Introduction
Rhabdomyosarcoma (RMS) is the most common pediatric
soft tissue sarcoma. RMS comprises 7–8% of all solid malignant
tumors in children and represents approximately two-thirds of all
infant sarcomas diagnosed (1).
There are 2 distinct histopathological subtypes of this malignancy,
embryonal RMS (ERMS) and alveolar RMS (ARMS) (2). In contrast to ERMS, ARMS is
characterized by specific translocations, i.e., t(2;13) (q35;q14)
in 55% of cases and t(1;13) (p36;q14) in 22% of cases (1). Current treatment options include
chemotherapy, complete surgical resection and radiotherapy
(3). However, the prognosis for
patients with advanced-stage RMS is quite poor (4). The main problems with clinical
treatments include metastatic invasion, local tumor recurrence and
multidrug resistance. Therefore, more specific, effective and less
toxic therapies are required.
Numerous novel anticancer agents are currently in
early phase clinical trials. Of these, immunotherapy with specific
monoclonal antibodies (mAbs) seems to be a promising approach
(5). Alemtuzumab, ibritumomab,
rituximab and tositumomab are mAbs already approved for the
targeted treatment of white blood cells in leukemia (US Food and
Drug Administration). Depending on the level of vascularization,
solid tumors may be effectively targeted by bevacizumab, which
inhibits vascular endothelial growth factor-A (5).
Identification of the epidermal growth factor
receptor (EGFR) as an oncogene has led to the development and
approval of panitumumab for the treatment of metastatic colorectal
cancer and trastuzumab in breast cancer therapy (5). Cetuximab, a widely used anti-EGFR
antibody, consists of a chimeric mouse-human mAb directed against
the extracellular domain of EGFR. Cetuximab has been shown to be
particularly effective against colorectal cancer and head and neck
cancer (6–9) and works by blocking EGFR, leading to
inhibition of cell cycle progression (10,11),
angiogenesis, invasion and metastasis (12). Treatment with mAbs increases and
activates pro-apoptotic molecules in tumor cells (11,13)
and enhances cytotoxicity of topotecan (14). Moreover, cetuximab is able to induce
antibody-dependent cell cytotoxicity (ADCC) (15–18)
and is therefore suitable for immunotherapeutic use. Potential
targets for immunotherapy in RMS are not known. The expression of
EGFR has been demonstrated in RMS cell lines and tumors. Moreover,
previous studies have shown that EGFR expression is a marker for
ERMS, with high sensitivity and specificity.
In the present study, we described the distribution
of EGFR in human RMS and evaluated the therapeutic potential of
cetuximab in RMS patients exhibiting overexpression of EGFR,
investigating whether cetuximab affects EGFR-dependent apoptosis
and enhances the antitumor activity of currently used
chemotherapeutic agents in RMS.
Materials and methods
Cell culture and reagents
The ERMS cell lines RD (ATCC, Manassas, VA, USA),
RMS-YM and KYM-1 and the ARMS cell line Rh30 (DSMZ, Braunschweig,
Germany) were cultured in RPMI-1640 medium supplemented with 10%
fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and 1%
penicillin/streptomycin (Biochrom, Berlin, Germany) in a humidified
atmosphere containing 5% CO2 at 37°C. All cells were
mycoplasma negative. Cetuximab (Erbitux, Merck, Lyon, France) was
obtained from Bristol-Myers Squibb Co. Actinomycin D (Cosmegen;
Merck & Co., Inc., Whitehouse Station, NJ, USA) was obtained
from Banyu Pharmaceutical Co., Ltd. (Tokyo, Japan).
Flow cytometric analysis
Trypsinized cells were incubated for 30 min in FACS
buffer (PBS with 2% FBS, 2 mM EDTA, 0.005% NaN3; all
reagents were from Sigma-Aldrich, Munich, Germany) containing 10
μg/ml cetuximab (Merck, Darmstadt, Germany). Excess antibodies were
washed out with FACS buffer, and cells were labeled with
FITC-conjugated goat anti-human IgG (Chemicon, Hofheim, Germany).
Data were acquired with a FACSCalibur machine (Becton-Dickinson,
Heidelberg, Germany) and analyzed by FlowJo software (Tomy Digital
Biology, Co., Ltd., Tokyo, Japan). Controls were acquired using
rituximab (Roche, Mannheim, Germany) or by omitting cetuximab. To
examine the expression of the differentiation and epithelium marker
EGFR in RMS cells, the cells were washed and incubated with mouse
monoclonal antibodies targeting EGFR-PE (Becton-Dickinson) for 30
min at 4°C. Cells were then washed and counterstained with 1 μg/ml
propidium iodide to label the dead cells.
K-ras mutation assay
DNA was purified from RMS cells using the DNeasy
Blood and Tissue kit (Qiagen, Hilden, Germany). Sample DNA was
added to 8 separate reactions. These reaction mixes contained a
single primer set specific for either the wild-type sequence or 1
of 7 mutations in codons 12 and 13. Direct sequencing was conducted
using a BigDye Terminator cycle sequencing kit (Applied Biosystems,
Foster City, CA, USA) and analyzed on an ABI Prism 310 DNA Analyzer
automated sequencer (Applied Biosystems).
Cell viability assay
Cells were plated in 96-well microplates and
cultured for 12 h before exposure to various concentrations of
drugs. Cell viability was quantified using the WST-8 assay,
determined colorimetrically by measuring the optical density (OD)
at a wavelength of 450 nm using a Rainbow Sunrise (Wako Pure
Chemical Industries, Ltd., Osaka, Japan). The concentration
resulting in 50% growth inhibition (IC50) was calculated
for each treatment condition. Data were analyzed to determine the
combination index (CI), a well-established index of the interaction
between 2 drugs. CI values of <1, 1, and >1 indicate
synergistic, additive and antagonistic effects, respectively.
Determination of combination effects
The effects of actinomycin D and cetuximab on growth
inhibition were determined as described by Chou and Talalay
(19). Briefly, the log (fa/fu) was
plotted against the concentration for each compound, alone or in
combination, where fa was the fraction affected and fu was the
fraction unaffected (1-fa) of cells at each concentration. A CI
value <1 represented synergism between actinomycin D and
cetuximab, while values equal to or greater than 1 represented
additive and antagonistic effects, respectively. The CI was
calculated using the Chou-Talalay method in relation to the
fraction of cells affected.
Analysis of apoptosis by flow
cytometry
Cell death was determined through Annexin
V-FITC/propidium iodide staining using the TACS Annexin V-FITC
Apoptosis Detection Kit (R&D Systems, Minneapolis, MN, USA)
according to the manufacturer’s instructions. Following incubation,
cells were processed as indicated by the manufacturer and analyzed
using FITC and propidium iodide detectors in a FACSCalibur flow
cytometer (Becton-Dickinson). Data were analyzed in FlowJo software
(Tomy Digital Biology).
RNA isolation and real-time PCR
Total RNA was extracted from untreated cells using
the RNeasy Micro kit (Qiagen), and cDNA was synthesized using the
Transcriptor High Fidelity cDNA Synthesis kit (Roche) according to
the manufacturer’s instructions. Real-time reverse
transcription-PCR was carried out in an ABI PRISM 7300 Real-time
PCR system (Applied Biosystems). TaqMan gene expression assay
primers and probe mixes were used for glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) and EGFR (assay IDs Hs99999905_m1 and
Hs01076078_m1, respectively; Applied Biosystems). GAPDH was
detected using TaqMan primers and probes and was used as the
control gene. The thermal cycling reaction included incubation at
95°C for 10 min and 40 cycles of 95°C for 15 sec and 60°C for 60
sec. Relative target mRNA expression was determined using the ΔΔCt
method (value obtained by subtracting the Ct value of GAPDH mRNA
from the Ct value of the target mRNA). Data were calculated as the
ratio of target mRNA to GAPDH mRNA using the 2−ΔΔCt
method (20).
Statistical analysis
Determination of the statistical significance of
differences between the gene expression analysis groups was carried
out using the Student’s t-tests in GraphPad Prism 4.00 software
(GraphPad Software Inc., La Jolla, CA, USA). All numeric data are
expressed as the means ± SD. P-values <0.05 were considered to
indicate statistically significant differences.
Results
Expression of EGFR and mutational status
of K-ras
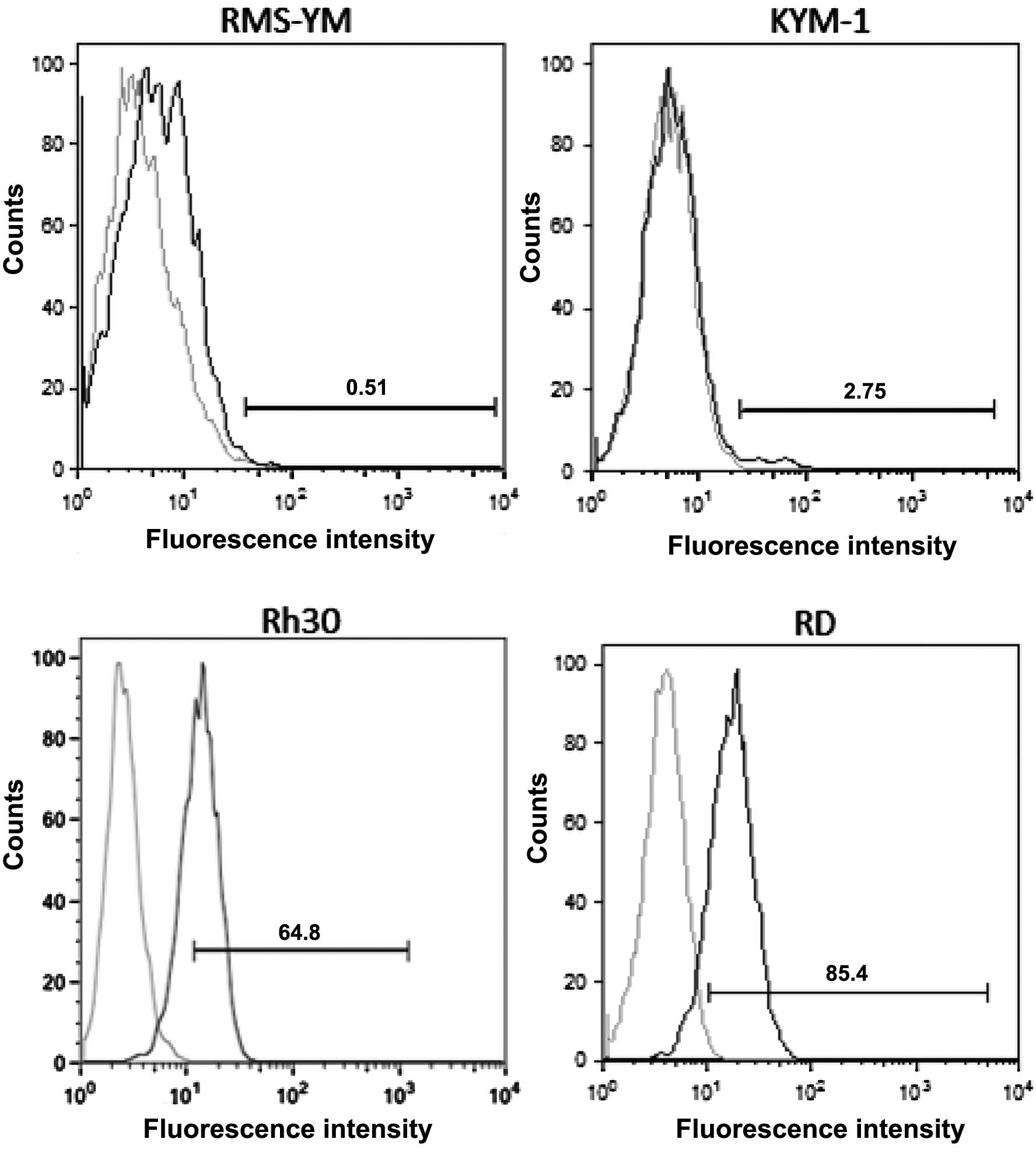
The RD and Rh30 cell lines had a large number of
EGFR-positive cells, whereas the KYM-1 and RMS-YM cell lines had a
small number of EGFR-positive cells (Fig. 1, Table
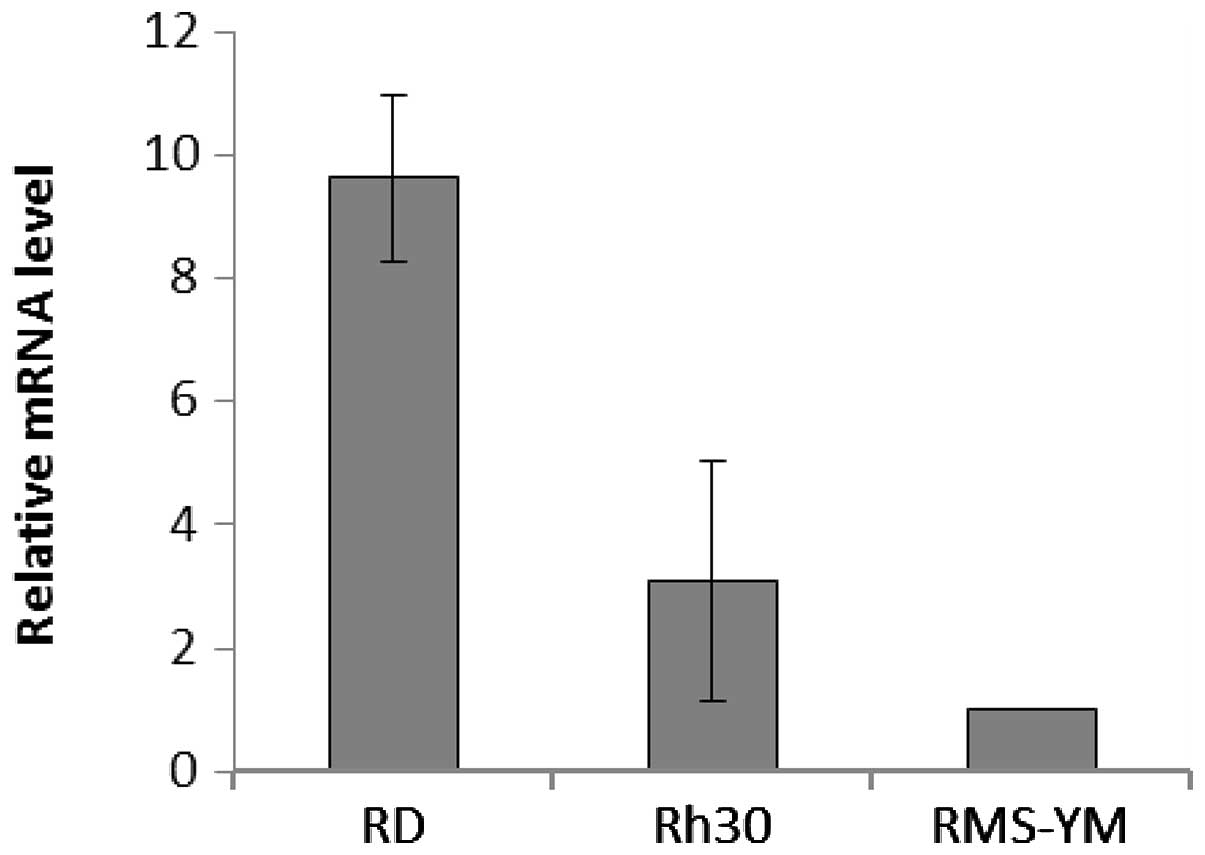
I). Real-time PCR analyses showed that EGFR was overexpressed
by 9.62±1.36- and 3.09±1.93-fold in RD and Rh30 cells,
respectively, compared with RMS-YM cells; EGFR was not detected in
KYM-1 cells in this assay (Fig. 2).
Collectively, these data suggest that EGFR is predominantly
expressed in RD and Rh30 cells. Sequencing of full-length cDNAs
revealed no mutations in codons 12 and 13 of K-ras, suggesting that
all 4 RMS cell lines expressed wild-type K-ras (data not
shown).
 | Table IPercentage of EGFR-positive cells in
the 4 RMS cell lines. |
Table I
Percentage of EGFR-positive cells in
the 4 RMS cell lines.
| Cell line | Means ± SD |
|---|
| RD | 70.0±0.91% |
| Rh30 | 65.0±0.78% |
| KYM-1 | 0.493±0.066% |
| RMS-YM | 15.9±0.32% |
Cetuximab inhibits cell growth in RMS
cell lines
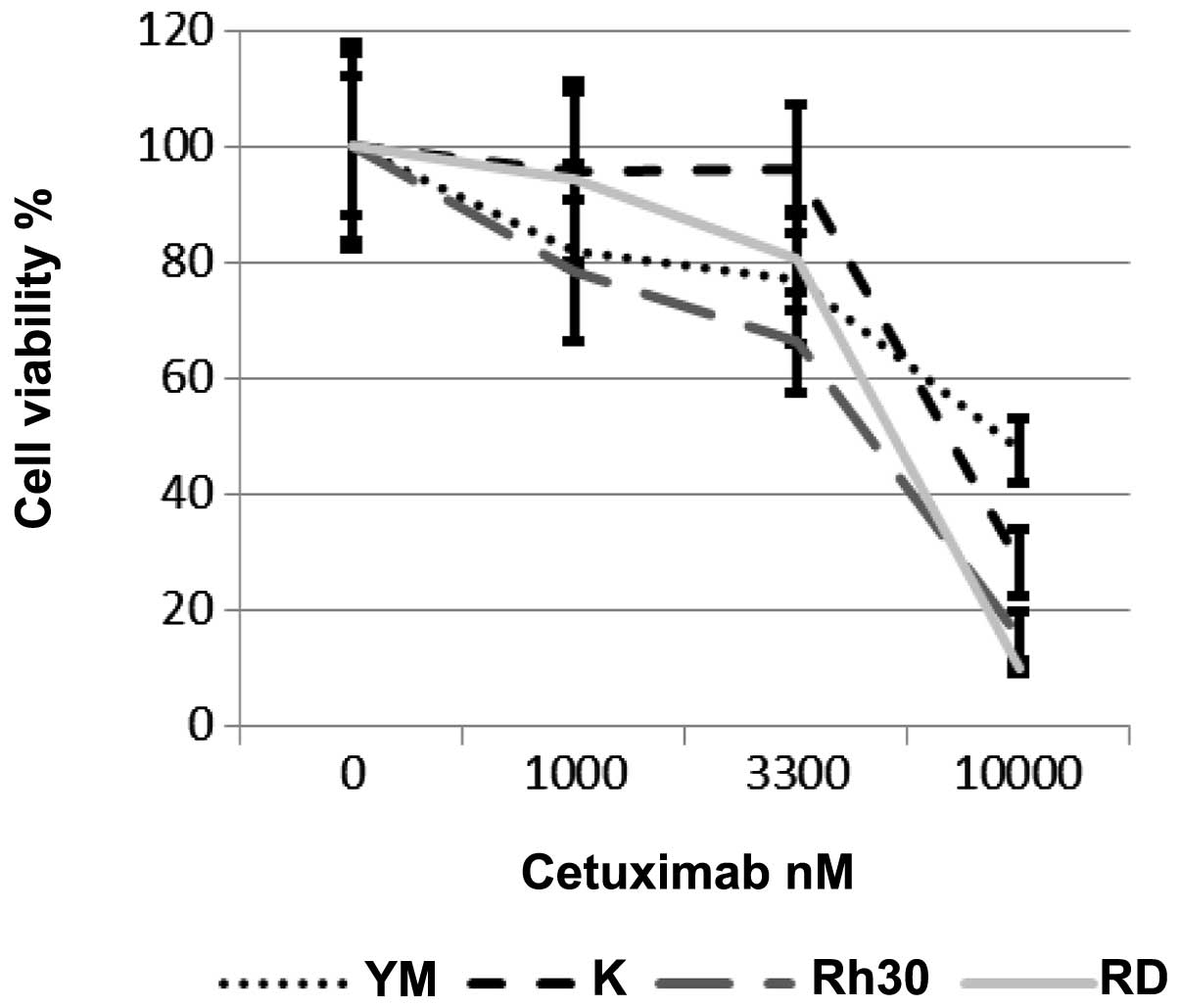
Next, we examined the effects of cetuximab on the
growth of RMS cells. Cetuximab dose-dependently inhibited the
growth of all 4 RMS cell lines, regardless of their EGFR-expression
status (Fig. 3). The
IC50 values of cetuximab in these 4 cell lines were 4.7
μM (Rh30), 5.3 μM (RD), 9.1 μM (RMS-YM) and 7.0 μM (KYM-1)
(Table II). IC50 value
were lower in EGFR-positive cells.
| Table IIThe IC50 values of
cetuximab in the 4 RMS cell lines. |
Table II
The IC50 values of
cetuximab in the 4 RMS cell lines.
| Cell line | IC50
nM |
|---|
| RD | 5333 |
| Rh30 | 4697 |
| KYM-1 | 6989 |
| RMS-YM | 9119 |
Cetuximab enhances actinomycin
D-dependent cytotoxicity in RMS cell lines expressing high levels
of EGFR
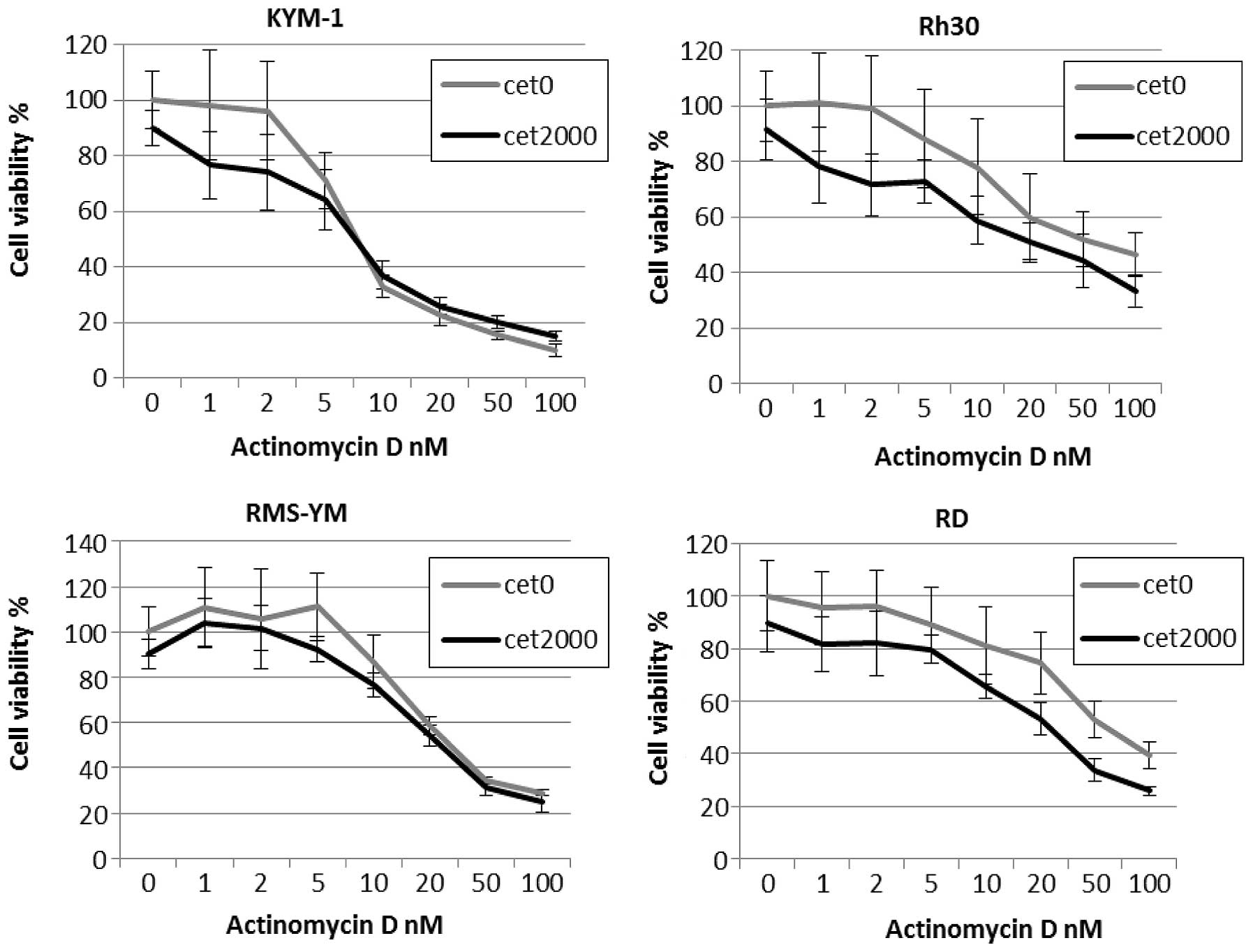
Incubation with 2 μmol/l cetuximab alone had only a
slight effect on the viability of the 4 RMS cell lines, while
combination treatment with cetuximab and actinomycin D enhanced
drug-induced cytotoxicity in EGFR-amplified cell lines (Fig. 4). This combination effect was
synergistic for cetuximab and actinomycin D in RD and Rh30 cells,
with CI values <1.0 for both cell lines. By contrast, the
combination effect was antagonistic for cetuximab and actinomycin D
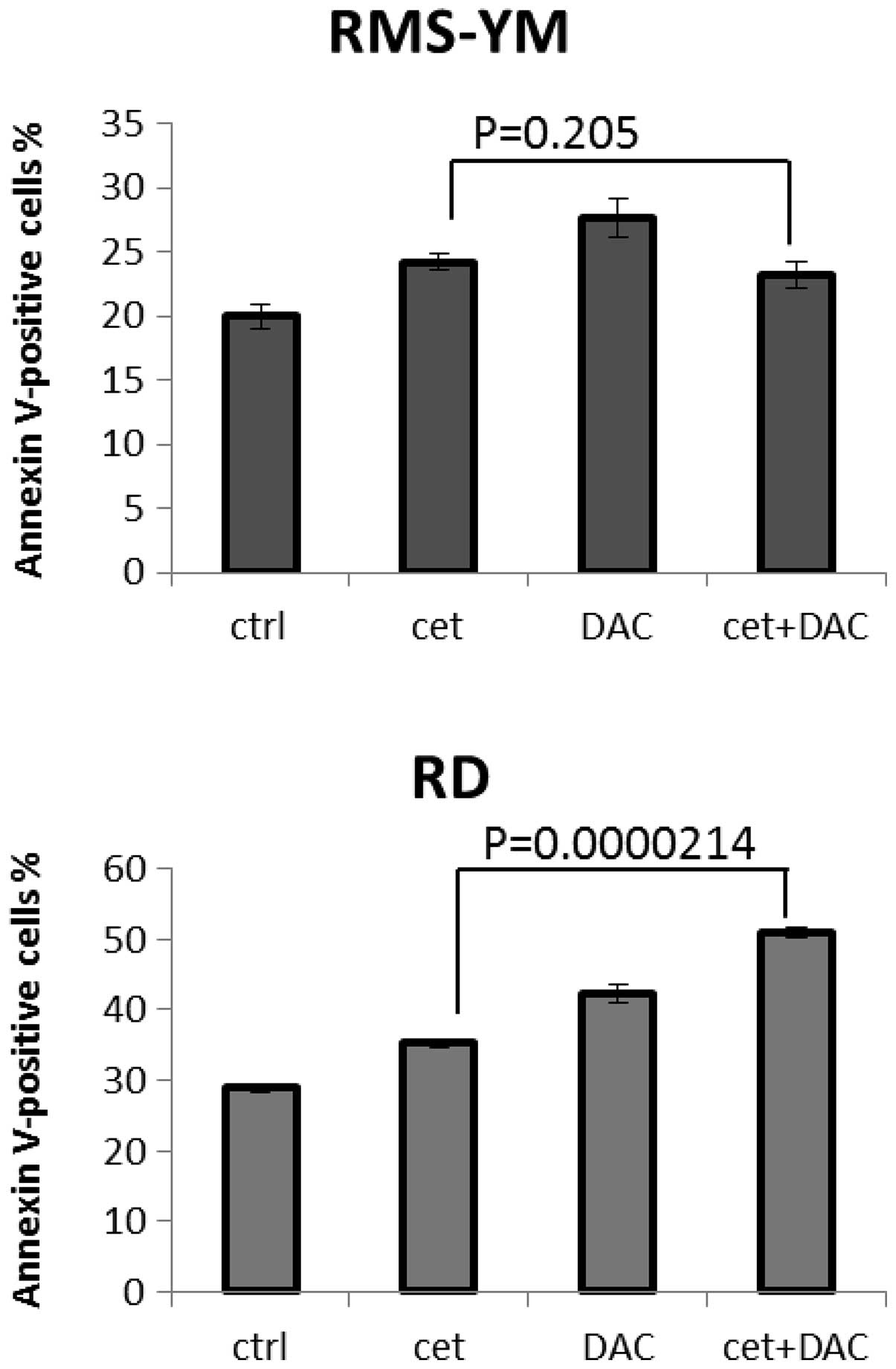
in RMS-YM and KYM-1 cells, with CI values >1.0 (Table III). Moreover, combination
treatment with cetuximab and actinomycin D induced apoptosis in
EGFR-positive RMS cells. At 72 h after treatment, a greater number
of Annexin V-positive cells were detected in RD cells treated with
both cetuximab and actinomycin D than in RD cells treated with
cetuximab alone (P=0.000214, t-test, statistical significance). The
increased apoptosis induced by treatment with these 2 agents was
less pronounced in RMS-YM cells than when treated with cetuximab
alone (P=0.205, t-test, not significant) (Fig. 5).
| Table IIICombination index values determined
for cetuximab-actinomycin D combinations for the 4 RMS cell
lines. |
Table III
Combination index values determined
for cetuximab-actinomycin D combinations for the 4 RMS cell
lines.
| Cell line | Combination
index |
|---|
| RD | 0.774 |
| Rh30 | 0.789 |
| KYM-1 | 1.265 |
| RMS-YM | 1.072 |
Discussion
Although inhibition of EGFR has shown promise as a
potential therapeutic treatment in several epithelial malignancies,
little is known about its effect on soft tissue tumors. The largest
case series reported in the literature demonstrated positive EGFR
staining in 60% of human adult soft tissue tumors (n=281) (21). Studies in RMS have shown that
expression of EGFR correlates with embryonal subtype (22). It is well established that embryonal
subtypes generally behave less aggressively than alveolar subtypes.
However, despite advances in the treatment of RMS, the overall
5-year failure-free survival rate does not exceed 80%, even among
patients with ERMS or early-stage disease. Novel approaches for the
treatment of RMS are required. In the present study, we chose
cetuximab, an mAB targeting EGFR that has already been approved for
therapeutic applications.
Array-based analysis revealed higher expression of
several genes, including BCL2L1 (23), CNR1 (24), CXCR4 (25), MET (26), MYCN (27,28),
PDGFR-A (29) and TFAP2(β)
(30,31), in ARMS compared with ERMS.
Conversely, EGFR (28), HMGA2
(26) and YB-1 (32) were upregulated in ERMS. Of these
gene products, CNR1, CXCR4, MET, PDGFR-A and EGFR are localized to
the cell membrane and may function as targets for therapeutic
antibodies. Our results showed that one ERMS and one ARMS cell
line, RD and Rh30, have high EGFR protein expression assessed by
flow cytometry, and high expression at the mRNA level by real-time
PCR, as previously reported (33).
Mutations in K-ras, which have been reported to
occur in colorectal cancer, are responsible for cetuximab
resistance in tumor cells. Prior to treatment with cetuximab, K-ras
mutations must be monitored (34).
However, in a study of RMS tissues, in which a response to blocking
antibodies such as cetuximab could be expected, K-ras mutations
were detected in only 2 out of 38 ERMS tissues and in no ARMS
tissues (n=12) (35). Additionally,
RD cells contain only an NRAS mutation (36), and our data showed no mutations in
K-ras for RD, RH30, KYM-1 and RMS-YM cells. Although mutations in
K-ras are rare in RMS, K-ras mutations should be evaluated prior to
treatment for effective treatment with cetuximab.
Actinomycin D is used in current standard treatments
for RMS, in combination with vincristine and cyclophosphamide.
Herrmann et al(33) reported
that cell-dependent cytotoxicity of peripheral blood mononuclear
cells to RD and Rh30 was enhanced specifically by cetuximab.
Herein, we evaluated the treatment effects of cetuximab alone as
well as the combination with cetuximab and antitumor reagent. A low
concentration of cetuximab in combination with actinomycin D had an
enhanced antitumor effect. The combination with cetuximab and
actinomycin D was synergistic in inhibiting cell growth, and
inducing cell apoptosis, with a CI of <1 in EGFR amplified
cells, RD and Rh30. By contrast, the combination was antagonistic
in RMS cells without EGFR amplified, with a CI of >1. Apoptotic
cells without EGFR amplified RMS, treated with cetuximab and
actinomycin D, were fewer than those treated with actinomycin D
alone (Fig. 5), suggesting that the
combination of cetuximab and standard chemotherapy including
actinomycin D may be a promising therapeutic strategy for patients
with EGFR amplified RMS, but not for patients without EGFR
amplified RMS. Previous studies reported that activation of EGFR
leads to downstream signaling that activates mitogenic and survival
pathways, such as the MAPK and Pi3-K/AKT pathways (37). Inhibition of these pathways by an
EGFR antagonist, such as cetuximab, can lead to induction of
apoptosis and anti-proliferative effects (38). These results suggest that
combination therapy may block the signaling pathways downstream of
EGFR.
In summary, we have shown that combination of
cetuximab and actinomycin D resulted in antitumor activity against
human RMS cell lines expressing high levels of EGFR, suggesting
that EGFR antagonists may be promising therapeutic interventions
for the treatment of RMS. Further animal studies and clinical
trials are required to evaluate the safety of EGFR antagonists.
Acknowledgements
The RMS cell lines were a kind gift from Dr Junko
Takita (Department of Pediatrics, Graduate School of Medicine,
University of Tokyo, Tokyo, Japan) and Dr Hajime Hosoi (Department
of Pediatrics, Graduate School of Medical Science, Kyoto
Prefectural University of Medicine, Kyoto, Japan). This study was
supported by a grant-in-aid for young scientists (B) from the
Ministry of Education, Culture, Sports, Science and Technology
(MEXT), Japan.
References
|
1
|
McDowell HP: Update on childhood
rhabdomyosarcoma. Arch Dis Child. 88:354–357. 2003. View Article : Google Scholar
|
|
2
|
Pappo AS, Shapiro DN, Crist WM and Maurer
HM: Biology and therapy of pediatric rhabdomyosarcoma. J Clin
Oncol. 13:2123–2139. 1995.PubMed/NCBI
|
|
3
|
Wolden SL, Anderson JR, Crist WM, Breneman
JC, Wharam MD Jr, Wiener ES, Qualman SJ and Donaldson SS:
Indications for radiotherapy and chemotherapy after complete
resection in rhabdomyosarcoma: a report from the Intergroup
Rhabdomyosarcoma Studies I to III. J Clin Oncol. 17:3468–3475.
1999.PubMed/NCBI
|
|
4
|
Koscielniak E, Morgan M and Treuner J:
Soft tissue sarcoma in children: prognosis and management. Paediatr
Drugs. 4:21–28. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma WW and Adjei AA: Novel agents on the
horizon for cancer therapy. CA Cancer J Clin. 59:111–137. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Capdevila J, Elez E, Macarulla T, Ramos
FJ, Ruiz-Echarri M and Tabernero J: Anti-epidermal growth factor
receptor monoclonal antibodies in cancer treatment. Cancer Treat
Rev. 35:354–363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cunningham D, Humblet Y, Siena S, Khayat
D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype
C, Chau I and Van Cutsem E: Cetuximab monotherapy and cetuximab
plus irinotecan in irinotecan-refractory metastatic colorectal
cancer. N Engl J Med. 351:337–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herbst RS, Arquette M, Shin DM, Dicke K,
Vokes EE, Azarnia N, Hong WK and Kies MS: Phase II multicenter
study of the epidermal growth factor receptor antibody cetuximab
and cisplatin for recurrent and refractory squamous cell carcinoma
of the head and neck. J Clin Oncol. 23:5578–5587. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vermorken JB: Squamous cell carcinoma of
the head and neck. J BUON. 7:311–317. 2002.PubMed/NCBI
|
|
10
|
Peng D, Fan Z, Lu Y, DeBlasio T, Scher H
and Mendelsohn J: Anti-epidermal growth factor receptor monoclonal
antibody 225 up-regulates p27KIP1 and induces
G1 arrest in prostatic cancer cell line DU145. Cancer
Res. 56:3666–3669. 1996.PubMed/NCBI
|
|
11
|
Wu X, Fan Z, Masui H, Rosen N and
Mendelsohn J: Apoptosis induced by an anti-epidermal growth factor
receptor monoclonal antibody in a human colorectal carcinoma cell
line and its delay by insulin. J Clin Invest. 95:1897–1905. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Perrotte P, Matsumoto T, Inoue K, Kuniyasu
K, Eve BY, Hicklin DJ, Radinsky R and Dinney CPN: Anti-epidermal
growth factor receptor antibody C225 inhibits angiogenesis in human
transitional cell carcinoma growing orthotopically in nude mice.
Clin Cancer Res. 5:257–265. 1999.PubMed/NCBI
|
|
13
|
Liu B, Fang M, Lu Y, Mendelsohn J and Fan
Z: Fibroblast growth factor and insulin-like growth factor
differentially modulate the apoptosis and G1 arrest induced by
anti-epidermal growth factor receptor monoclonal antibody.
Oncogene. 20:1913–1922. 2001. View Article : Google Scholar
|
|
14
|
Ciardiello F, Bianco R, Damiano V, De
Lorenzo S, Pepe S, De Placido S, Fan Z, Mendelsohn J, Bianco AR and
Tortora G: Antitumor activity of sequential treatment with
topotecan and anti-epidermal growth factor receptor monoclonal
antibody C225. Clin Cancer Res. 5:909–916. 1999.PubMed/NCBI
|
|
15
|
Kawaguchi Y, Kono K, Mimura K, Sugai H,
Akaike H and Fujii H: Cetuximab induce antibody-dependent cellular
cytotoxicity against EGFR-expressing esophageal squamous cell
carcinoma. Int J Cancer. 120:781–787. 2007. View Article : Google Scholar
|
|
16
|
Kimura H, Sakai K, Arao T, Shimoyama T,
Tamura T and Nishio K: Antibody-dependent cellular cytotoxicity of
cetuximab against tumor cells with wild-type or mutant epidermal
growth factor receptor. Cancer Sci. 98:1275–1280. 2007. View Article : Google Scholar
|
|
17
|
Kurai J, Chikumi H, Hashimoto K, Yamaguchi
K, Yamasaki A, Sako T, Touge H, Makino H, Takata M, Miyata M,
Nakamoto M, Burioka N and Shimizu E: Antibody-dependent cellular
cytotoxicity mediated by cetuximab against lung cancer cell lines.
Clin Cancer Res. 13:1552–1561. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naramura M, Gillies SD, Mendelsohn J,
Reisfeld RA and Mueller BM: Therapeutic potential of chimeric and
murine anti-(epidermal growth factor receptor) antibodies in a
metastasis model for human melanoma. Cancer Immunol Immunother.
37:343–349. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sato O, Wada T, Kawai A, Yamaguchi U,
Makimoto A, Kokai Y, Yamashita T, Chuman H, Beppu Y, Tani Y and
Hasegawa T: Expression of epidermal growth factor receptor, ERBB2
and KIT in adult soft tissue sarcomas: a clinicopathologic study of
281 cases. Cancer. 103:1881–1890. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ganti R, Skapek SX, Zhang J, Fuller CE, Wu
J, Billups CA, Breitfeld PP, Dalton JD, Meyer WH and Khoury JD:
Expression and genomic status of EGFR and ErbB-2 in alveolar and
embryonal rhabdomyosarcoma. Mod Pathol. 19:1213–1220. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Margue CM, Bernasconi M, Barr FG and
Schäfer BW: Transcriptional modulation of the anti-apoptotic
protein BCL-XL by the paired box transcription factors PAX3 and
PAX3/FKHR. Oncogene. 19:2921–2929. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Laé M, Ahn EH, Mercado GE, Chuai S, Edgar
M, Pawel BR, Olshen A, Barr FG and Ladanyi M: Global gene
expression profiling of PAX-FKHR fusion-positive alveolar and
PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. J Pathol.
212:143–151. 2007.PubMed/NCBI
|
|
25
|
Tomescu O, Xia SJ, Strezlecki D,
Bennicelli JL, Ginsberg J, Pawel B and Barr FG: Inducible
short-term and stable long-term cell culture systems reveal that
the PAX3-FKHR fusion oncoprotein regulates CXCR4, PAX3, and PAX7
expression. Lab Invest. 84:1060–1070. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Taulli R, Scuoppo C, Bersani F, Accornero
P, Forni PE, Miretti S, Grinza A, Allegra P, Schmitt-Ney M,
Crepaldi T and Ponzetto C: Validation of Met as a therapeutic
target in alveolar and embryonal rhabdomyosarcoma. Cancer Res.
66:4742–4749. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mercado GE, Xia SJ, Zhang C, Ahn EH,
Gustafson DM, Laé M, Ladanyi M and Barr FG: Identification of
PAX3-FKHR-regulated genes differentially expressed between alveolar
and embryonal rhabdomyosarcoma: focus on MYCN as a biologically
relevant target. Genes Chromosomes Cancer. 47:510–520. 2008.
View Article : Google Scholar
|
|
28
|
Williamson D, Lu YJ, Gordon T, Sciot R,
Kelsey A, Fisher C, Poremba C, Anderson J, Pritchard-Jones K and
Shipley J: Relationship between MYCN copy number and expression in
rhabdomyosarcomas and correlation with adverse prognosis in the
alveolar subtype. J Clin Oncol. 23:880–888. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Taniguchi E, Nishijo K, McCleish AT,
Michalek JE, Grayson MH, Infante AJ, Abboud HE, Legallo RD, Qualman
SJ, Rubin BP and Keller C: PDGFR-A is a therapeutic target in
alveolar rhabdomyosarcoma. Oncogene. 27:6550–6560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Davicioni E, Anderson MJ, Finckenstein FG,
Lynch JC, Qualman SJ, Shimada H, Schofield DE, Buckley JD, Meyer
WH, Sorensen PHB and Triche TJ: Molecular classification of
rhabdomyosarcoma - genotypic and phenotypic determinants of
diagnosis: a report from the Children’s Oncology Group. Am J
Pathol. 174:550–564. 2009.PubMed/NCBI
|
|
31
|
Ebauer M, Wachtel M, Niggli FK and Schäfer
BW: Comparative expression profiling identifies an in vivo target
gene signature with TFAP2B as a mediator of the survival function
of PAX3/FKHR. Oncogene. 26:7267–7281. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oda Y, Kohashi K, Yamamoto H, Tamiya S,
Kohno K, Kuwano M, Iwamoto Y, Tajiri T, Taguchi T and Tsuneyoshi M:
Different expression profiles of Y-box-binding protein-1 and
multidrug resistance-associated proteins between alveolar and
embryonal rhabdomyosarcoma. Cancer Sci. 99:726–732. 2008.
View Article : Google Scholar
|
|
33
|
Herrmann D, Seitz G, Warmann SW, Bonin M,
Fuchs J and Armeanu-Ebinger S: Cetuximab promotes immunotoxicity
against rhabdomyosarcoma in vitro. J Immunother. 33:279–286. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lièvre A, Bachet J-B, Le Corre D, Boige V,
Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, Rougier
P, Penault-Llorca F and Laurent-Puig P: KRAS mutation status is
predictive of response to cetuximab therapy in colorectal cancer.
Cancer Res. 66:3992–3995. 2006.PubMed/NCBI
|
|
35
|
Wellcome Trust Sanger Institute. Catalogue
of Somatic Mutations in Cancer. http://www.sanger.ac.uk/genetics/CGP/cosmic/.
Accessed June 4, 2012
|
|
36
|
Chen Y, Takita J, Hiwatari M, Igarashi T,
Hanada R, Kikuchi A, Hongo T, Taki T, Ogasawara M, Shimada A and
Hayashi Y: Mutations of the PTPN11 and RAS genes in
rhabdomyosarcoma and pediatric hematological malignancies. Genes
Chromosomes Cancer. 45:583–591. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bancroft CC, Chen Z, Yeh J, Sunwoo JB, Yeh
NT, Jackson S, Jackson C and Van Waes C: Effects of pharmacologic
antagonists of epidermal growth factor receptor, PI3K and MEK
signal kinases on NF-kappaB and AP-1 activation and IL-8 and VEGF
expression in human head and neck squamous cell carcinoma lines.
Int J Cancer. 99:538–548. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Raymond E, Faivre S and Armand JP:
Epidermal growth factor receptor tyrosine kinase as a target for
anticancer therapy. Drugs. 60(Suppl 1): 15–23. 2000. View Article : Google Scholar : PubMed/NCBI
|