Introduction
Pancreatic cancer is one of the most lethal human
cancers and continues to be a major health problem. Despite
considerable efforts made in the past 50 years, conventional
therapeutic approaches, such as surgery, radiation, chemotherapy,
or combinations of these modalities, have little impact on the
course of this aggressive neoplasm (1). Additionally, current treatments or
therapies used to treat human cancer have low selectivity or
specificity against tumor cells and often result in systemic
toxicity that damages normal healthy tissues. Thus, novel
strategies with enhanced therapeutic efficiency and selectivity for
treating pancreatic cancer are needed.
Stem cells have recently gained considerable
attention from researchers given their possible clinical use for
developing new cancer treatments. While traditional chemotherapy
involves the administration of drugs, genetically engineered stem
cells (GESTECs) can be used to induce the in vivo production
of therapeutic agents (2–7). This technique enables the replacement
of damaged genes or insertion of additional genes with new
functions. For example, human neural stem cells (hNSCs) were found
to have therapeutic potential and tumor tropism for treating
malignant tumors in the human brain including medulloblastomas and
gliomas (8–10). This supports the possibility of
using hNSCs as a carrier to deliver genes to cells in tumor sites
for tumor-specific enzyme/prodrug systems with concomitant prodrug
administration (11).
HB1.F3 cells are immortalized hNSCs derived from the
human fetal brain at 15 weeks of gestation using the amphotropic
replication-incompetent retroviral vector, v-myc (12,13).
Clonal HB1.F3.CD cells are derived from parental HB1.F3 cells
transfected with the Escherichia coli (E. coli)
cytosine deaminase (CD) gene (12).
Additionally, clonal HB1.F3.CD. IFN-β cells are derived from
parental HB1.F3.CD cells, and express both the E. coli CD
and human interferon-β (IFN-β) genes (5). This type of clonally isolated
multipotent HB1.F3 cells has the ability to self-renew and
differentiate into cells of neuronal and glial lineages both in
vivo and in vitro(12).
The CD/5-fluorocytosine (5-FC) system is a gene-directed
enzyme/prodrug therapy (GEPT) (14–18) in
which the non-toxic prodrug 5-FC is converted into a cytotoxic
metabolite, 5-fluorouracil (5-FU) (19,20).
5-FU inhibits DNA synthesis in cells and results in cytotoxicity
(21,22). This GEPT system has been
experimentally tested against several types of tumors including
colorectal and prostate cancers, demonstrating that GEPT appears to
be an effective therapy for these tumors (23–26).
In our previous studies, diverse sources of stem cells, neural,
amniotic-fluid and amniotic membrane, were employed to express
therapeutic genes for selectively targeting different types of
human cancers including primary and metastatic cancers (2–4,7,26–28).
In the present study, we aimed to ascertain whether
2 types of therapeutic stem cells, HB1.F3.CD and HB1.F3.CD.IFN-β,
can selectively migrate toward human pancreatic cancer cells. As a
cancer cell model, human pancreatic carcinoma cells, PANC-1, were
employed to investigate whether therapeutic stem cells migrate and
target this type of cancer in in vitro and in vivo
models. We also evaluated the therapeutic value of these cells in
an enzyme/prodrug system that could serve as a novel pancreatic
cancer therapy. The antitumor and tumor-tropic properties of
GESTECs provide the potential for treating invasive tumors without
any side-effects (8–10,29).
By selectively delivering therapeutic genes to tumor cells, GESTECs
expressing CD and IFN-β may have synergistic antitumor effects
against pancreatic cancer cells.
Materials and methods
Cell culture
The PANC-1 epithelioid cells line derived from human
pancreatic carcinoma of ductal cell origin was obtained from the
Korean Cell Line Bank (Seoul, Korea). The cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%
(v/v) fetal bovine serum (FBS) (both from HyClone Laboratories,
Inc., Logan, UT, USA), 1% penicillin/streptomycin (Cellgro
Mediatech, Inc., Manassas, VA, USA), 1% HEPES (Invitrogen Life
Technologies, Carlsbad, CA, USA), and 0.1% anti-mycoplasma
plasmocin (InvivoGen, San Diego, CA, USA) at 37°C in a humidified
atmosphere of 5% CO2-95% air. HB1.F3, HB1.F3.CD and
HB1.F3.CD.IFN-β cells, along with human dermal fibroblasts (HDFs),
were also cultured in the same media as indicated above. All cells
were trypsinized with 0.05% trypsin/0.02% EDTA (PAA Laboratories,
Austria) in Mg2+/Ca2+-free HBSS.
Reverse transcription (RT)-PCR
To confirm the expression of CD and/or the IFN-β
gene in the HB1.F3.CD and HB1.F3.CD.IFN-β cells, RT-PCR was
performed. The presence of these chemoattractant molecules and
their interaction with specific receptors, such as stem cell factor
(SCF)/c-KIT, CXC chemokine receptor 4 (CXCR4), and vascular
endothelial growth factor (VEGF)/VEGFR1 and VEGFR2 receptors, in
the GESTECs and related ligands in PANC-1 cells were detected by
RT-PCR.
RNA extraction was performed using TRIzol reagent
(Invitrogen Life Technologies). One microgram of total RNA was
reversely transcribed into complementary DNA (cDNA) using murine
leukemia virus reverse transcriptase (MMLV-RT; iNtRON
Biotechnology, Sungnam, Korea), 10 pM dNTPs (Bioneer Co., Daejeon,
Korea), 200 pM nonamer random primers (Takara Bio, Inc., Shiga,
Japan), 0.5 μl RNase inhibitor (iNtRON Biotechnology), and 5X RT
buffer. The cDNA prepared from this procedure was amplified by PCR
performed with 0.2 μmol/l of each reverse and forward primer, 2.5
units of Taq polymerase, 0.2 mmol/l dNTP and 10X PCR buffer (all
from iNtRON Biotechnology). PCR reaction for these chemoattractant
factors (ligands and receptors) and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) as a positive control was carried out in a
PTC-100 thermal cycler (MJ Research Inc., Waltham, MA, USA) with 30
cycles of denaturation at 95°C for 30 sec, annealing at 58°C for 30
sec, and extension at 72°C for 30 sec. The PCR products were
separated on a 1.5% agarose gel containing ethidium bromide (EtBr),
and the results of electrophoresis were analyzed using Gel Doc 2000
(Bio-Rad Laboratories, Hercules, CA, USA). The sequences of the
reverse and forward primers along with the predicted product sizes
are shown in Table I.
 | Table ISequences of the oligonucleotide
primers used in this study and the predicted PCR product sizes. |
Table I
Sequences of the oligonucleotide
primers used in this study and the predicted PCR product sizes.
| mRNA | Oligo-sequences
(5′-3′) | Expected size
(bp) |
|---|
| CD | Forward:
GCGCGAGTCACCGCCAGCCACACCACGGC | 559 |
| Reverse:
GTTTGTATTCGATGGCTTCTGGCTGC | |
| SCF | Forward:
ACTTGGATTCTCACTTGCATTT | 505 |
| Reverse:
CTTTCTCAGGACTTAATGTTGAAG | |
| c-KIT | Forward:
GCCCACAATAGATTGGTATTT | 570 |
| Reverse:
AGCATCTTTACAGCGACAGTC | |
| CXCR4 | Forward:
CTCTCCAAAGGAAAGCGCAGGTGGACAT | 558 |
| Reverse:
AGACTGTACACTGTAGGTGCTGAAATCA | |
| IFN-β | Forward:
AAAGAAGCAGCAATTTTCAG | 296 |
| Reverse:
TTTCTCCAGTTTTTCTTCCA | |
| VEGF | Forward:
AAGCCATCCTGTGTGCCCCTGATG | 377 |
| Reverse:
GCTCCTTCCTCCTGCCCGGCTCAC | |
| VEGFR2 | Forward:
ACGCTGACATGTACGGTCTAT | 438 |
| Reverse:
GCCAAGCTTGTACCATGTGAG | |
| GAPDH | Forward:
ATGTTCGTCATGGGTGTGAACCA | 351 |
| Reverse:
TGGCAGGTTTTTCTAGACGGCAG | |
Cell viability assay
To investigate the effects of 5-FC and 5-FU on
PANC-1 cells, a cell viability assay was conducted. PANC-1 cells
were seeded in 96-well plates (5,000 cells/well) and cultured in
100 μl culture medium supplemented with 5% FBS. Next, 5-FC and 5-FU
(Sigma-Aldrich, St. Louis, MO, USA) were serially diluted with
phosphate-buffered saline (PBS) (final concentration 0.1, 0.3, 0.5,
1 and 5 mmol/l) and were used to treat the cells for 3 days. A
3-(29)-2,5-diphenyltetrazolium
bromide (MTT) assay was performed to measure cell viability on day
5. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
(MTT) solution (10 μl of a 10 mg/ml stock) was added to each well,
and the plates were incubated for 4 h at 37°C. Supernatants were
then removed, and 100 μl of dimethyl sulfoxide (DMSO, 99.0%; Junsei
Chemical Co. Ltd., Tokyo, Japan) was added to each well to dissolve
the resultant formazan crystals. Cell viability of the PANC-1 cells
was measured at 540 nm using an ELISA plate reader (Molecular
Devices, Sunnyvale, CA, USA) in duplicates.
To measure cell growth inhibition resulting from
treatment with 5-FC, PANC-1 cells (1,250 cells/well) were seeded in
96-well plates and cultured in 100 μl cell culture medium with 5%
FBS. After seeding, HB1.F3, HB1.F3.CD or HB1.F3.CD.IFN-β cells
(3,750 cells/well) in 100 μl cell culture medium containing 5% FBS
were added to the cultures and incubated for 24 h. Next, the cells
were treated with different concentrations (0.1, 0.3, 0.5, 1 and 5
mmol/l) of 5-FC for 3 days. A MTT assay was performed in duplicate
to measure cell viability on day 5.
In vitro migration assay
To ascertain whether GESTECs are capable of
migrating toward pancreatic cancer cells, PANC-1 cells and HDFs
(1×105 cells/500 μl/well) were seeded in 24-well plates
in DMEM containing 10% FBS and incubated for 6 h at 37°C. The cells
were then incubated in fresh 2% FBS containing DMEM for 24 h in
37°C. Transwell plates (8 μm; BD Biosciences, Franklin Lakes, NJ,
USA) coated with fibronectin (250 μg/ml; Sigma-Aldrich) were placed
in empty 24-well plates and incubated overnight in 37°C. Using a
general protocol, 2 μM of
chloromethylbenzamido-1,1′-dioctadecyl-3,3,3′-tetramethyl-indocarbocyanine
perchlorate (CM-DiI; Invitrogen Life Technologies) was used to
label the HB1.F3, HB1.F3.CD or HB1.F3.CD.IFN-β cells
(1×105 cells/well) which were plated in the upper
chambers of the Transwell plates (11,26).
The cells were then cultured in DMEM supplemented with 2% FBS for
24 h at 37°C. The PANC-1 cells and HDFs were subsequently stained
with 200 ng/ml 4′,6-diamidino-2-phenylindole solution (DAPI;
Invitrogen Life Technologies) in the dark for 10 min at 37°C. Each
well was then washed with PBS, and the upper side of the Transwell
membrane was scraped to remove cells that had not migrated into the
membrane. Cells stained with CM-DiI and DAPI were examined by
fluorescence microscopy (IX71 inverted microscope; Olympus, Tokyo,
Japan).
Pancreatic cancer xenograft mouse
models
All animal experimental procedures were approved by
the Animal Care Committee of Chungbuk National University.
Twenty-one 6-week-old male BALB/c nude mice were purchased from the
Central Animal Laboratory (SLC, Shizuoka, Japan). The mice were
acclimated to a controlled environment (22–24°C with 40–60%
relative humidity, a 12-h light/dark cycle, and frequent
ventilation) for 1 week prior to the experiments. After this
period, PANC-1 cells (2×106 cells/mouse) were injected
into the subcutaneous dorsal thoracic region of each mouse. The
resulting tumor volume was measured using a caliper every week and
was calculated using the formula: (0.5236 × length × width ×
height).
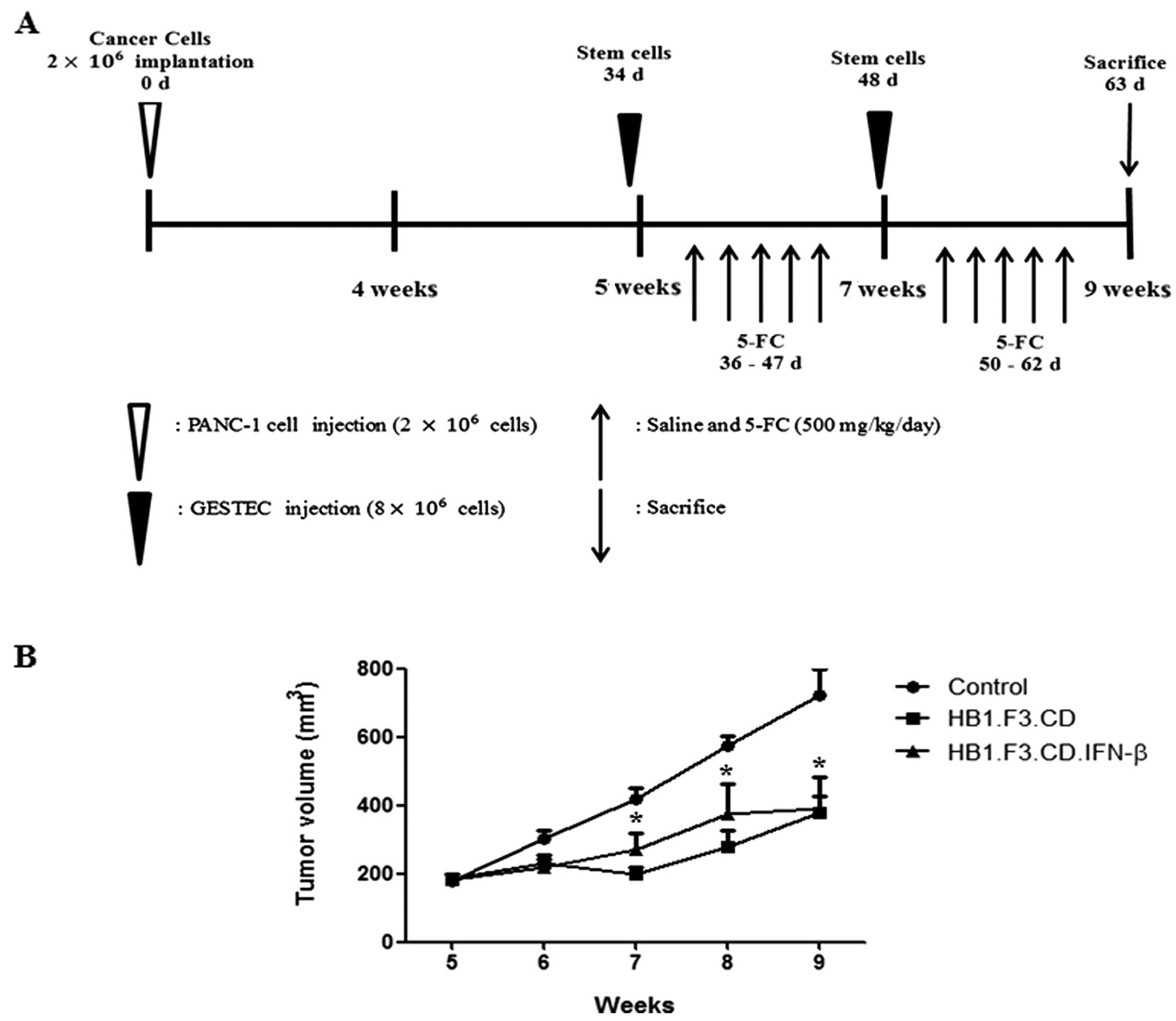
In vivo antitumor effect of GESTECs
To evaluate the therapeutic effects of the GESTECs,
the xenograft mice were divided into 3 groups that were i) treated
with 0.85% saline as a negative control, ii) inoculated with
HB1.F3.CD cells (8×106 cells/mouse/2 weeks) in the
presence of 5-FC (500 mg/kg/day), or iii) inoculated with
HB1.F3.CD.IFN-β cells (8×106 cells/mouse/2 weeks) in the
presence of 5-FC (500 mg/kg/day). The experiment was conducted for
4 weeks after GESTECs injection. After tumor volumes reached 200
mm3, GESTECs stained with CM-DiI were injected in areas
surrounding the tumor mass. Injections were performed twice a week
during the experimental period. Two days after injection of the
GESTECs, the control group received intraperitoneal injections of
normal saline (100 μl), while the other mice with GESTECs were
injected with 5-FC (500 mg/kg/day in 100 μl saline) every day
during the experimental period. One day after the last 5-FC
administration, the mice were sacrificed and tumors were
excised.
Histopathology
Tumors excised from the mice during necropsy were
fixed in a 10% normal formalin solution (Sigma-Aldrich). The fixed
samples were cut into 4- to 6-mm sections, embedded in paraffin,
and cut into 3-μm sections using a microtome (Leica, Wetzlar,
Germany). The sections were mounted on slides and stained with
hematoxylin and eosin (H&E) (Sigma-Aldrich). The stained slides
were then examined by light microscopy using a BX51 microscope
(Olympus).
Fluorescence analysis
To observe localization of the GESTECs in the
xenograft tumor mass, DAPI staining as a counterstaining was
performed on prepared tumor section slides. After rehydration, the
slides were fixed in a 10% normal formalin (Sigma-Aldrich) solution
for 10 min, washed twice in PBS and incubated with DAPI for 10 min
at 37°C. The stained slides were examined with an IX71 inverted
microscope.
Statistical analysis
Data from each experiment are presented as the means
± standard deviation (SD) or standard error of the mean (SEM) in
in vitro and in vivo. Differences between each group
were evaluated with one-way ANOVA and Tukey’s test using Prism
GraphPad (v5.0; GraphPad software, San Diego, CA, USA). P-values
<0.05 were considered to indicate statistically significant
results.
Results
Identification of CD and IFN-β gene
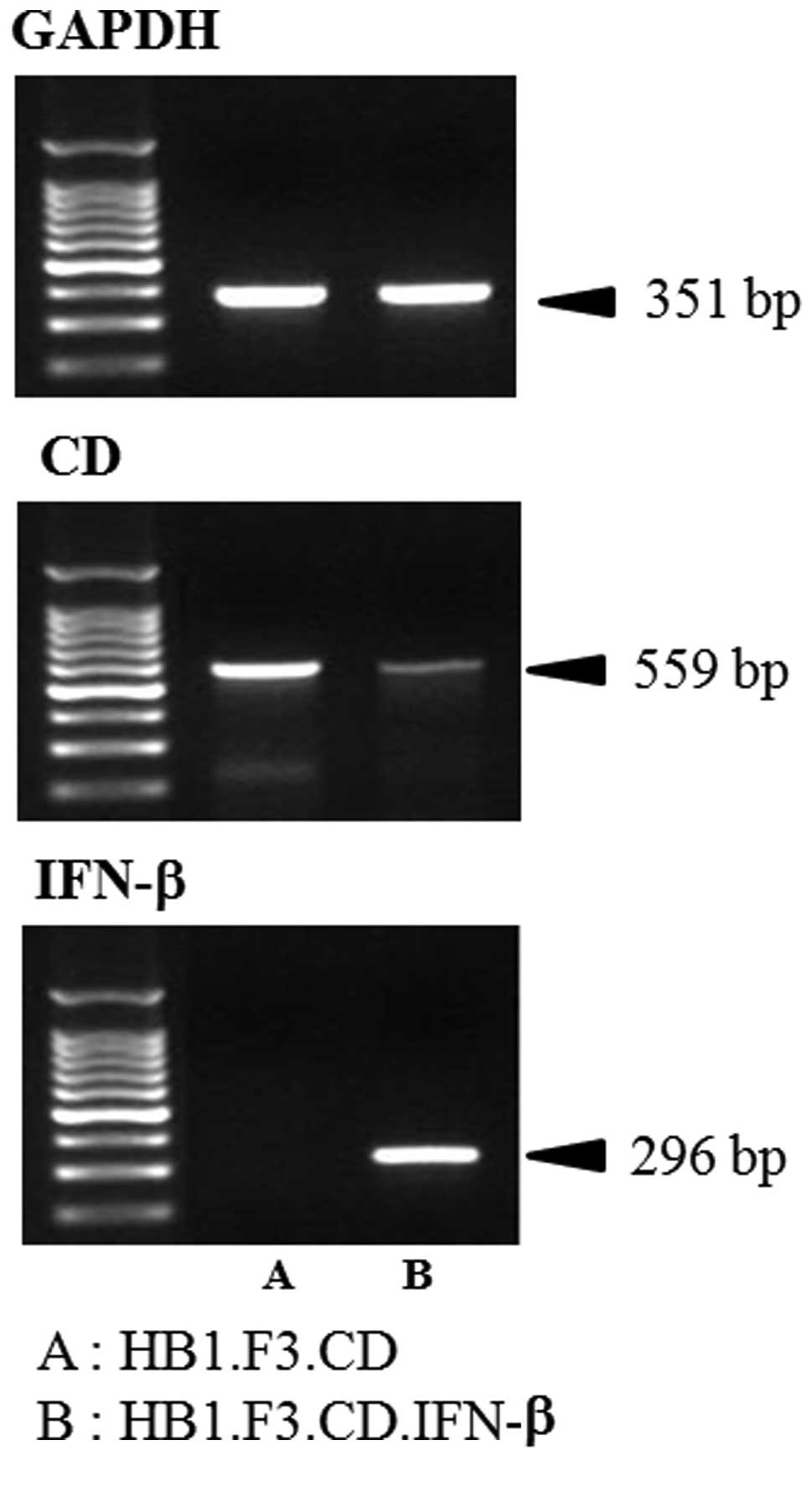
expression in the GESTECs
Expression of CD and IFN-β genes in the HB1.F3.CD
and HB1.F3.CD.IFN-β cells was confirmed by RT-PCR. cDNA synthesized
from GESTECs RNA was amplified. The CD gene (559 bp) was identified
in both HB1.F3.CD and HB1.F3.CD.IFN-β cells as shown in Fig. 1. In addition, the IFN-β gene (296
bp) was observed in the HB1.F3.CD.IFN-β cells but not in the
HB1.F3.CD cells (Fig. 1). GAPDH
(351 bp) was used as an internal control.
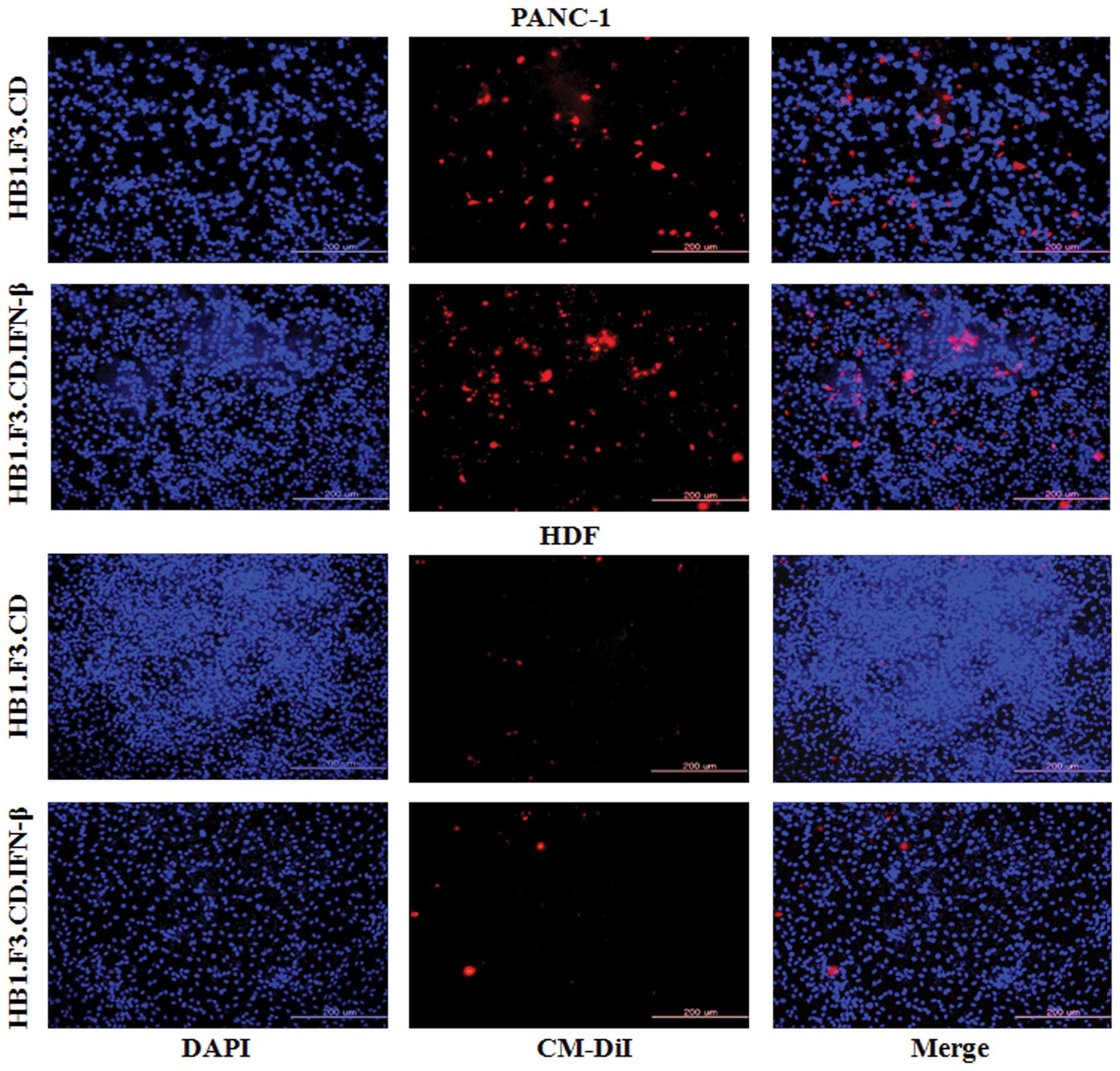
In vitro migration of GESTECs toward
pancreatic cancer cells
To measure the ability of GESTECs to migrate toward
PANC-1 pancreatic cancer cells, a modified Transwell migration
assay was performed. HB1.F3.CD and HB1.F3.CD.IFN-β cells stained
with CM-DiI and DAPI were examined by fluorescence microscopy
(Fig. 2). Compared to DAPI-stained
HDFs as a control, the migratory capacity of GESTECs toward PANC-1
cells was significantly greater (Fig.
2). This result indicates that therapeutic stem cells are
capable of migration towards human PANC-1 pancreatic cancer
cells.
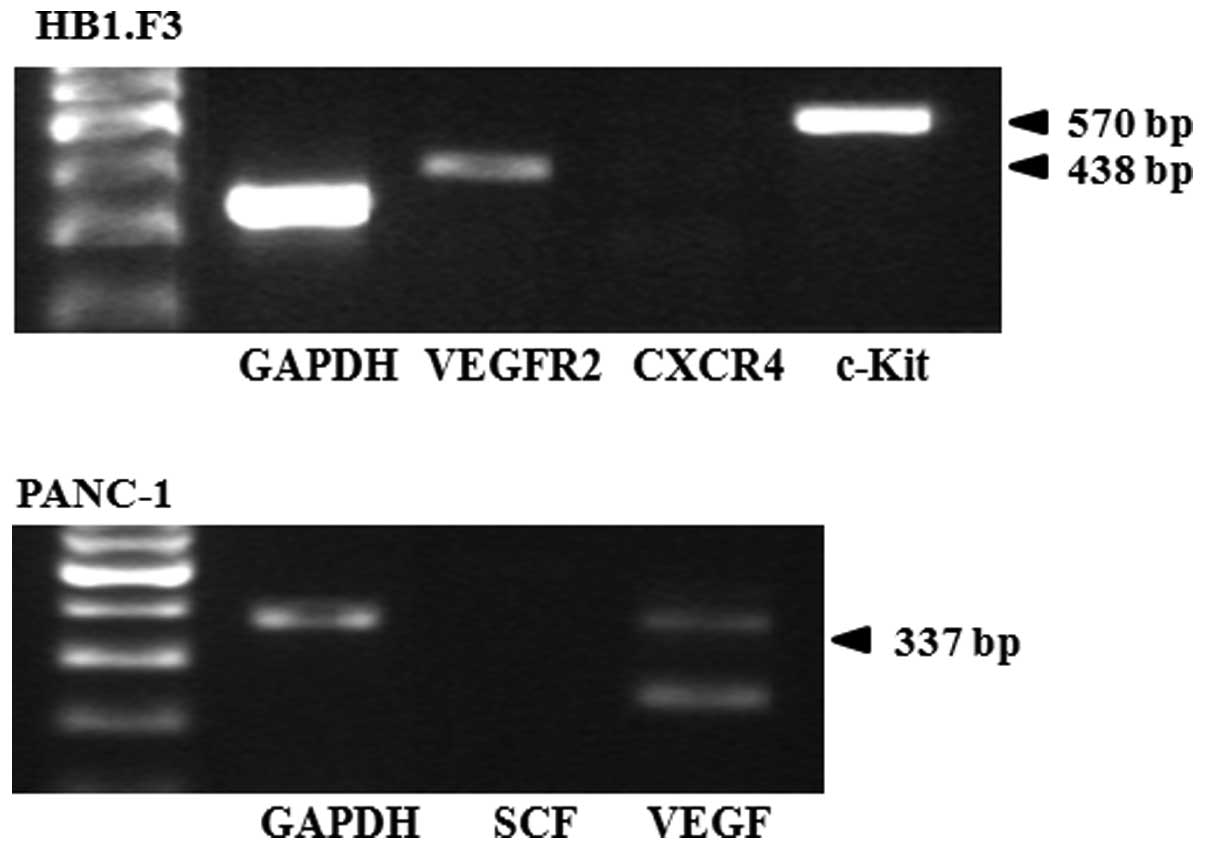
Identification of chemoattractant factors
in PANC-1 cells and GESTECs
To determine whether the PANC-1 cells and GESTECs
express chemoattractant molecules, RT-PCR reaction for several
chemoattractant ligands and their respective receptors was
performed. In the GESTECs, expression of the chemoattractant
receptors VEGFR2 and c-KIT was observed while CXCR4 gene expression
was not found (Fig. 3). In the
PANC-1 cells, VEGF (a chemoattractant ligand) was expressed but SCF
was not (Fig. 3).
Cell viability assay
Cell viability was assessed using two methods.
First, the cytotoxic effects of 5-FC and 5-FU on PANC-1 cells were
assessed. Although treatment with 5-FC did not affect cancer cell
growth, 5-FU noticeably decreased cell proliferation by 85% at a
concentration of 0.5 mmol/l (Fig.
4A). Secondly, the cytotoxic effect of 5-FC at different
concentrations (0.1–5 mmol/l) was examined on PANC-1 cells
co-cultured with GESTECs. PANC-1 cell growth was markedly inhibited
by co-culturing with HB1.F3.CD or HB1.F3.CD.IFN-β cells in the
presence of 5-FC. When the number of GESTECs was fixed, cell
proliferation of PANC-1 was decreased by increasing concentrations
of 5-FC (0.1–5 mmol/l) (Fig. 4B).
When the cytotoxic effects of HB1.F3.CD and HB1.F3.CD.IFN-β were
compared, no statistically significant difference was detected
(Fig. 4B).
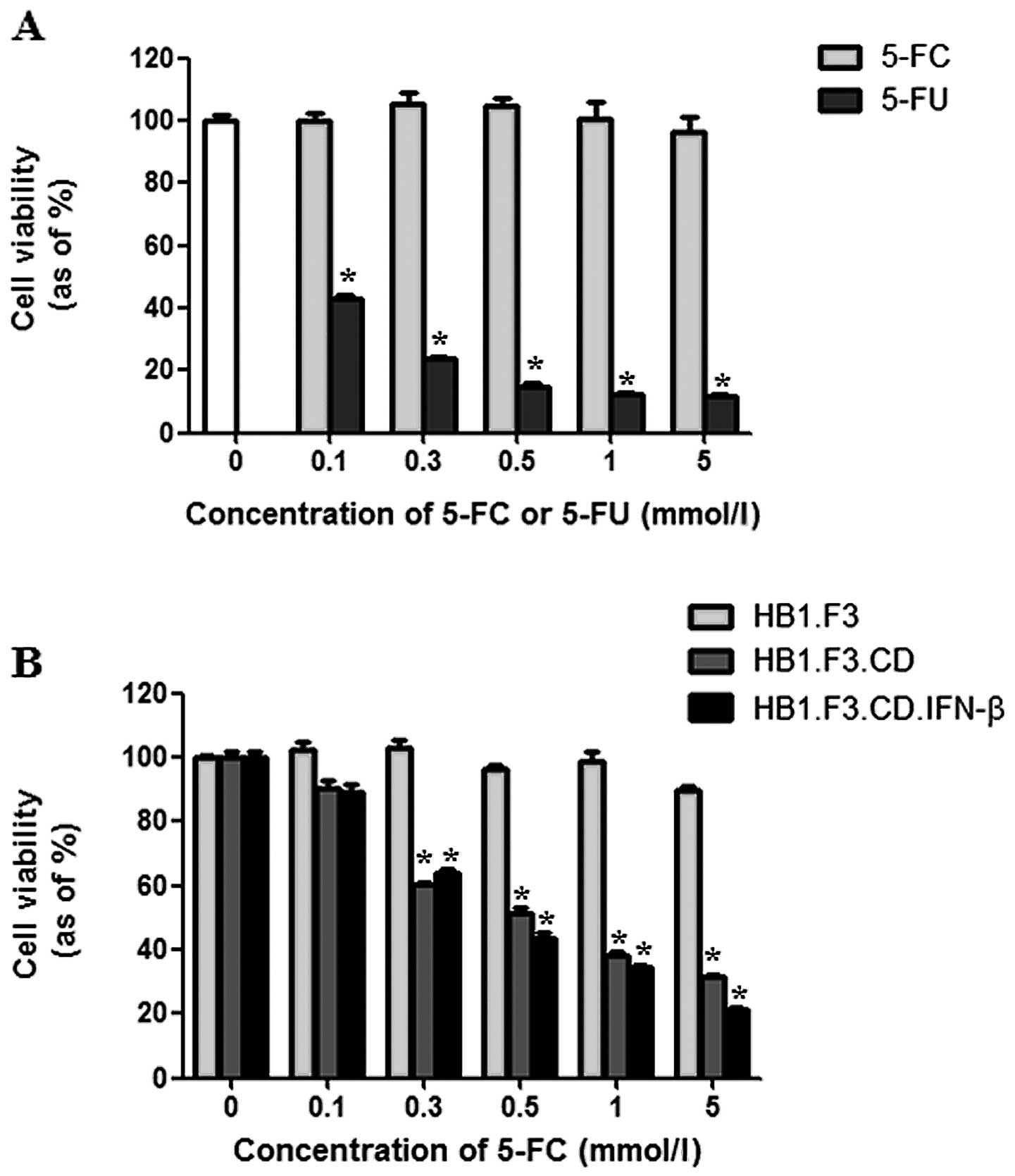 | Figure 4Anticancer effects of GESTECs in
vitro. PANC-1 cells were seeded in 96-well plates and
co-cultured with HB1.F3, HB1.F3.CD or HB1.F3.CD.IFN-β cells
(5×103 cells/well) for 24 h. The cells were then treated
with different concentrations of 5-FC or 5-FU (0, 0.1, 0.3, 0.5, 1
and 5 mmol/l). After 3 days, cell viability was measured with an
MTT assay. (A) The cytotoxic effect of 5-FC or 5-FU at different
concentrations was measured in the PANC-1 cells. (B) PANC-1 cells
(1.25×103 cells/well) were co-cultured with GESTECs
(3.75×103 cells/well) in the presence of increasing
concentrations of 5-FC (0, 0.1, 0.3, 0.5, 1 and 5 mmol/l).
*P<0.05 vs 5-FC or HB1.F3. |
Tumor mass formation in a mouse xenograft
model
PANC-1 cells were injected into the subcutaneous
dorsal thoracic region of 21 BALB/c nude male mice, and the stem
cells were administered into these mice following tumor formation
in the presence of a prodrug, 5-FC (Fig. 5A). Tumor volume (V) was measured
using a caliper every week on the subcutaneous dorsal thoracic area
of each mouse using the formula: V = (0.5236 × length × width ×
height). After tumor formation, pancreatic tumor growth in the mice
was significantly inhibited up to 50% by HB1.F3.CD or
HB1.F3.CD.IFN-β cells in the presence of 5-FC (500 mg/kg/day)
(Fig. 5B). This result indicated
that HB1.F3.CD and HB1.F3.CD.IFN-β cells had effective in
vivo therapeutic activity in the presence of the prodrug 5-FC.
However, a significant synergistic effect of simultaneous CD and
IFN-β expression was not observed when the effect of the
HB1.F3.CD.IFN-β cells were compared with that of the HB1.F3.CD
cells in the in vitro cell viability assay (Fig. 5B).
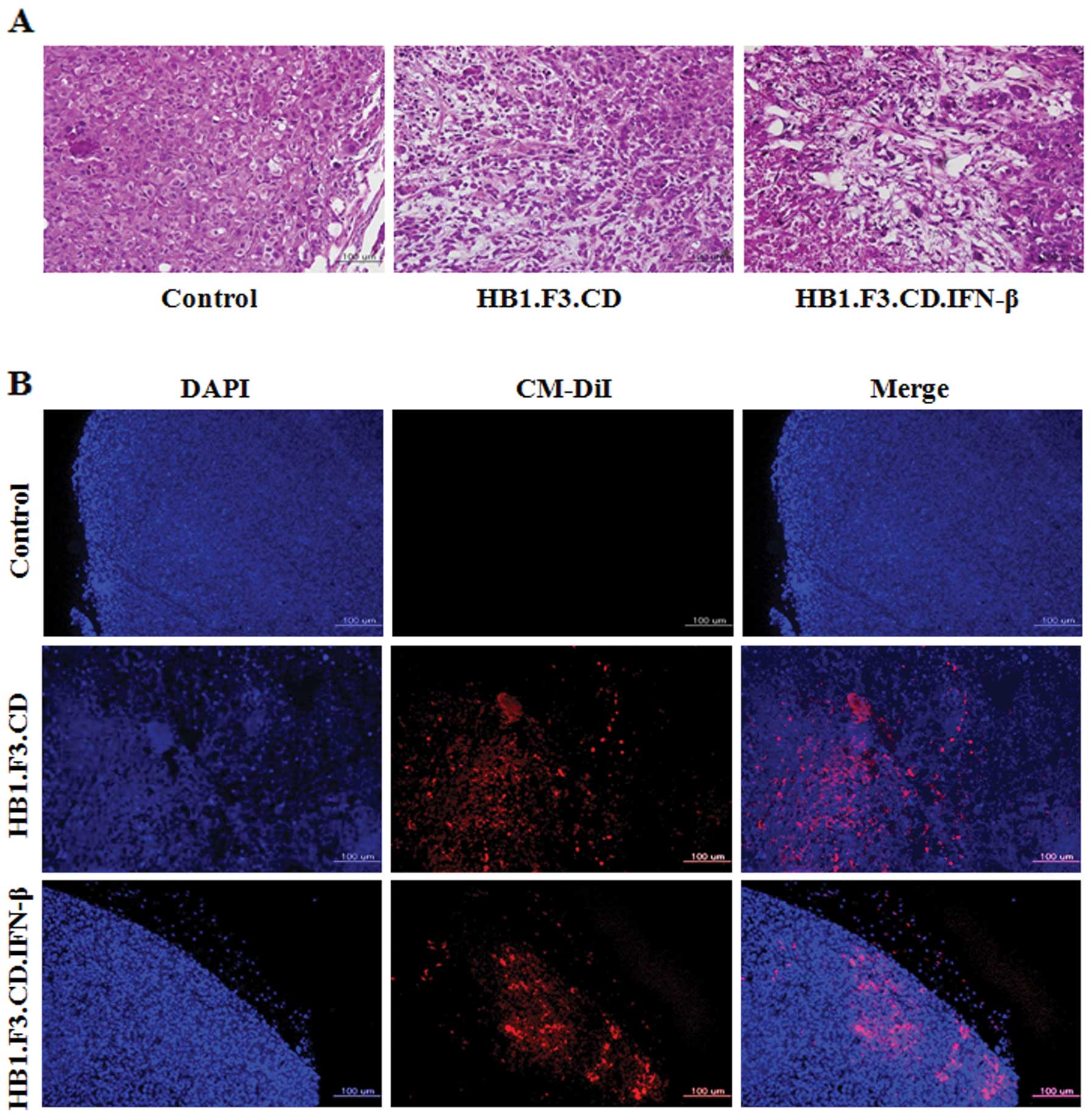
Histological analysis of the pancreatic
tumor mass
To identify changes in histological features of the
pancreatic tumor masses from the mice, a histopathological analysis
was performed on tumor tissues stained with H&E. Typical tumor
morphology, such as active mitosis and a high nucleus/cytoplasm
ratio, was observed in tissues from the control mice that were not
treated with GESTECs (Fig. 6A). In
contrast, the pancreatic tumor masses from mice treated with
HB1.F3.CD or HB1.F3.CD.IFN-β cells in the presence of 5-FC showed
extensive necrosis characterized by pyknosis and destruction of
cell morphology (Fig. 6A).
Fluorescent analysis to detect GESTEC
migration
To explore the mechanisms underlying tumor growth
inhibition by GESTECs, a fluorescence analysis was performed using
GESTECs stained with CM-DiI and DAPI-stained PANC-1. As observed
with fluorescence microscopy (Fig.
6B), CM-DiI-stained GESTECs (red) migrated toward the PANC-1
cells (blue). This result indicates that the GESTECs effectively
migrated toward the pancreatic tumor mass. This migration of
therapeutic stem cells expressing a suicide gene and IFN-β to the
sites of pancreatic tumor formation may induce the antitumor
activity in the presence of a prodrug, 5-FC.
Discussion
Novel strategies that overcome the limits of
conventional cancer therapies such as radiotherapy, chemotherapy
and immunotherapy are being developed. One of these modalities,
stem cell therapy using a GEPT system, has received much attention
as an innovative cancer therapy (30). The CD/5-FC system is a GEPT
(14–18) involving the conversion of a
non-toxic prodrug 5-FC into a cytotoxic metabolite, 5-FU (19,20).
5-FU inhibits DNA synthesis in cells, resulting in cytotoxicity
(21,22). This CD/5-FC GEPT system has been
experimentally tested against several types of tumors including
colorectal and prostate lesions (23–25).
However, GEPT systems have not yet been used clinically for
treating pancreatic cancer since the efficacy of gene delivery
vectors (i.e., adenovirus and lentivirus) remains questionable.
Recent studies have found that immortalized GESTECs
have advantages that may be useful for gene therapy and
neurological cell replacement therapeutic approaches for treating
neurological diseases and injuries (12). These GESTECs were shown to
selectively migrate toward brain tumors and reduced tumor growth
both in in vitro cell models and in vivo mouse models
(9,10). A previous study demonstrated that
when HB1.F3.CD cells expressing the E. coli CD gene are
administered along with 5-FC in an mouse model, tumor size is
decreased but this effect was not observed with 5-FC or HB1.F3 CD
cells alone (10). Another study
reported that GESTECs expressing a fusion gene (HB1.F3.CD.IFN-β)
have antitumor effects unlike the HB1.F3 parental cell line
(31).
In the present study, tumor-tropic effect of
HB1.F3.CD and HB1.F3.CD.IFN-β cells toward PANC-1 cells was
verified by a migration assay. The therapeutic effects of our GEPT
system specific for pancreatic cancer were examined in
vitro. Based on the results of our in vitro experiments,
the therapeutic effect of the GEPT system was evaluated in a
xenograft mouse model injected with pancreatic cancer cells.
In a modified Transwell assay, GESTECs migrated
toward PANC-1 cancer cells but not non-tumorigenic HDFs. Although
the molecular mechanism underlying the tumor-tropism of GESTECs has
not been clearly elucidated, a major factor appears to be
chemoattractant molecules secreted by the pancreatic cancer cells.
Our results showed that the chemoattractant ligands and their
corresponding receptors, i.e., VEGF and VEGFR2 were expressed in
PANC-1 cells and GESTECs, suggesting a migratory capacity of
therapeutic stem cells toward human pancreatic cancer cells. These
chemoattractants are presumed to enable GESTECs to selectively
migrate toward pancreatic cancer cells and deliver therapeutic
genes.
Expression of CD and IFN-β genes in the HB1.F3.CD
and HB1.F3.CD.IFN-β cells was confirmed. These cells were found to
exert a high cytotoxic effect on the PANC-1 pancreatic cancer cells
in the presence of 5-FC. But no significant difference in cell
viability between HB1.F3.CD and HB1.F3.CD.IFN-β was observed. In a
similar study of liver cancer, HB1.F3.CD.IFN-β cells were found to
have a greater inhibitory effect on cancer growth compared to the
HB1.F3.CD cells, indicating that HB1.F3.CD.IFN-β cells have a
synergistic cytotoxic effect in the CD/5-FC system (26,32).
However, PANC-1 cells may not express IFN receptors. In this case,
HB1.F3.CD.IFN-β cells expressing CD and IFN-β would not be likely
to have a synergistic effect (33).
Finally, the therapeutic efficacy of GESTECs was
verified in a xenograft mouse model bearing tumors resulting from
injection with PANC-1 pancreatic cancer cells. Treatment with
HB1.F3.CD or HB1.F3.CD.IFN-β cells resulted in the inhibition of
pancreatic tumor growth up to 50% in the presence of 5-FC compared
to the control. However, no differences between mice receiving
HB1.F3.CD and HB1.F3.CD.IFN-β cell were observed similar to the
in vitro experimental results. Fluorescence microscopy was
used to observe the migration of GESTECs toward the pancreatic
tumor mass. Notably, HB1.F3.CD and HB1.F3.CD.IFN-β cells were found
to have tumor-tropic migration capabilities in vivo.
According to the histopathological analysis, typical tumor
formation and morphology were observed in the mice without GESTECs.
In contrast, the mice treated with HB1.F3.CD or HB1.F3.CD.IFN-β
cells showed extensive tumor necrosis associated with pyknosis and
destruction of cell morphology in the presence of the prodrug 5-FC.
This result implies that GESTECs effectively migrated toward the
pancreatic tumor mass and exerted their anticancer effects through
our GEPT system.
In summary, this present study demonstrated that
GESTECs expressing CD and/or the IFN-β gene can selectively migrate
toward human pancreatic cancer cells in in vitro and mouse
xenograft models. This GEPT system using therapeutic stem cells had
an anti-proliferative effect on pancreatic cancer cells in
vitro. Furthermore, treatment of xenograft mice with GESTECs
expressing the CD suicide gene and IFN-β in the presence of the
prodrug 5-FC had a significant antitumor effect. Taken together,
these results suggest that stem cell therapy using a GEPT system
expressing a suicide gene and IFN-β may have potential as a
clinical therapeutic tool for treating patients suffered from
pancreatic cancer.
Acknowledgements
This study was supported by a grant from the
Next-Generation BioGreen 21 Program (No. PJ009599), Rural
Development Administration, Republic of Korea. In addition, this
study was also supported by Priority Research Centers Program
through the NRF funded by the Ministry of Education, Science and
Technology (2011-0031403).
References
|
1
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar
|
|
2
|
Kang NH, Yi BR, Lim SY, Hwang KA, Baek YS,
Kang KS and Choi KC: Human amniotic membrane-derived epithelial
stem cells display anticancer activity in BALB/c female nude mice
bearing disseminated breast cancer xenografts. Int J Oncol.
40:2022–2028. 2012.
|
|
3
|
Yi BR, Choi KJ, Kim SU and Choi KC:
Therapeutic potential of stem cells expressing suicide genes that
selectively target human breast cancer cells: Evidence that they
exert tumoricidal effects via tumor tropism (Review). Int J Oncol.
41:798–804. 2012.
|
|
4
|
Yi BR, Kim SU, Kim YB, Lee HJ, Cho MH and
Choi KC: Antitumor effects of genetically engineered stem cells
expressing yeast cytosine deaminase in lung cancer brain metastases
via their tumor-tropic properties. Oncol Rep. 27:1823–1828.
2012.
|
|
5
|
Studeny M, Marini FC, Champlin RE,
Zompetta C, Fidler IJ and Andreeff M: Bone marrow-derived
mesenchymal stem cells as vehicles for interferon-β delivery into
tumors. Cancer Res. 62:3603–3608. 2002.
|
|
6
|
Zhang JF, Wei F, Wang HP, Li HM, Qiu W,
Ren PK, Chen XF and Huang Q: Potent anti-tumor activity of
telomerase-dependent and HSV-TK armed oncolytic adenovirus for
non-small cell lung cancer in vitro and in vivo. J Exp Clin Cancer
Res. 29:522010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yi BR, Kang NH, Hwang KA, Kim SU, Jeung EB
and Choi KC: Antitumor therapeutic effects of cytosine deaminase
and interferon-β against endometrial cancer cells using genetically
engineered stem cells in vitro. Anticancer Res. 31:2853–2861.
2011.
|
|
8
|
Aboody KS, Brown A, Rainov NG, Bower KA,
Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM,
Breakefield XO and Snyder EY: Neural stem cells display extensive
tropism for pathology in adult brain: evidence from intracranial
gliomas. Proc Natl Acad Sci USA. 97:12846–12851. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aboody KS, Bush RA, Garcia E, Metz MZ,
Najbauer J, Justus KA, Phelps DA, Remack JS, Yoon KJ, Gillespie S,
Kim SU, Glackin CA, Potter PM and Danks MK: Development of a
tumor-selective approach to treat metastatic cancer. PLoS One.
1:e232006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim SK, Kim SU, Park IH, Bang JH, Aboody
KS, Wang KC, Cho BK, Kim M, Menon LG, Black PM and Carroll RS:
Human neural stem cells target experimental intracranial
medulloblastoma and deliver a therapeutic gene leading to tumor
regression. Clin Cancer Res. 12:5550–5556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim KY, Kim SU, Leung PC, Jeung EB and
Choi KC: Influence of the prodrugs 5-fluorocytosine and CPT-11 on
ovarian cancer cells using genetically engineered stem cells:
tumor-tropic potential and inhibition of ovarian cancer cell
growth. Cancer Sci. 101:955–962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim SU: Human neural stem cells
genetically modified for brain repair in neurological disorders.
Neuropathology. 24:159–171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim SU, Nakagawa E, Hatori K, Nagai A, Lee
MA and Bang JH: Production of immortalized human neural crest stem
cells. Methods Mol Biol. 198:55–65. 2002.PubMed/NCBI
|
|
14
|
Evoy D, Hirschowitz EA, Naama HA, Li XK,
Crystal RG, Daly JM and Lieberman MD: In vivo adenoviral-mediated
gene transfer in the treatment of pancreatic cancer. J Surg Res.
69:226–231. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hirschowitz EA, Ohwada A, Pascal WR, Russi
TJ and Crystal RG: In vivo adenovirus-mediated gene transfer of the
Escherichia coli cytosine deaminase gene to human colon
carcinoma-derived tumors induces chemosensitivity to
5-fluorocytosine. Hum Gene Ther. 6:1055–1063. 1995.PubMed/NCBI
|
|
16
|
Kanai F, Lan KH, Shiratori Y, Tanaka T,
Ohashi M, Okudaira T, Yoshida Y, Wakimoto H, Hamada H, Nakabayashi
H, Tamaoki T and Omata M: In vivo gene therapy for
α-fetoprotein-producing hepatocellular carcinoma by
adenovirus-mediated transfer of cytosine deaminase gene. Cancer
Res. 57:461–465. 1997.
|
|
17
|
Lan KH, Kanai F, Shiratori Y, Okabe S,
Yoshida Y, Wakimoto H, Hamada H, Tanaka T, Ohashi M and Omata M:
Tumor-specific gene expression in carcinoembryonic antigen -
producing gastric cancer cells using adenovirus vectors.
Gastroenterology. 111:1241–1251. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Z, Shanmugam N, Katayose D, Huber B,
Srivastava S, Cowan K and Seth P: Enzyme/prodrug gene therapy
approach for breast cancer using a recombinant adenovirus
expressing Escherichia coli cytosine deaminase. Cancer Gene
Ther. 4:113–117. 1997.PubMed/NCBI
|
|
19
|
Austin EA and Huber BE: A first step in
the development of gene therapy for colorectal carcinoma: cloning,
sequencing, and expression of Escherichia coli cytosine
deaminase. Mol Pharmacol. 43:380–387. 1993.PubMed/NCBI
|
|
20
|
Mullen CA, Kilstrup M and Blaese RM:
Transfer of the bacterial gene for cytosine deaminase to mammalian
cells confers lethal sensitivity to 5-fluorocytosine: a negative
selection system. Proc Natl Acad Sci USA. 89:33–37. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Etienne MC, Cheradame S, Fischel JL,
Formento P, Dassonville O, Renée N, Schneider M, Thyss A, Demard F
and Milano G: Response to fluorouracil therapy in cancer patients:
the role of tumoral dihydropyrimidine dehydrogenase activity. J
Clin Oncol. 13:1663–1670. 1995.PubMed/NCBI
|
|
22
|
Pinedo HM and Peters GF: Fluorouracil:
biochemistry and pharmacology. J Clin Oncol. 6:1653–1664.
1988.PubMed/NCBI
|
|
23
|
Chung-Faye GA, Chen MJ, Green NK, Burton
A, Anderson D, Mautner V, Searle PF and Kerr DJ: In vivo gene
therapy for colon cancer using adenovirus-mediated, transfer of the
fusion gene cytosine deaminase and uracil
phosphoribosyltransferase. Gene Ther. 8:1547–1554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Crystal RG, Hirschowitz E, Lieberman M,
Daly J, Kazam E, Henschke C, Yankelevitz D, Kemeny N, Silverstein
R, Ohwada A, Russi T, Mastrangeli A, Sanders A, Cooke J and Harvey
BG: Phase I study of direct administration of a replication
deficient adenovirus vector containing the E. coli cytosine
deaminase gene to metastatic colon carcinoma of the liver in
association with the oral administration of the pro-drug
5-fluorocytosine. Hum Gene Ther. 8:985–1001. 1997.PubMed/NCBI
|
|
25
|
Freytag SO, Khil M, Stricker H, Peabody J,
Menon M, DePeralta-Venturina M, Nafziger D, Pegg J, Paielli D,
Brown S, Barton K, Lu M, Aguilar-Cordova E and Kim JH: Phase I
study of replication-competent adenovirus-mediated double suicide
gene therapy for the treatment of locally recurrent prostate
cancer. Cancer Res. 62:4968–4976. 2002.PubMed/NCBI
|
|
26
|
Yi BR, Hwang KA, Kang NH, Kim SU, Jeung
EB, Kim HC and Choi KC: Synergistic effects of genetically
engineered stem cells expressing cytosine deaminase and
interferon-β via their tumor tropism to selectively target human
hepatocarcinoma cells. Cancer Gene Ther. 19:644–651. 2012.
|
|
27
|
Kang NH, Hwang KA, Yi BR, Lee HJ, Jeung
EB, Kim SU and Choi KC: Human amniotic fluid-derived stem cells
expressing cytosine deaminase and thymidine kinase inhibits the
growth of breast cancer cells in cellular and xenograft mouse
models. Cancer Gene Ther. 19:412–419. 2012. View Article : Google Scholar
|
|
28
|
Kim KY, Yi BR, Lee HR, Kang NH, Jeung EB,
Kim SU and Choi KC: Stem cells with fused gene expression of
cytosine deaminase and interferon-β migrate to human gastric cancer
cells and result in synergistic growth inhibition for potential
therapeutic use. Int J Oncol. 40:1097–1104. 2012.PubMed/NCBI
|
|
29
|
Schmidt NO, Przylecki W, Yang W, Ziu M,
Teng Y, Kim SU, Black PM, Aboody KS and Carroll RS: Brain tumor
tropism of transplanted human neural stem cells is induced by
vascular endothelial growth factor. Neoplasia. 7:623–629. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nawa A, Tanino T, Luo C, Iwaki M, Kajiyama
H, Shibata K, Yamamoto E, Ino K, Nishiyama Y and Kikkawa F: Gene
directed enzyme prodrug therapy for ovarian cancer: could GDEPT
become a promising treatment against ovarian cancer? Anticancer
Agents Med Chem. 8:232–239. 2008. View Article : Google Scholar
|
|
31
|
Lee DH, Ahn Y, Kim SU, Wang KC, Cho BK,
Phi JH, Park IH, Black PM, Carroll RS, Lee J and Kim SK: Targeting
rat brainstem glioma using human neural stem cells and human
mesenchymal stem cells. Clin Cancer Res. 15:4925–4934. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yi BR, OSN, Kang NH, Hwang KA, Kim SU,
Jeung EB, Kim YB, Heo GJ and Choi KC: Genetically engineered stem
cells expressing cytosine deaminase and interferon-β migrate to
human lung cancer cells and have potentially therapeutic anti-tumor
effects. Int J Oncol. 39:833–839. 2011.
|
|
33
|
Saidi RF, Williams F, Ng J, Danquah G,
Mittal VK, ReMine SG and Jacobs MJ: Interferon receptors and the
caspase cascade regulate the antitumor effects of interferons on
human pancreatic cancer cell lines. Am J Surg. 191:358–363. 2006.
View Article : Google Scholar : PubMed/NCBI
|