Introduction
Deleted in liver cancer-1 (DLC-1) is a tumor
suppressor gene, identified through representational difference
analysis, and has been found to be localized at the short arm of
chromosome 8, at p21.3, where deletions are frequently found in
several types of human cancer, such as nasopharyngeal carcinoma
(1), breast cancer (2), colorectal cancer (3), lung cancer (4), hepatocellular carcinoma (HCC)
(5) and medulloblastoma (6). Promoter hypermethylation, an
epigenetic mechanism leading to the silencing of the gene
expression (7,8), may be the reason for the absence of
DLC-1. Previous studies indicated that promoter hypermethylation of
DLC-1 is found in HCC, colon, breast and prostate cancer (9). Methylation of DLC-1 is also common in
hematologic malignancies; it has been found in 87% of non-Hodgkin’s
lymphomas (NHLs) and multiple myeloma tumors or tumor cell lines,
but not in benign hyperplasia. It has also been proposed that DLC-1
methylation status in NHLs could be used as a diagnostic marker
(10). Normally, there are three
mechanisms leading to gene silencing: mutation, genomic deletion
and promoter methylation. Mutation, as a cause of DLC-1
inactivation, is effectively ruled out due to the low rate of
happening (11). This suggests that
either genomic deletion or promoter methylation is the primary
cause for altered expression or inactivation of the DLC-1 gene
(12). Though hypermethylation is
responsible for the silencing of the DLC-1 gene in a limited
portion of breast cancer cases, transfection of the gene into DLC-1
deficient T-47D cells raised the DLC-1 mRNA level and resulted in
inhibition of cell growth and reduced colony formation capacity,
which indicate a role of DLC-1 in breast cancer carcinogenesis
(13). Nevertheless, cells derived
from other types of cancer such as lung, liver or ovarian cancer
are also highly sensitive to reactivation of DLC-1 function
(11,14). Recent studies have shown that
restoration of the DLC-1 in breast cancer cells resulted in the
inhibition of migration and invasion. In addition, Chan et
al(15) reported that DLC-1
expression reduced the migration and invasiveness of SMMC-7721 HCC
cells. There is a strong potential for an effective therapy based
on DLC-1 transfer to tumor cells. However, DLC-1 has been little
examined in colon cancer; thus, studies on DLC-1 expression and the
mechanism of DLC-1 gene inactivation may be necessary to elucidate
the possible role of DLC-1 in colorectal tumor development. In the
present study, we explored the involvement of DLC-1 promoter
hypermethylation in colorectal tumors. Additionally, we
investigated the effects of DLC-1 on colon cancer cell
proliferation by RNA interference (RNAi) techniques.
Materials and methods
Colon cancer cell lines
Human colon cancer cell lines, Caco-2, HT-29 and
LoVo, were obtained from the Shanghai Institute of Cell Biology,
Chinese Academy of Sciences. Cells were cultured in RPMI-1640
(Sigma, St. Louis, MO, USA) and antibiotics, supplemented with 10%
fetal bovine serum and 100 U/ml of penicillin and streptomycin at
37°C in a humidified 5% CO2 atmosphere and stored at
−20°C.
DNA/RNA extraction
By standard methods, genomic DNA was isolated by
digestion with Proteinase K followed by phenol:chloroform (1:1)
extraction and ethanol precipitation from Caco-2, HT-29 and LoVo
cell lines. Total-RNA extraction was performed using TRIzol reagent
(Invitrogen Life Technologies, Groningen, The Netherlands)
according to the manufacturer’s instructions. To avoid the genomic
DNA contamination, RNA samples were treated with DNase I
(RNase-free) (Takara Bio, Inc., Shiga, Japan) for 20 min at 37°C.
The enzyme was heat inactivated and the RNA was ethanol
precipitated following phenol chloroform extraction. Following
precipitation, the RNA was resuspended in 10 μl of
diethylpyrocarbonate (DEPC)-treated water and stored at −80°C.
Reverse transcription (RT) reaction
RNA (5 μg) was used as template in the first strand
complementary DNA (cDNA) synthesis in a 20 μl reaction volume,
which consisted of oligo-dT primer (0.2 μg/reaction), dNTP (0.5 mM
of each), avian myeloblastosis virus reverse transcriptase (AMV RT;
Promega Corporation, Madison, WI, USA) (20 U/reaction), RNasin
(Takara Bio) (20 U/reaction). The reaction was incubated at 42°C
for 60 min, followed by heating at 99°C for 5 min. Reactions in
which pure water replaced the RNA were used as RT-negative
controls.
Polymerase chain reaction (PCR)
amplification and electrophoresis
All primers and amplification conditions used in
this study are listed in Table I.
Single round β-actin amplification was used to demonstrate RNA
integrity and the RT performance. The reaction mixture consisted of
the template (50–100 μg of cDNA), dNTP (0.2 mM of each), Taq DNA
polymerase (Promega Corporation) (0.625 U/reaction) and a selected
primer pair (20 pmol/primer/reaction) in a total volume of 25 μl.
The final products were analyzed by electrophoresis on 2% agarose
gels containing ethidium bromide.
 | Table IThe primers used and conditions of PCR
amplification. |
Table I
The primers used and conditions of PCR
amplification.
| Primer | 5′-3′ sequence | PCR conditions | Size of amplified
products (bp) |
|---|
| DLC-1 (RT-PCR) |
GGACACCATGATCCTAACAC
CTCATCCTCGTCTGAATCGT | 94/3 min (94/30 sec,
52/30 sec, 72/40 sec) ×42, 72/7 min | 252 |
| β-actin (RT-PCR) |
CGTGGCCTTAGCTGTGCT
TGTGCATAAAGTGTAAGTGTATAAGCA | 94/3 min (94/30 sec,
50/30 sec, 72/40 sec) ×42, 72/7 min | 457 |
| DLC-1 (unmethylation
specific primers) |
TTTTTTAAAGATTGAAATGAGGGAGTG
AAACCCAACAAAAAAACCCAACTAACA | 94/3 min (94/30 sec,
52/30 sec, 72/40 sec) ×45, 72/7 min | 172 |
| DLC-1 (methylation
specific primers) |
TTTAAAGATCGAAACGAGGGAGCG
CCCAACGAAAAACCCGACTAACG 72/40 sec) | 94/3 min (94/30 sec,
52/30 sec, ×45, 72/7 min | 178 |
| Bisulfite treated DNA
(PCR primer) |
GTTTTTAGTTAGGATATGGT
CTTCTTTCTACACATCAAACA | 94/3 min (94/30 sec,
52/30 sec, 72/40 sec) ×45, 72/7 min | 292 |
| β-actin |
CCCTGGACTTCGAGCAAGAGAT
GTTTTCTGCGCAAGTTAGG | 95/2 min (95/1 min,
60/1 min, 72/1 min) ×35 | 531 |
Sodium bisulfite treatment
Genomic DNA (2 μg) was denatured with 0.3 M NaOH for
10 min at 37°C and mixed with 30 μl of 10 mM hydroquinone (Sigma),
510 μl of 3.0 M NaHSO3 (pH 5.0; Sigma), covered with
paraffin oil, then deaminated in the dark for 16 h at 50°C. To use,
10 mM hydroquinone and 3.0 M NaHSO3 (pH 5.0) were
freshly prepared and mixed. Bisulfite-treated DNA was purified
using a Wizard DNA Clean-Up System (Promega Corporation).
Subsequently, purified DNA samples were desulfonated with 0.3 M
NaOH for 5 min at room temperature, neutralized with ammonium
acetate, ethanol precipitated and resuspended in 20 μl Tris-EDTA
buffer.
Methylation-specific PCR (MSP) and
sequencing
To determine whether the loss of DLC-1 expression
was associated with promoter hypermethylation, we used MSP to
detect the methylation status of the 5′CpG island.
Bisulfite-treated DNA was amplified by PCR with methylation
status-specific primer pairs. The primer sequences for the MSP and
amplification conditions used in this study are listed in Table I. The sodium bisulfite reaction
converts unmethylated cytosine in DNA to uracil while leaving the
methylcytosine unchanged, so that methylated and unmethylated
alleles can be distinguished by MSP. Bisulfite-treated DNA (8 μl)
was subjected to PCR using bisulfite treated DNA PCR primer (the
primer can amplify both the sequences methylated and unmethylated)
(Table I), a 292 bp fragment,
particularly for the upstream region of the basic promoter of DLC-1
was amplified and PCR product was sequenced.
Short hairpin RNA (shRNA) preparation and
plasmid construction
In order to construct two plasmids, two pairs of
shRNA sequence were designed, one is according to the DLC-1
sequence in the GenBank (AF035119), as pGCsiDLC-1, another sequence
of shRNA has no homology with human sequence, as a control group.
Each pair contained a unique 19-nt double-stranded sequence that is
separated by a loop of 9-nt sequence (ttcaagaga). Following
purification and restriction digestion, the oligonucleotides were
ligated into plasmid pGCsi with polymerase III U6 promoter,
purchased from GeneChem Inc, China. After amplifying the plasmids
into E.coli, the selected clones with the shRNA insert were
selected and purified with plasmid purification kit (Promega
Corporation). The oligonucleotide sequences of siRNA are listed in
Table II.
| Table IIOligonucleotide sequences of
siRNA. |
Table II
Oligonucleotide sequences of
siRNA.
| Group | Sequence of siRNA
nucleotides | Sites |
|---|
| pGCsil-DLC-1 |
5′-GGAACTGAAGAGACGCAAT-3′ | 1596–1614 |
| Control |
5′-TTCTCCGAACGTGTCACGT-3′ | - |
Transfection assay
To generate DLC-1 transfected DLC-1 cells, 3 μg of
plasmid DNA were transfected in 1×105 cells in a 60-mm
dish using Lipofectamine® 2000 (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer’s
instructions. Selection for transfected cells was carried out in a
medium containing 400 μg/ml G418 (Geneticin; Life Technologies,
Inc., USA).
Western blot analysis
Experiments were conducted in three groups, the
pGCsiDLC-1, the mock and the control group. Following transfection
for 24 and 48 h, LoVo cells (10×106) were harvested and
lysed in 60 μl cell lysis reagent, containing 50 mmol/l Tris-HCl
(pH 8.0), 150 mmol/l NaCl, 100 μg/ml phenylmethylsulfonyl fluoride
(PMSF) and 1% TritonX-100. Equal amounts of total protein were
separated by 5% sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene
fluoride (PVDF). After blocking in a 5% non-fat dry milk solution
in washing buffer containing 10 mmol/l Tris (pH 7.6), 150 mmol/l
NaCl and 0.05% Tween-20, membranes were incubated for 1 h at room
temperature with human monoclonal anti-DLC-1 (1:200; clone 3; BD
Biosciences Pharmingen, San Diego, CA, USA) as primary antibodies,
washed 3 times, incubated again with bovine anti-mouse IgG
(1:2,500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) as
second antibody. β-tublin (1:300) staining served as the internal
standard for all membranes.
MTT assay
The transfected cells were plated in 96-well
microtiter plates at a density of 1×104 cells/well.
Cells were further cultured for 24, 36 and 48 h respectively, after
which the medium was replaced with 100 μl of fresh serum-free
medium containing 50 μg MTT. Four hours later, the color reaction
was quantified with an ELISA plate reader at a test wavelength of
570 nm and a reference wavelength of 630 nm. The experiment was
repeated three times independently.
Colony formation assay
The cells stably transfected with DLC-1 and control
cells were seeded into six-well plates at a density of
1×104 cells/well, the culture medium contained 500 μg/ml
G418 (Geneticin; Life Technologies, Inc). After culturing for two
weeks, the G418-resistant colonies were washed twice with
phosphate-buffered saline (PBS), stained with Giemsa, and the
colony formation efficiency was tested. For soft agar colony
assays, the pGCsil-DLC-1, the mock and the control group were mixed
with RPMI-1640 complete medium containing 0.4% agar and placed over
0.6% of basal agar in six-well plates. Cells were grown for two
weeks and colonies were visualized microscopically and
photographed.
Results
Detection of DLC-1 expression in colon
cancer cell lines
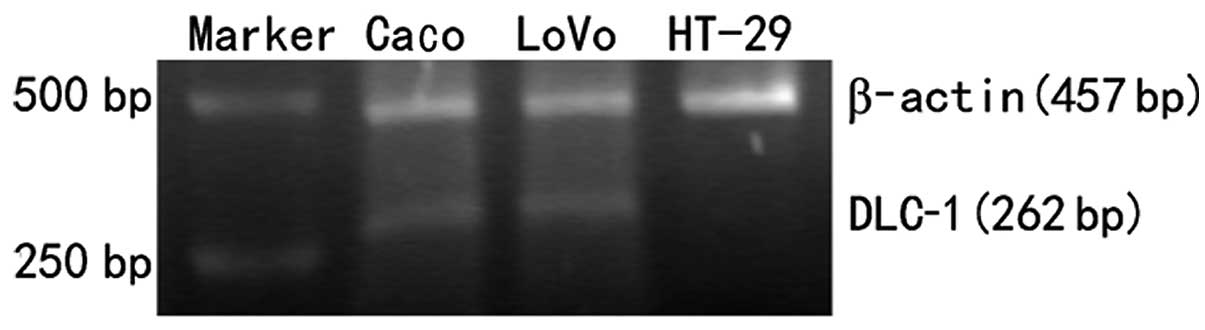
RT-PCR was performed in Caco-2, LoVo and HT-29 cells
to examine the expression of DLC-1 in colon cancer cell lines. To
validate the PCR results, the same cDNA was amplified for β-actin.
All the cell lines were positive for β-actin, which indicates both
RNA preparation and cDNA synthesis were successful. Caco-2 and LoVo
colon cancer cell lines were all positive for DLC-1 mRNA and a band
of 262 bp, but low levels or absence of DLC-1 mRNA were observed in
HT-29 cells (Fig. 1). In order to
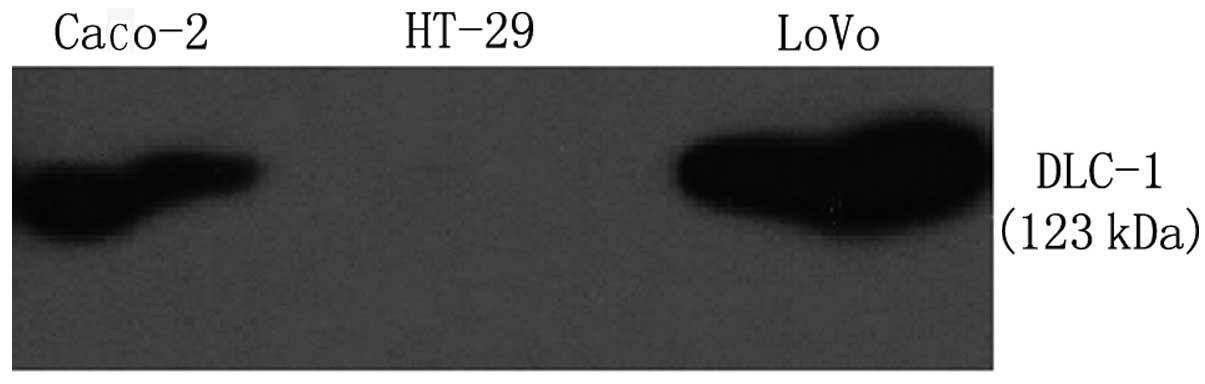
detect the protein expression of DLC-1 in these cells, western blot
analysis was performed and the results were in agreement with the
results obtained from RT-PCR. Loss of DLC-1 protein expression was
detected in HT-29, Caco-2 and LoVo revealed a fragment of 123 kDa
(Fig. 2).
Methylation of the DLC-1 gene promoter in
colon cancer cell lines
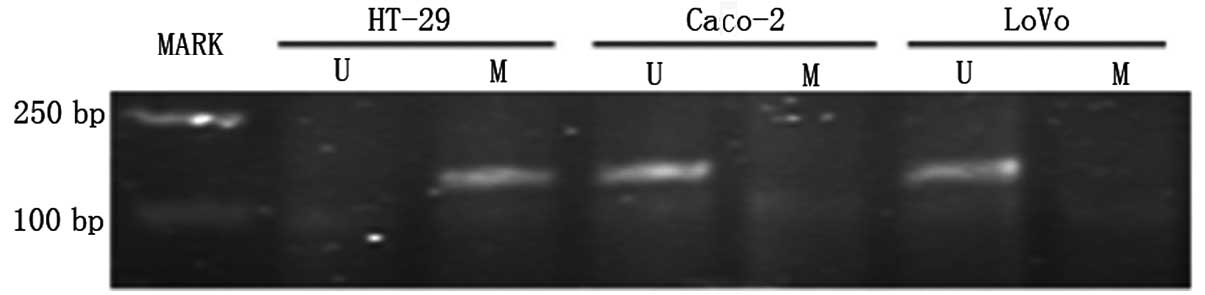
To determine whether the loss of DLC-1 expression
was mediated by promoter hypermethylation in human colon cancer
cell lines, MSP was performed on Caco-2, LoVo and HT-29 cells.
Representative results are shown in Fig. 2. All cells exhibited the bands that
correspond to either unmethylated (178 bp) or methylated (172 bp)
CpG island. PCR with methylated primers showed that HT-29 has
methylated forms of DLC-1, which showed no expression or low levels
of DLC-1 expression. On the other hand, the unmethylated band was
observed in Caco-2 and LoVo cells (Fig.
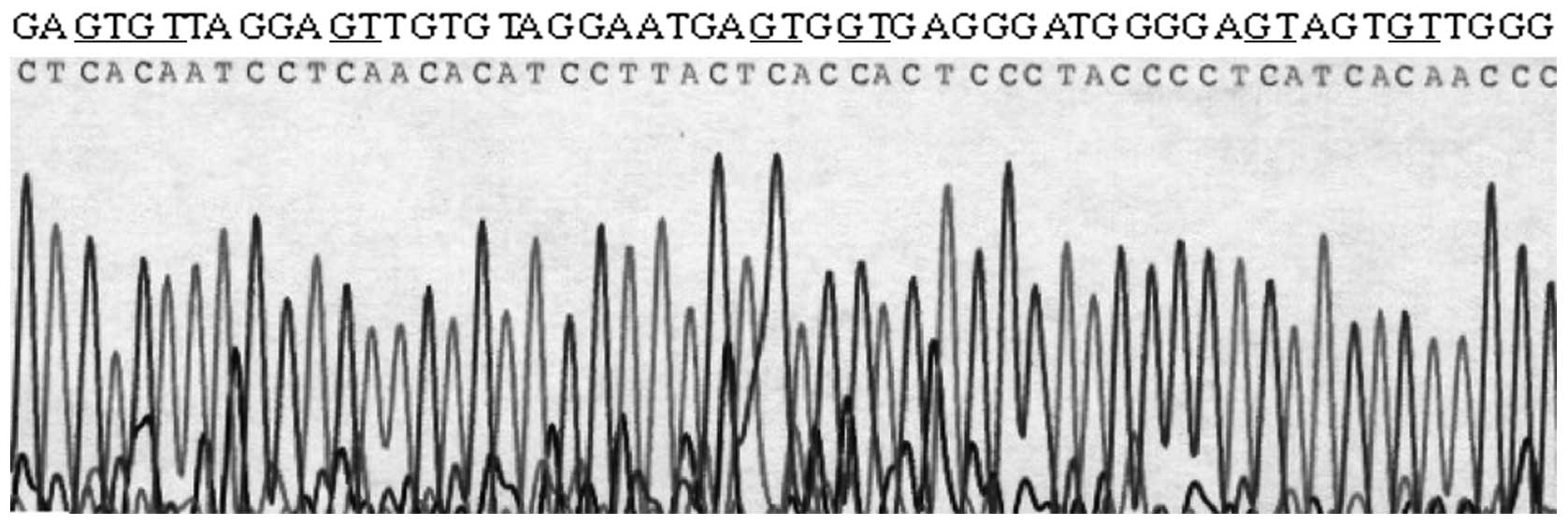
3). To further confirm the methylation status as well as to
determine whether the loss of DLC-1 expression was mediated by
promoter hypermethylation in colon cancer cell lines, a 292 bp
fragment of the DLC-1 promoter region was sequenced on LoVo and
HT-29 cells. In agreement with the results obtained from MSP study,
6 CpG dinucleotides in LoVo cell were unmethylated, whereas HT-29
cells showed extensive hypermethylation at these CpG dinucleotides
(Fig. 4).
Inhibition of DLC-1 gene expression by
shRNA expression vectors
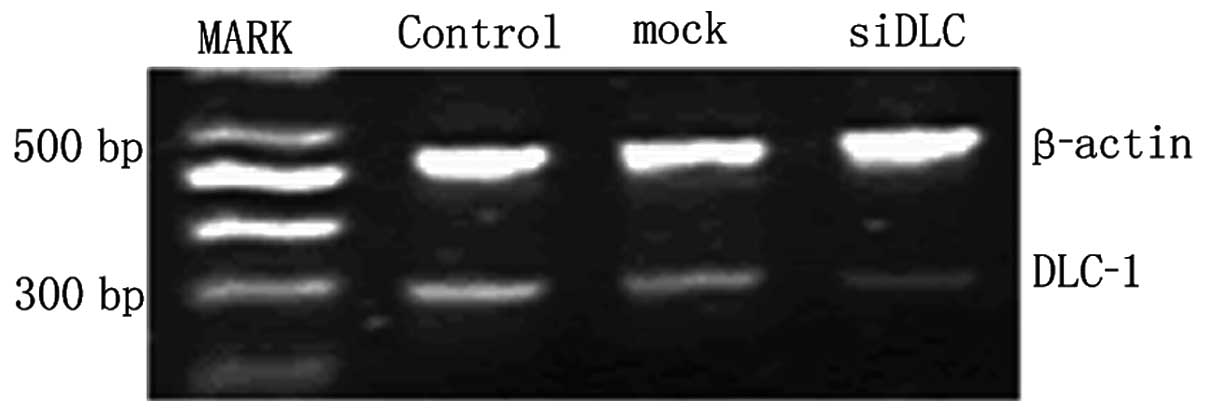
The knockdown efficiencies of DLC-1 specific shRNAs
in LoVo cells were analyzed by semiquantitative RT-PCR. Relative
DLC-1 mRNA levels were normalized by internal control β-actin after
transfection. Compared with the mock and the control group, the
DLC-1 mRNA expression was reduced in the pGCsiDLC-1 LoVo cells, but
it was not different in the mock and the control group (Fig. 5). The expression of DLC-1 protein
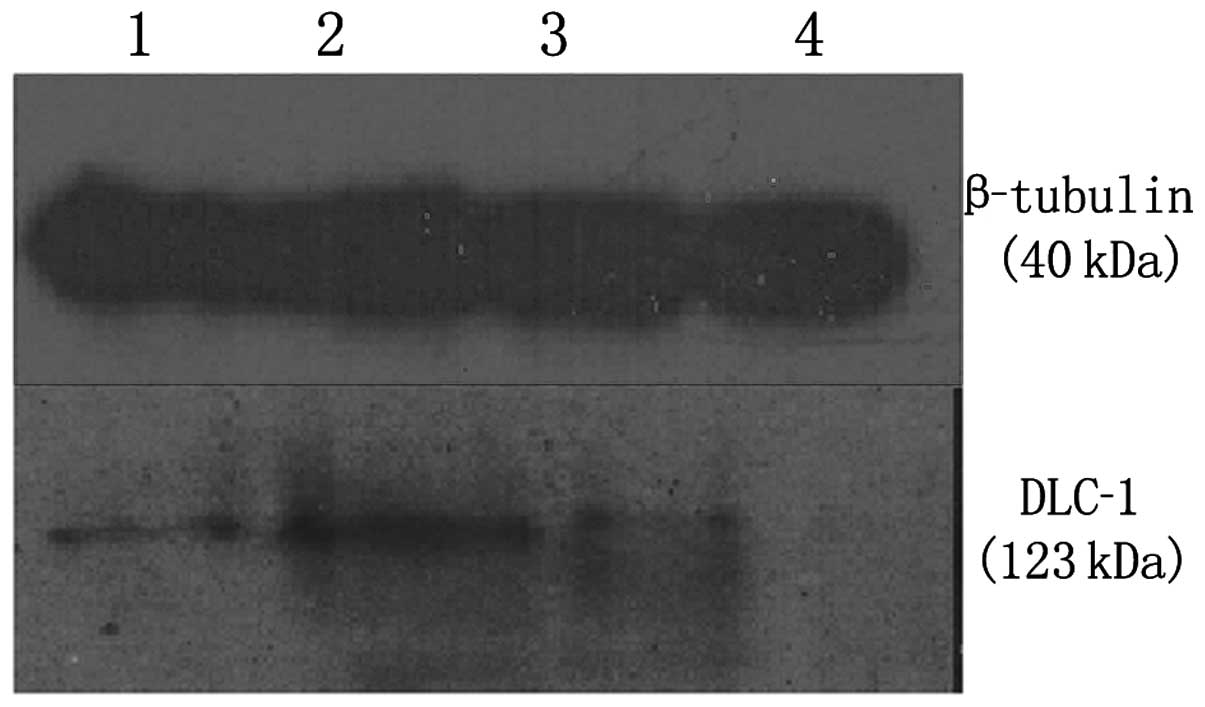
can reflect post-transcription level of the DLC-1 gene. The
knockdown efficiencies of DLC-1 protein in LoVo cells were analyzed
by western blot analysis. β-tubulin protein expression was used to
normalize the expression of DLC-1. The results showed DLC-1 protein
expression to be downregulated following transfection, particularly
after 48 h (Fig. 6).
Effects of DLC-1 expression on cell
growth in LoVo cells
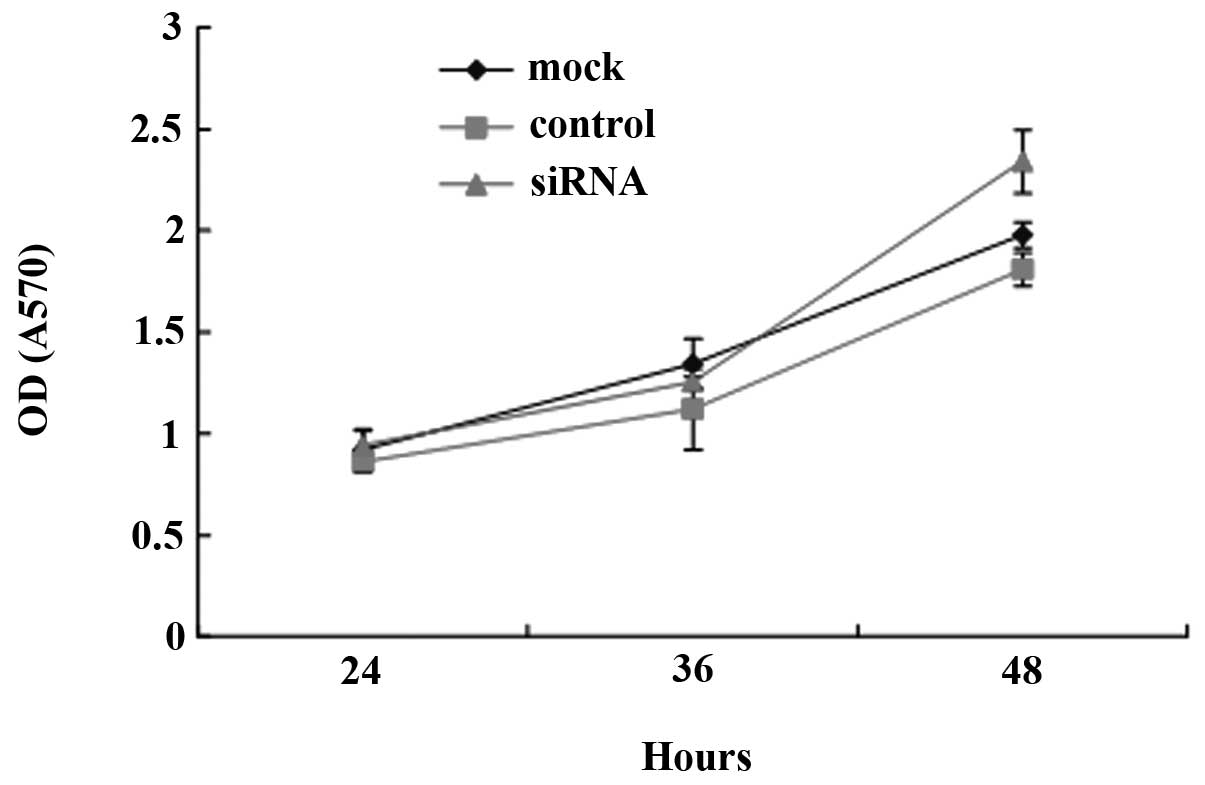
To determine whether decreased DLC-1 expression may
change the rate of LoVo cell proliferation, DLC-1 RNAi recombinant
vector was transfected into LoVo cells to downregulate DLC-1
expression and MTT assay was carried out to test cell
proliferation. Fig. 7 shows that
after the cells were transfected with pGCsiDLC-1, the proliferation
rate of the LoVo cells increased significantly at 48 h compared to
the control and the mock group (P<0.01). No significant
difference of growth inhibition was observed between the mock and
the control group. To further assess the time of DLC-1 inhibitory
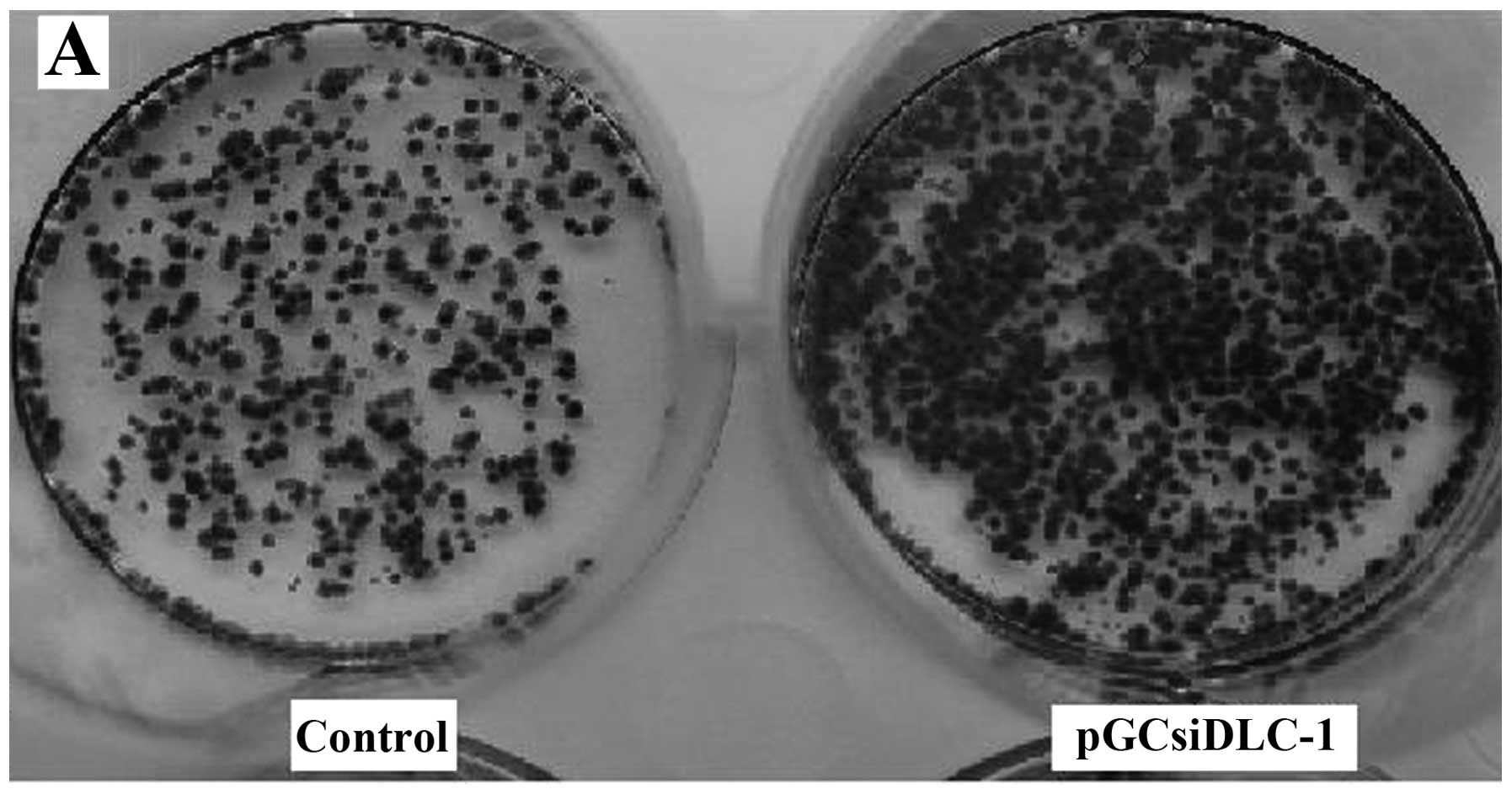
effects, we tested the effect of DLC-1 on colony formation of LoVo
cells; cells transfected with pGCsiDLC-1 and control vector were
cultured in G418 for two weeks and the colony number was counted on
Giemsa-stained dishes. A significant increase both in colony number
and size of G418-resistant colonies was observed in transfection
LoVo cell lines compared to the control group (Fig. 8A) and for soft agar assays, cells
transfected with pGCsiDLC-1 formed colonies more quickly than the
mock and the control group (Fig.
8B).
Discussion
A candidate tumor suppressor gene referred to as
DLC-1 was isolated from human HCC by PCR-based subtractive
hybridization approach (16).
Determination of the DLC-1 cDNA sequence shows that it is the human
homologue of rat p122, which has been found to act as a Rho GTPase
protein (RhoGAP) (17). DLC-1
contains a RhoGAP domain, and two other functional motifs, a
sterile alpha motif (SAM) domain and a StaR-related lipid transfer
(START) domain. Overexpression of p122 in cultured cells induces
morphological changes in adherent cells and the detachment of cells
from the substratum (18). It was
originally reported that the human DLC-1 gene is a regulator of the
Rho family of small GTPases, indicating that the DLC-1 gene
controls actin cytoskeleton organization, membrane trafficking,
gene expression, cell proliferation, malignant transformation and
metastasis (19). The length of the
human DLC-1 gene is 50 kb and consists of 14 exons, the sequence at
the 5′end of the first exon is GC-rich and typical of a CpG island,
it has a 63% G+C content, that encompasses the transcription start
site and harbors several potential transcription factor-binding
sites (20). Results have shown
that the 5′CpG island of the DLC-1 gene is unmethylated in DNA from
normal cells but is methylated in DNA from several tumor cell lines
that have been shown to lack detectable levels of DLC-1 mRNA.
Almost all CpG sites within DLC-1 CpG island were methylated in
gastric cancer cells (21,22). This methylation promotes the binding
of proteins that recognize methyl-CpGs and lead to alterations in
chromatin structure that repress transcription.
In this study, we first investigated the expression
of the DLC-1 in colon cancer cell lines. Levels of DLC-1 expression
were reduced or undetectable in HT-29, while high levels of DLC-1
were detected in Caco-2; RT-PCR and western blot analysis
demonstrated the expression of the DLC-1 gene in LoVo was positive
both at the mRNA and the protein level. The findings agree with
those reported by Ullmannova and Popescu (23). Since mounting evidence demonstrated
that promoter hypermethylation of certain tumor-suppressor genes
can inhibit gene transcription, the methylation status of the
promoter region of the DLC-1 gene was examined by using MSP and
sodium genomic sequencing in the three colon cancer cell lines.
These findings confirmed that the promoter of DLC-1 is
hypermethylated in the HT-29 colon cancer cell line, which does not
have detectable levels of DLC-1 mRNA and protein expression. On the
contrary, no methylation was found in LoVo and Caco-2 cells which
are positive for DLC-1 expression. It is agreed that with the loss
of mRNA expression, epigenetic silencing by mechanisms such as
aberrant methylation of the DLC-1 promoter might be responsible,
particularly in colon cancer. Mutations of DLC-1 in other types of
cancer are low (24,25). DLC-1 inactivation caused by
methylation was not rare in colon cancer cell lines; thus, promoter
hypermethylation might be a major mechanism responsible for the
silencing of DLC-1 in a variety of solid tumors and hematological
malignancies.
The investigation by Yuan et al(26) demonstrated that transfer of DLC-1
into three DLC-1 negative human non-small cell lung carcinoma cell
lines caused a significant inhibition in cell proliferation and a
decrease in colony formation. DLC-1 restoration in DLC-1 negative
SNU-368 human HCC cells resulted in inhibition of cell
proliferation and migration, and induction of cell morphological
changes. In addition, significant reduction of tumorigenicity was
shown in nude mice, which indicates DLC-1 plays the role of a tumor
suppressor gene. To understand whether DLC-1 expression affects
colon cancer cell growth, eukaryotic expression plasmid vectors of
shRNA specific for the DLC-1 gene were designed and generated. The
plasmid vectors were transfected into LoVo cells by cation
liposomes to inhibit DLC-1 expression in the LoVo cell line which
highly expresses the DLC-1 gene. Data also showed that lack of
DLC-1 expression resulted in the inhibition of cell growth and
reduced colony formation. Forty-eight hours after the cells were
transfected, the proliferation rate of the LoVo cells increased
significantly compared to that of the control and the mock
transfection group (P<0.01); a significant increase in both
colony number and size of colonies was also observed in transfected
cells. Collectively, our observations suggest that hypermethylation
is responsible for abrogating the function of the DLC-1 gene in
colon cancer and indicate an inhibitory effect on colon cancer cell
growth in vitro.
Acknowledgements
This study was supported by grants from the Shanghai
Bureau of Health, P.R. China (no. 2010052).
References
|
1
|
Zhang X, Li W, Li H, et al: Genomic
methylation profiling combined with gene expression microarray
reveals the aberrant methylation mechanism involved in
nasopharyngeal carcinoma taxol resistance. Anticancer Drugs.
23:856–864. 2012. View Article : Google Scholar
|
|
2
|
Muehlich S, Hampl V, Khalid S, et al: The
transcriptional coactivators megakaryoblastic leukemia 1/2 mediate
the effects of loss of the tumor suppressor deleted in liver cancer
1. Oncogene. 31:3913–3923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Low JS, Tao Q, Ng KM, et al: A novel
isoform of the 8p22 tumor suppressor gene DLC1 suppresses tumor
growth and is frequently silenced in multiple common tumors.
Oncogene. 30:1923–1935. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castro M, Grau L, Puerta P, et al:
Multiplexed methylation profiles of tumor suppressor genes and
clinical outcome in lung cancer. J Transl Med. 8:862010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zimonjic DB and Popescu NC: Role of DLC1
tumor suppressor gene and MYC oncogene in pathogenesis of human
hepatocellular carcinoma: Potential prospects for combined targeted
therapeutics (Review). Int J Oncol. 41:393–406. 2012.
|
|
6
|
Pang JC, Chang Q, Chung YF, et al:
Epigenetic inactivation of DLC-1 in supratentorial primitive
neuroectodermal tumor. Hum Pathol. 36:36–43. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Flanagan S, Lee M, Li CC, et al: Promoter
methylation analysis of IDH genes in human gliomas. Front Oncol.
2:1932012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Uhm KO, Lee ES, Lee YM, et al: Aberrant
promoter CpG islands methylation of tumor suppressor genes in
cholangiocarcinoma. Oncol Res. 17:151–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Park SW, Durkin ME, Thorgeirsson SS and
Popescu NC: DNA variants of DLC-1, a candidate tumor suppressor
gene in human hepatocellular carcinoma. Int J Oncol. 23:133–137.
2003.PubMed/NCBI
|
|
10
|
Shi H, Guo J, Duff DJ, et al: Discovery of
novel epigenetic markers in non-Hodgkin’s lymphoma. Carcinogenesis.
28:60–70. 2007.PubMed/NCBI
|
|
11
|
Liao YC, Shih YP and Lo SH: Mutations in
the focal adhesion targeting region of deleted in liver cancer-1
attenuate their expression and function. Cancer Res. 68:7718–7722.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sabbir MG, Wigle N, Loewen S, et al:
Identification and characterization of Dlc1 isoforms in the mouse
and study of the biological function of a single gene trapped
isoform. BMC Biol. 8:172010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Teramoto A, Tsukuda K, Yano M, et al: Less
frequent promoter hypermethylation of DLC-1 gene in primary
breast cancers. Oncol Rep. 12:141–144. 2004.
|
|
14
|
Syed V, Mukherjee K, Lyons-Weiler J, et
al: Identification of ATF-3, caveolin-1, DLC-1, and NM23-H2 as
putative antitumorigenic, progesterone-regulated genes for ovarian
cancer cells by gene profiling. Oncogene. 24:1774–1787. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chan LK, Ko FC, Sze KM, et al:
Nuclear-targeted deleted in liver cancer 1 (DLC1) is less efficient
in exerting its tumor suppressive activity both in vitro and in
vivo. PLoS One. 6:e255472011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hers I, Wherlock M, Homma Y, et al:
Identification of p122RhoGAP (deleted in liver cancer-1) Serine 322
as a substrate for protein kinase B and ribosomal S6 kinase in
insulin-stimulated cells. J Biol Chem. 281:4762–4770. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Durkin ME, Yuan BZ, Zhou X, et al: DLC-1:
a Rho GTPase-activating protein and tumour suppressor. J Cell Mol
Med. 11:1185–1207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim TY, Vigil D, Der CJ and Juliano RL:
Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in
regulation of the cytoskeleton and cell motility. Cancer Metastasis
Rev. 28:77–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hall EH, Daugherty AE, Choi CK, et al:
Tensin1 requires protein phosphatase-1alpha in addition to RhoGAP
DLC-1 to control cell polarization, migration, and invasion. J Biol
Chem. 284:34713–34722. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu JB, Wu XM, Cai J, et al: CpG island
methylator phenotype and Helicobacter pylori infection
associated with gastric cancer. World J Gastroenterol.
18:5129–5134. 2012.PubMed/NCBI
|
|
21
|
Wang MX, Wang HY, Zhao X, et al: Molecular
detection of B-cell neoplasms by specific DNA methylation
biomarkers. Int J Clin Exp Pathol. 3:265–279. 2010.PubMed/NCBI
|
|
22
|
Liu JB, Zhang YX, Zhou SH, et al: CpG
island methylator phenotype in plasma is associated with
hepatocellular carcinoma prognosis. World J Gastroenterol.
17:4718–4724. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ullmannova V and Popescu NC: Expression
profile of the tumor suppressor genes DLC-1 and DLC-2 in solid
tumors. Int J Oncol. 29:1127–1132. 2006.PubMed/NCBI
|
|
24
|
Feng XL, Zhou W, Li H, et al: The DLC-1
-29A/T polymorphism is not associated with nasopharyngeal carcinoma
risk in Chinese population. Genet Test. 12:345–349. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Calvisi DF, Pascale RM and Feo F:
Dissection of signal transduction pathways as a tool for the
development of targeted therapies of hepatocellular carcinoma. Rev
Recent Clin Trials. 2:217–236. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan BZ, Jefferson AM, Baldwin KT, et al:
DLC-1 operates as a tumor suppressor gene in human non-small cell
lung carcinomas. Oncogene. 23:1405–1411. 2004. View Article : Google Scholar : PubMed/NCBI
|