugated
with horseradish peroxidase (1:1,000 dilution) for 2 h at room
temperature. Proteins bands were detected via enhanced
chemiluminescence (Kirkegaard & Perry Laboratories,
Gaithersburg, MD, USA). Images were scanned by an AlphaImager
densitometer (Alpha Innotech Corp., San Leandro, CA, USA). β-actin
was used as a loading control. Membranes were incubated with
anti-β-actin antibody (1:10,000 dilution) for 30 min at room
temperature, washed, and anti-mouse secondary antibody (1:10,000
dilution) for another 30 min at room temperature.
To determine the effect of PKC inhibition on JNK
phosphorylation, MCF-7/COX-2 cells were plated at 4×105
cells/well in 6-well plates in DMEM/F-12 medium containing 5% FBS.
Two days later, cells were treated with the PKC inhibitor Gö6976
(0, 25 and 50 nM) for 8 h. Untreated and treated cells were
harvested and lysed. Western blot analysis was performed as
described above.
Extraction of nuclear proteins
Nuclear proteins were extracted as previously
described (23). MCF-7 and
MCF-7/COX-2 cells were plated at 4×105 cells/well in
6-well plates in DMEM/F-12 medium containing 5% FBS. Cells were
harvested, and cell pellets were lysed with 250 μl buffer (10 mM
HEPES, 10 mM KCl, 0.5% Nonidet P-40, pH 7.9) on ice for 15 min.
Nuclei were pelleted by centrifugation at 13,000 rpm for 1 min, and
nuclear proteins were extracted with 50 μl nuclear extraction
buffer (20 mM HEPES, 400 mM NaCl, pH 7.9). Nuclear protein
concentrations were determined using the Bio-Rad DC protein assay.
Nuclear proteins (50 μg) were electrophoresed on 12% polyacrylamide
gels (Bio-Rad Laboratories), transferred to nitrocellulose
membranes (Bio-Rad Laboratories), and western blot analysis for
c-Jun was performed (primary antibody was added at a 1:500
dilution). Histone H3 was used as a loading control. Membranes were
incubated with the anti-Histone H3 antibody (1:5,000 dilution) for
30 min at room temperature, washed, and with the anti-mouse
secondary antibody (1:5,000 dilution) for another 30 min at room
temperature.
Matrigel invasion assay
Matrigel invasion assay was preformed as previously
described (13,23,24) by
counting the number of cells that invaded through Transwell inserts
coated with the Matrigel artificial basement membrane. Six-well
plate Transwell inserts with 8-μm pore-size polycarbonate filters
(Thermo Fisher Scientific, Middleton, VA, USA) were coated with
Matrigel (0.7 mg/ml) and placed at room temperature for 40 min.
MCF-7/COX-2 cells (4×105 in 500 μl) were pretreated with
the JNK inhibitor SP600125 or the ERK inhibitor PD98059 (0, 5 or 10
μM) in DMEM/F-12 medium containing 5% FBS for 30 min before being
added to the Matrigel-coated Transwell inserts. Seventy-two hours
later, cells that invaded through the Matrigel onto the lower side
of the filter were fixed, stained with Hema-3 and photographed. The
invaded cells from each filter were counted in five fields under a
light microscope at magnification ×40. The invasiveness of
MCF-7/COX-2 cells was expressed as the mean number of cells that
had invaded to the lower side of the filter. The experiments were
performed in triplicate wells.
CellTiter 96 Aqueous non-radioactive
proliferation assay
The inhibitory effects of tamoxifen on MCF-7/COX-2
cells were studied as previously described (10). MCF-7/COX-2 cells were plated at
1,000 cells/well in 96-well plates in 0.1 ml of DMEM/F-12 medium
supplemented with 5% FBS. The next day, the medium was replaced
with DMEM/F-12 medium supplemented with 5% charcoal-stripped serum
(CSS). Twenty-four hours later, cells were pretreated with SP600125
(0, 5 and 10 μM) before being treated with various concentrations
of tamoxifen for 5 days. At the end of the incubation, cell
proliferation was determined by the Promega (Madison, WI, USA)
CellTiter 96 Aqueous non-radioactive proliferation (MTS) assay and
was expressed as the percentage of proliferating cells relative to
the untreated cells.
Results
COX-2 overexpression increases
phosphorylated JNK levels and nuclear c-Jun levels
High levels of COX-2 or PGE2, have been
associated with activation of MAPKs (19–21)
and Akt (22). We sought to
determine whether MAPK and/or Akt are utilized by COX-2 to induce
breast cancer cell invasion and tamoxifen resistance. First, we
determined whether COX-2 overexpression alters the phosphorylation
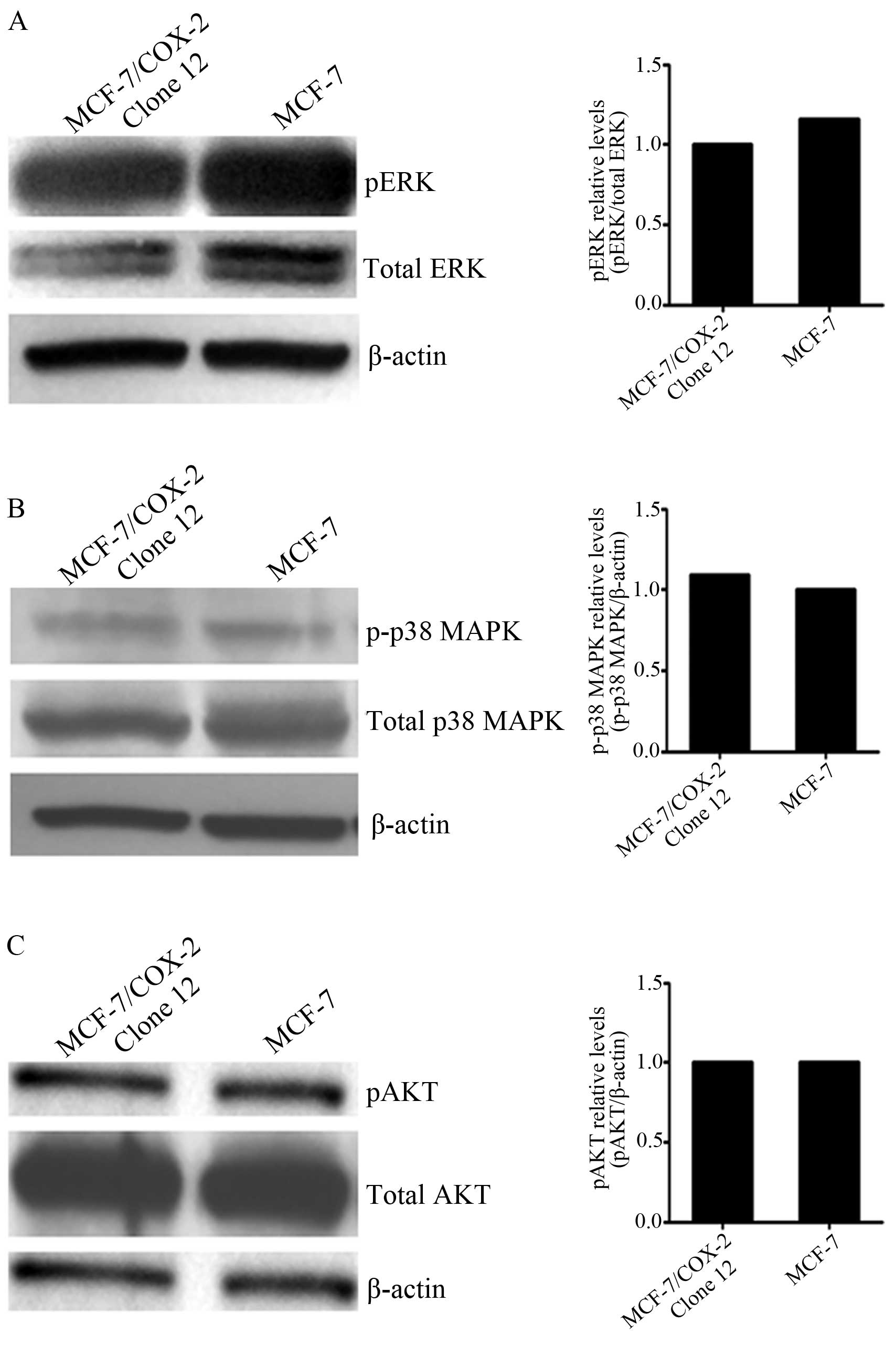
levels of the MAPK family or that of Akt. Phosphorylation levels of
ERK (Fig. 1A), p38 MAPK (Fig. 1B) and Akt (Fig. 1C) were very similar between the
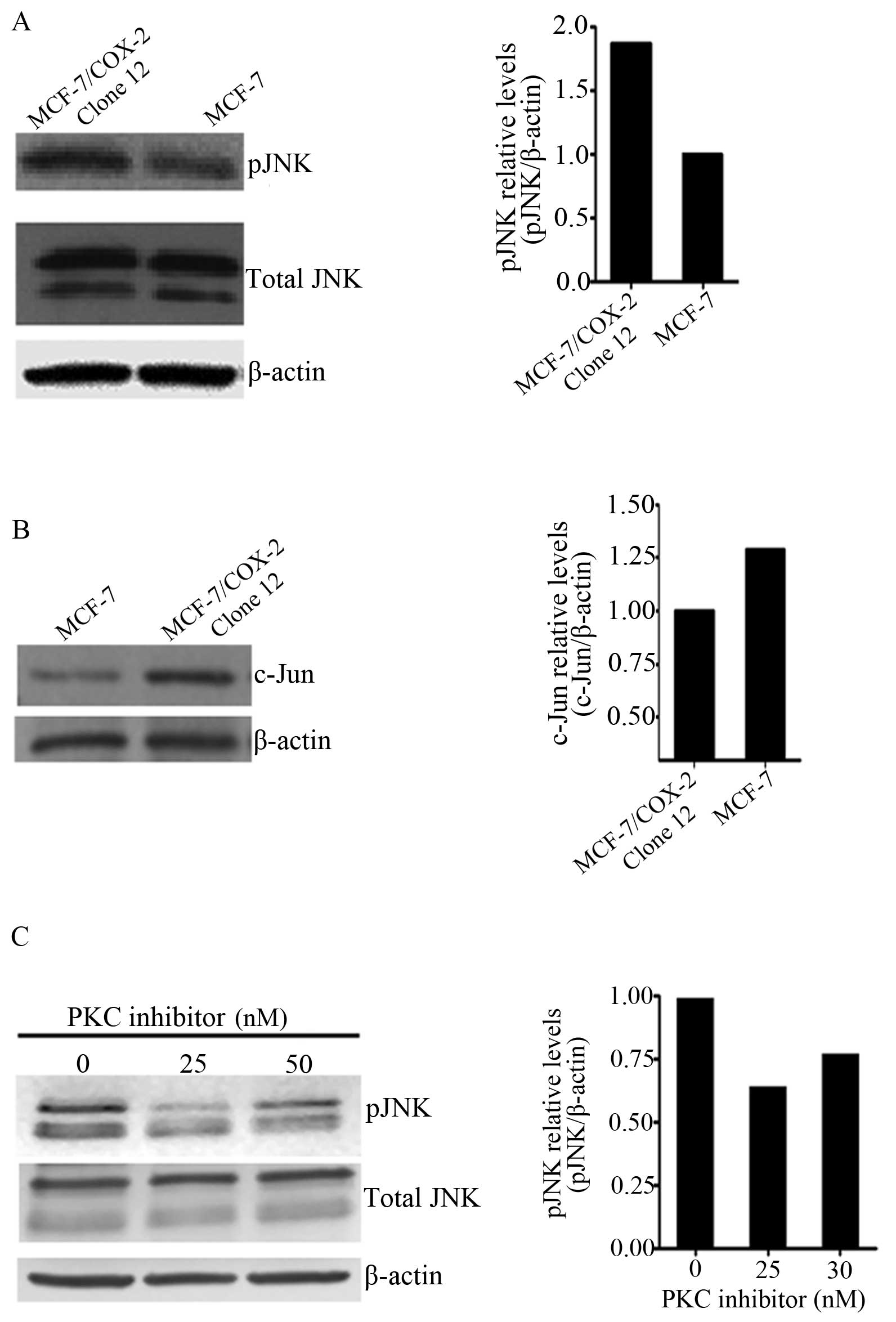
MCF-7/COX-2 and MCF-7 parental cells. However, higher JNK
phosphorylation levels were observed in MCF-7/COX-2 cells when
compared with levels in the MCF-7 cells (Fig. 2A). This corresponded to higher
nuclear c-Jun levels in MCF-7/COX-2 cells than levels in MCF-7
cells (Fig. 2B). Previously we
showed that PKC plays an essential role in mediating COX-2-induced
invasion and tamoxifen resistance in MCF-7 breast cancer cells
(10). To determine whether JNK is
downstream of PKC in the COX-2 pathway, we treated MCF-7/COX-2
cells with the PKC inhibitor Gö6976 and utilized western blotting
to analyze JNK phosphorylation levels. Inhibition of PKC resulted
in decreased JNK phosphorylation in the MCF-7/COX-2 cells (Fig. 2C). These data indicate that PKC
regulates COX-2-induced JNK activation.
Inhibition of JNKs decreases the
invasiveness of MCF-7/COX-2 cells
Since we observed that COX-2 stimulates JNK
phosphorylation, we hypothesize that COX-2 utilizes JNKs to induce
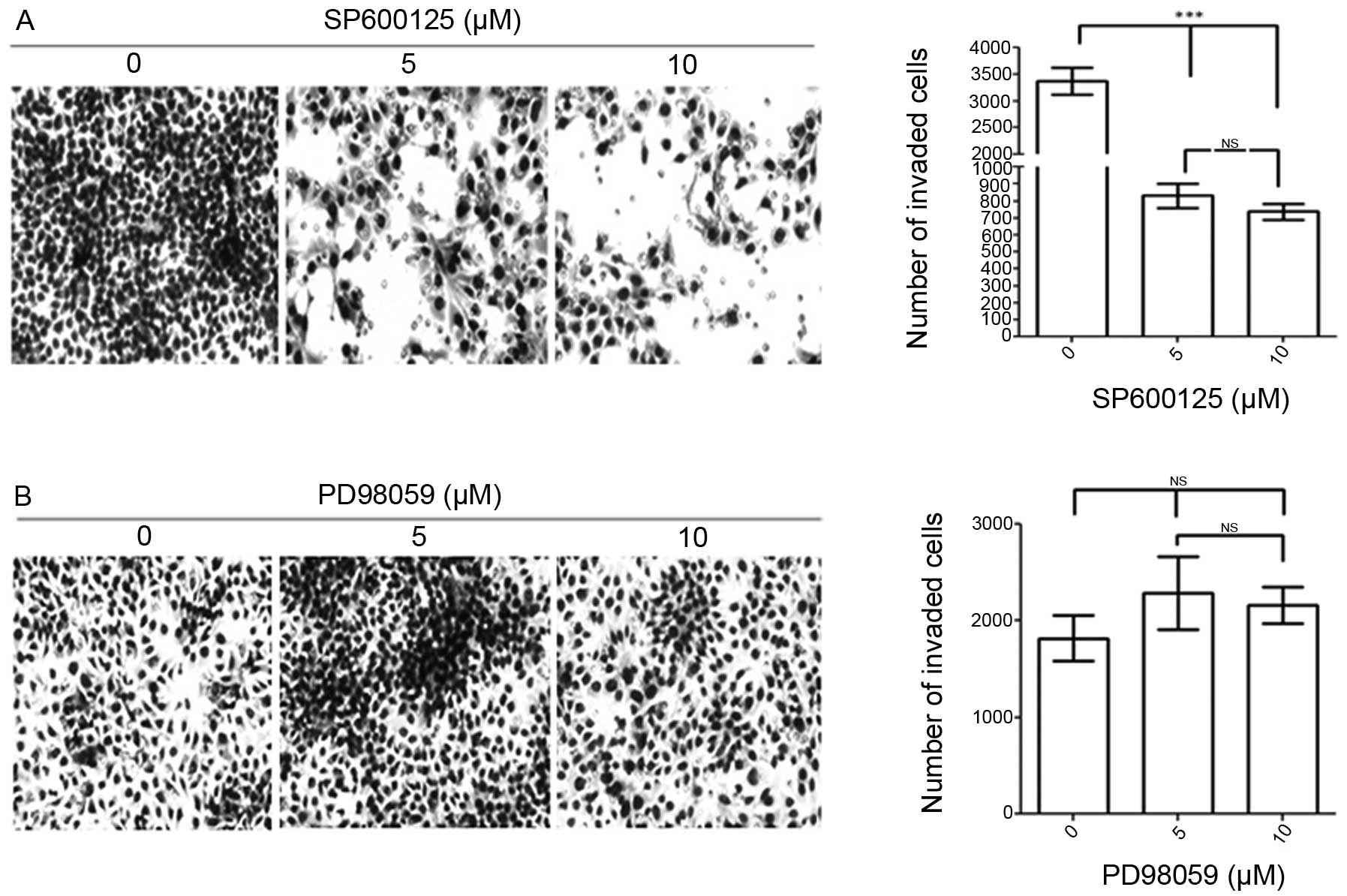
invasion. MCF-7/COX-2 breast cancer cells were pretreated with
SP600125, a chemical inhibitor against JNKs, to determine whether
JNK inhibition would decrease COX-2-mediated invasion. At 5 and 10
μM concentration, SP600125 decreased MCF-7/COX-2 invasive activity
by ~75 and 80%, respectively (Fig.
3A). On the other hand, since COX-2 did not increase ERK
phosphorylation, we did not expect ERK to mediate COX-2-induced
invasion. Indeed, when we treated MCF-7/COX-2 cells with PD98059, a
chemical inhibitor against ERK, PD98059 did not affect MCF-7/COX-2
invasive activity (Fig. 3B). These
data indicate that JNKs, not ERK, mediate COX-2-induced breast
cancer cell invasion.
Inhibition of JNKs does not affect
tamoxifen sensitivity in MCF-7/COX-2 cells
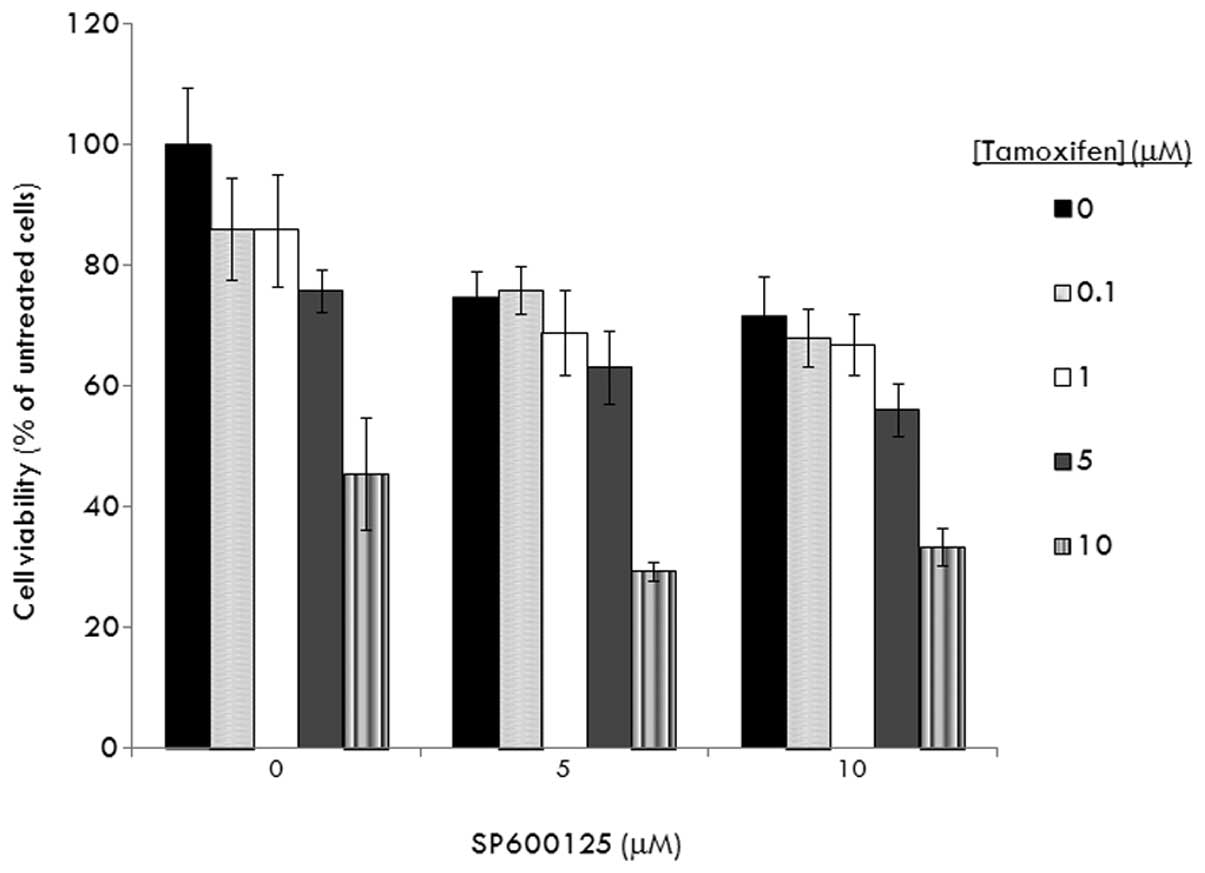
We determined whether JNKs are also essential for
COX-2 to induce tamoxifen resistance. As shown in our previous
report (10), MCF-7/COX-2 cells
were very insensitive to tamoxifen. At 5 μM concentration,
tamoxifen inhibited MCF-7/COX-2 cell growth by only 20% (Fig. 4). In the presence of 5 and 10 μM
SP600125, tamoxifen (at 5 μM) inhibited MCF-7/COX-2 cell growth by
30 and 40%, respectively (P>0.05) (Fig. 4). These data indicate that JNKs do
not mediate COX-2-induced tamoxifen resistance.
Discussion
In the present study, we demonstrated that COX-2
utilizes PKC to stimulate JNK activity, which is essential for
COX-2 to induce breast cancer cell invasion. JNKs have been shown
to mediate the invasive activity of breast cancer cells induced by
various proteins, including interleukin-8 (13), leptin (23) and transglutaminase (25). Activated JNKs can translocate to the
nucleus where they regulates transcription factors such as c-Jun,
ATF-2, Elk-1, p53 and c-Myc, resulting in enhanced expression
and/or activity of proteases, such as urokinase plasminogen
activator (13),
metalloproteinase-2 (23), or
reduced expression of programmed cell death 4 (26), a candidate tumor suppressor gene,
thereby enhancing the ability of breast cancer cells to invade
through the basement membrane. Wang et al(27) also demonstrated that JNKs mediate
epithelial-mesenchymal transition, survival and proliferation of
breast cancer cells. These pro-tumorigenic properties of JNKs may
explain why elevated levels of phosphorylated JNKs have been
correlated with a poorer prognosis in breast cancer patients
(28,29).
Tamoxifen is the most widely used drug for breast
cancer treatment. Unfortunately, many patients with advanced
ERα-positive disease fail to respond to tamoxifen, and many
responsive patients acquire resistance to tamoxifen, leading to
disease progression. Although upregulation of JNK activity, c-Jun
phosphorylation and AP-1 DNA binding activity have been found in
ERα-positive breast tumors with the acquired tamoxifen resistance
phenotype (30,31), JNKs have not been shown to mediate
tamoxifen resistance. Here, we demonstrated that inhibition of JNKs
did not resensitize COX-2-overexpressing breast cancer cells to
tamoxifen. Our data suggest that either JNKs are not involved in
regulating tamoxifen sensitivity, or that inhibition of JNKs alone
is not sufficient to reverse tamoxifen resistance. Indeed several
mechanisms, including loss or modification of ERα expression,
regulation of signal transduction pathways, altered expression of
specific microRNAs, balance of co-regulatory proteins and genetic
polymorphisms involved in tamoxifen metabolism, could contribute to
the development of tamoxifen resistance (32–34).
In conclusion, we found that COX-2 increased the
activity of JNKs to mediate invasion, but not tamoxifen resistance,
in MCF-7 breast cancer cells. In breast cancer, COX-2 expression is
a predictor of poor disease-free and overall survival (4–9) and
has been implicated as a marker of high metastatic potential
(11,12). Pharmacological inhibition aiming at
JNKs may have potential therapeutic benefit in patients with
ERα-positive COX-2-overexpressing breast tumors by reducing tumor
invasiveness and metastatic potential.
Acknowledgements
The present study was supported by the Susan G.
Komen Breast Cancer Foundation.
References
|
1
|
Eberhart CE, Coffey RJ, Radhika A,
Giardiello FM, Ferrenbach S and DuBois RN: Up-regulation of
cyclooxygenase 2 gene expression in human colorectal adenomas and
adenocarcinomas. Gastroenterology. 107:1183–1188. 1994.PubMed/NCBI
|
|
2
|
Soslow RA, Dannenberg AJ, Rush D, et al:
COX-2 is expressed in human pulmonary, colonic, and mammary tumors.
Cancer. 89:2637–2645. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kirschenbaum A, Liu X, Yao S and Levine
AC: The role of cyclooxygenase-2 in prostate cancer. Urology.
58:127–131. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ristimaki A, Sivula A, Lundin J, et al:
Prognostic significance of elevated cyclooxygenase-2 expression in
breast cancer. Cancer Res. 62:632–635. 2002.PubMed/NCBI
|
|
5
|
Denkert C, Winzer KJ, Muller BM, et al:
Elevated expression of cyclooxygenase-2 is a negative prognostic
factor for disease-free survival and overall survival in patients
with breast carcinoma. Cancer. 97:2978–2987. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
O’Connor JK, Avent J, Lee RJ, Fischbach J
and Gaffney DK: Cyclooxygenase-2 expression correlates with
diminished survival in invasive breast cancer treated with
mastectomy and radiotherapy. Int J Radiat Oncol Biol Phys.
58:1034–1040. 2004.PubMed/NCBI
|
|
7
|
Spizzo G, Gastl G, Wolf D, et al:
Correlation of COX-2 and Ep-CAM overexpression in human invasive
breast cancer and its impact on survival. Br J Cancer. 88:574–578.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Surowiak P, Materna V, Matkowski R, et al:
Relationship between the expression of cyclooxygenase 2 and
MDR1/P-glycoprotein in invasive breast cancers and their prognostic
significance. Breast Cancer Res. 7:R862–R870. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Holmes MD, Chen WY, Schnitt SJ, et al:
COX-2 expression predicts worse breast cancer prognosis and does
not modify the association with aspirin. Breast Cancer Res Treat.
130:657–662. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tari AM, Simeone AM, Li YJ,
Gutierrez-Puente Y, Lai S and Symmans WF: Cyclooxygenase-2 protein
reduces tamoxifen and N-(4-hydroxyphenyl)retinamide inhibitory
effects in breast cancer cells. Lab Invest. 85:1357–1367. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Costa C, Soares R, Reis-Filho J, Leitao D,
Amendoeira I and Schmitt F: Cyclo-oxygenase 2 expression is
associated with angiogenesis and lymph node metastasis in human
breast cancer. J Clin Pathol. 55:429–434. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ranger GS, Thomas V, Jewell A and Mokbel
K: Elevated cyclooxygenase-2 expression correlates with distant
metastases in breast cancer. Anticancer Res. 24:2349–2351.
2004.PubMed/NCBI
|
|
13
|
Simeone AM, Nieves-Alicea R, McMurtry VC,
Colella S, Krahe R and Tari AM: Cyclooxygenase-2 uses the protein
kinase C/interleukin-8/urokinase-type plasminogen activator pathway
to increase the invasiveness of breast cancer cells. Int J Oncol.
30:785–792. 2007.PubMed/NCBI
|
|
14
|
Prosperi JR, Mallery SR, Kigerl KA, Erfurt
AA and Robertson FM: Invasive and angiogenic phenotype of MCF-7
human breast tumor cells expressing human cyclooxygenase-2.
Prostaglandins Other Lipid Mediat. 73:249–264. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singh B, Berry JA, Shoher A, Ramakrishnan
V and Lucci A: COX-2 overexpression increases motility and invasion
of breast cancer cells. Int J Oncol. 26:1393–1399. 2005.PubMed/NCBI
|
|
16
|
Connolly EM, Harmey JH, O’Grady T, et al:
Cyclo-oxygenase inhibition reduces tumour growth and metastasis in
an orthotopic model of breast cancer. Br J Cancer. 87:231–237.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kundu N and Fulton AM: Selective
cyclooxygenase (COX)-1 or COX-2 inhibitors control metastatic
disease in a murine model of breast cancer. Cancer Res.
62:2343–2346. 2002.PubMed/NCBI
|
|
18
|
Roche-Nagle G, Connolly EM, Eng M,
Bouchier-Hayes DJ and Harmey JH: Antimetastatic activity of a
cyclooxygenase-2 inhibitor. Br J Cancer. 91:359–365.
2004.PubMed/NCBI
|
|
19
|
Ogunwobi O, Mutungi G and Beales IL:
Leptin stimulates proliferation and inhibits apoptosis in Barrett’s
esophageal adenocarcinoma cells by cyclooxygenase-2-dependent,
prostaglandin-E2-mediated transactivation of the epidermal growth
factor receptor and c-Jun NH2-terminal kinase
activation. Endocrinology. 147:4505–4516. 2006.PubMed/NCBI
|
|
20
|
Leone V, di Palma A, Ricchi P, et al:
PGE2 inhibits apoptosis in human adenocarcinoma Caco-2
cell line through Ras-PI3K association and cAMP-dependent kinase A
activation. Am J Physiol Gastrointest Liver Physiol. 293:G673–G681.
2007.PubMed/NCBI
|
|
21
|
Repasky GA, Zhou Y, Morita S and Der CJ:
Ras-mediated intestinal epithelial cell transformation requires
cyclooxygenase-2-induced prostaglandin E2 signaling. Mol
Carcinog. 46:958–970. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Z, Lai GH and Sirica AE:
Celecoxib-induced apoptosis in rat cholangiocarcinoma cells
mediated by Akt inactivation and Bax translocation. Hepatology.
39:1028–1037. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McMurtry V, Simeone AM, Nieves-Alicea R
and Tari AM: Leptin utilizes Jun N-terminal kinases to stimulate
the invasion of MCF-7 breast cancer cells. Clin Exp Metastasis.
26:197–204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gonzalez-Villasana V, Nieves-Alicea R,
McMurtry V, Gutierrez-Puente Y and Tari AM: Programmed cell death 4
inhibits leptin-induced breast cancer cell invasion. Oncol Rep.
27:861–866. 2012.PubMed/NCBI
|
|
25
|
Li Z, Xu X, Bai L, Chen W and Lin Y:
Epidermal growth factor receptor-mediated tissue transglutaminase
overexpression couples acquired tumor necrosis factor-related
apoptosis-inducing ligand resistance and migration through c-FLIP
and MMP-9 proteins in lung cancer cells. J Biol Chem.
286:21164–21172. 2011. View Article : Google Scholar
|
|
26
|
Nieves-Alicea R, Colburn NH, Simeone AM
and Tari AM: Programmed cell death 4 inhibits breast cancer cell
invasion by increasing tissue inhibitor of metalloproteinases-2
expression. Breast Cancer Res Treat. 114:203–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang J, Kuiatse I, Lee AV, Pan J, Giuliano
A and Cui X: Sustained c-Jun-NH2-kinase activity
promotes epithelial-mesenchymal transition, invasion, and survival
of breast cancer cells by regulating extracellular signal-regulated
kinase activation. Mol Cancer Res. 8:266–277. 2010.
|
|
28
|
Davidson B, Konstantinovsky S, Kleinberg
L, et al: The mitogen-activated protein kinases (MAPK) p38 and JNK
are markers of tumor progression in breast carcinoma. Gynecol
Oncol. 102:453–461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yeh YT, Hou MF, Chung YF, et al: Decreased
expression of phosphorylated JNK in breast infiltrating ductal
carcinoma is associated with a better overall survival. Int J
Cancer. 118:2678–2684. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Johnston SR, Lu B, Scott GK, et al:
Increased activator protein-1 DNA binding and c-Jun
NH2-terminal kinase activity in human breast tumors with
acquired tamoxifen resistance. Clin Cancer Res. 5:251–256.
1999.PubMed/NCBI
|
|
31
|
Schiff R, Reddy P, Ahotupa M, et al:
Oxidative stress and AP-1 activity in tamoxifen-resistant breast
tumors in vivo. J Natl Cancer Inst. 92:1926–1934. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garcia-Becerra R, Santos N, Diaz L and
Camacho J: Mechanisms of resistance to endocrine therapy in breast
cancer: focus on signaling pathways, miRNAs and genetically based
resistance. Int J Mol Sci. 14:108–145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sweeney EE, McDaniel RE, Maximov PY, Fan P
and Jordan VC: Models and mechanisms of acquired antihormone
resistance in breast cancer: significant clinical progress despite
limitations. Horm Mol Biol Clin Investig. 9:143–163.
2012.PubMed/NCBI
|
|
34
|
Bianco S and Gevry N: Endocrine resistance
in breast cancer: from cellular signaling pathways to epigenetic
mechanisms. Transcription. 3:165–170. 2012. View Article : Google Scholar : PubMed/NCBI
|