Introduction
Autophagy is primarily a protective process for the
cell (1). Basal levels of autophagy
play a critical role in maintaining normal cellular homeostasis by
recycling intracellular components (1,2).
Currently, the role of autophagy has been extended to human disease
and physiology (3). Autophagy was
first associated with cancer through the identification and
characterization of the Beclin-1 gene, which is thought to be a
tumor suppressor (4). Subsequently,
several autophagy genes have been determined to play a role in
tumorigenesis (3,5). Atg proteins are essential for
autophagy (6). The C-terminus of
newly synthesized microtubule-associated protein light chain 3
(LC3), a well-known Atg protein, is cleaved to generate LC3-I.
Subsequently, 22 amino acids from the C-terminus are removed to
produce LC3-II, which is recruited to form autophagosomes and
serves as a marker of autophagy (7).
Mitochondria are essential and delicate organelles
in eukaryotic cells. They function as chemical factories for key
metabolic reactions and energy generation and as a communication
site for diverse signaling pathways (8). Mitochondrial damage may result in
dysfunctional mitochondrial proteins and mitochondrial DNA (mtDNA),
sometimes leading to cell death by promoting the intrinsic
apoptotic pathways (9). Therefore,
accurate control of mitochondrial quality and quantity is necessary
for energy metabolism homeostasis and other essential cellular
processes. Multiple lines of evidence indicate that the selective
degradation of mitochondria by autophagy controls mitochondrial
number and health (10,11). Mitochondrial autophagy (termed
mitophagy) plays a vital role in selectively degrading superfluous
or severely damaged mitochondria (12,13).
Since mitochondria have their own genome,
mitochondrial gene knockout cells are utilized to investigate
interactions between nuclear and mitochondrial genomes in
mitochondrial disease (14). During
the establishment of human lung cancer cell lines lacking mtDNA, a
progressive depopulation of mitochondria was observed. In the
process, autophagy was determined to be over-activated. In the
present study, we demonstrated that ethidium bromide (EtBr)-induced
selective degradation of mitochondria occurred via autophagy. This
process was regulated by the phosphatidylinositol 3-kinase
(PI3K)-Beclin-1 signaling pathway. EtBr-induced mitochondrial
autophagy reduced lung cancer cell growth in vitro and in
vivo.
Materials and methods
Cell culture
The A549, SPC-A1 and H322 human lung cancer cell
lines were obtained from the American Type Culture Collection
(ATCC). Cells were cultured in ATCC-recommended medium supplemented
with 10% FBS and 100 ng/ml penicillin and streptomycin. The medium
was replaced every other day. For nutrient deprivation, cells were
incubated in medium without serum or glucose for 18 h. For EtBr
treatment, cells were exposed to 250 ng/ml EtBr (Biosharp, Korea)
for 7 days. The medium was supplemented with 50 μg/ml uridine and
100 μg/ml pyruvate.
Laser scanning confocal microscopy
Cells were loaded with 200 nM tetramethylrhodamine
methyl ester (TMRM, T668) or 200 nM LysoTracker Red (LTR, L7528;
both from Invitrogen) for 20 min. In other experiments, cells were
co-loaded with 200 nM MitoTracker Green (MTG, M7514; Invitrogen)
and 200 nM red-fluorescing LTR for 20 min. After fluorescence
loading, cells were washed thrice with fresh phosphate-buffered
saline (PBS). Confocal images were captured at 2 μm intervals using
a Leica laser scanning confocal microscope with a Plan Apochromat
oil immersion objective lens. The images were merged using LAS AF
Lite software (Leica, Germany). Image analysis was performed with
Image-Pro Plus5.1 software (IPP, USA).
Transmission electron microscopy
(TEM)
Cells were fixed in ice-cold electron
microscopy-grade glutaraldehyde, rinsed with PBS, post-fixed with
1% OsO4 in 0.1% potassium ferricyanide, dehydrated in a
graded series of ethanol and embedded in Epon. Ultrathin sections
were cut with a diamond knife, stained with 2% uranyl acetate and
Reynold’s lead citrate and examined using a Philips EM420
transmission electron microscope.
GFP-LC3 transfection
Cells were transiently transfected with pEGFP-C1-LC3
(Yingrun Biotechnology, China) using Lipofectamine 2000
(Invitrogen). After 24 h, cells were cultured in nutrient-deprived
medium or treated with EtBr. Cells were fixed in 4% formaldehyde
for 20 min, washed with PBS, stained with DAPI and observed under a
Leica laser scanning confocal microscope.
Western blot analysis
Cells were lysed with SDS lysis buffer (Beyotime,
China) containing a protease inhibitor. The protein concentration
was measured by the BCA method (Beyotime). An equivalent amount of
each denatured protein sample was separated by 12% SDS-PAGE and
electrophoretically transferred onto polyvinylidene difluoride
membranes. After blocking with 5% non-fat milk for 1 h at room
temperature, LC3B (ab48394; Abcam) and Beclin-1 (Epitomics, USA)
antibodies were incubated with the membranes at 4°C overnight. The
membranes were subsequently washed for 30 min in TBS-Tween 20,
incubated with an HRP-conjugated secondary antibody (Beyotime) for
1 h, and observed using chemiluminescence (Beyotime).
Cell proliferation, clonogenic and
migration assays
Cells treated with EtBr for 1, 3, 5 or 7 days were
trypsinized and counted. For cell proliferation assays, cells were
seeded in 96-well plates (0.5×104/well) and incubated
overnight. WST-1 (10 μl/well; Beyotime) was added and the cells
were incubated at 37°C for 1–2 h. Differences in absorbance were
measured using a microplate reader. For clonogenic assays, cells
were seeded in 6-well plates (103/well) and then
incubated in selective growth medium (supplemented with 50 μg/ml
uridine and 100 μg/ml pyruvate) for 10 days. Cells were fixed with
methanol for 15 min and stained with 0.1% crystal violet for 10
min. Colonies were visualized and counted using light
microscopy.
For cell migration assays, cells (1×104;
100 μl) were seeded in the upper chambers of 24-well Transwell
plates (8-μm pore size; Corning, NY, USA), and medium supplemented
with 12% FBS (800 μl) was added to the bottom chambers. After 20–22
h, non-migrated cells on the upper side of the filter membrane were
gently removed. Cells that had migrated to the lower side of the
insert membrane were fixed with methanol for 15 min and stained
with 1% crystal violet for 10 min. The migrated cells were counted
in five random fields using a microscope.
In vivo analysis of tumor growth
The animal studies were performed in accordance with
the guidelines of the Third Military Medical University Animal Care
and Use Committee. NOD/SCID mice (5 mice/group) were injected
subcutaneously in the left flank with 1×106 EtBr-treated
cells suspended in 200 μl PBS. Tumor volume was measured with
calipers twice a week for 6 weeks, after which the mice were
sacrificed. Tumors were removed and photographed.
Statistical analysis
Data are presented as the mean ± SD and the 95%
confidence interval. Independent t-tests were utilized to evaluate
the data. Statistical significance was defined as P<0.05.
Results
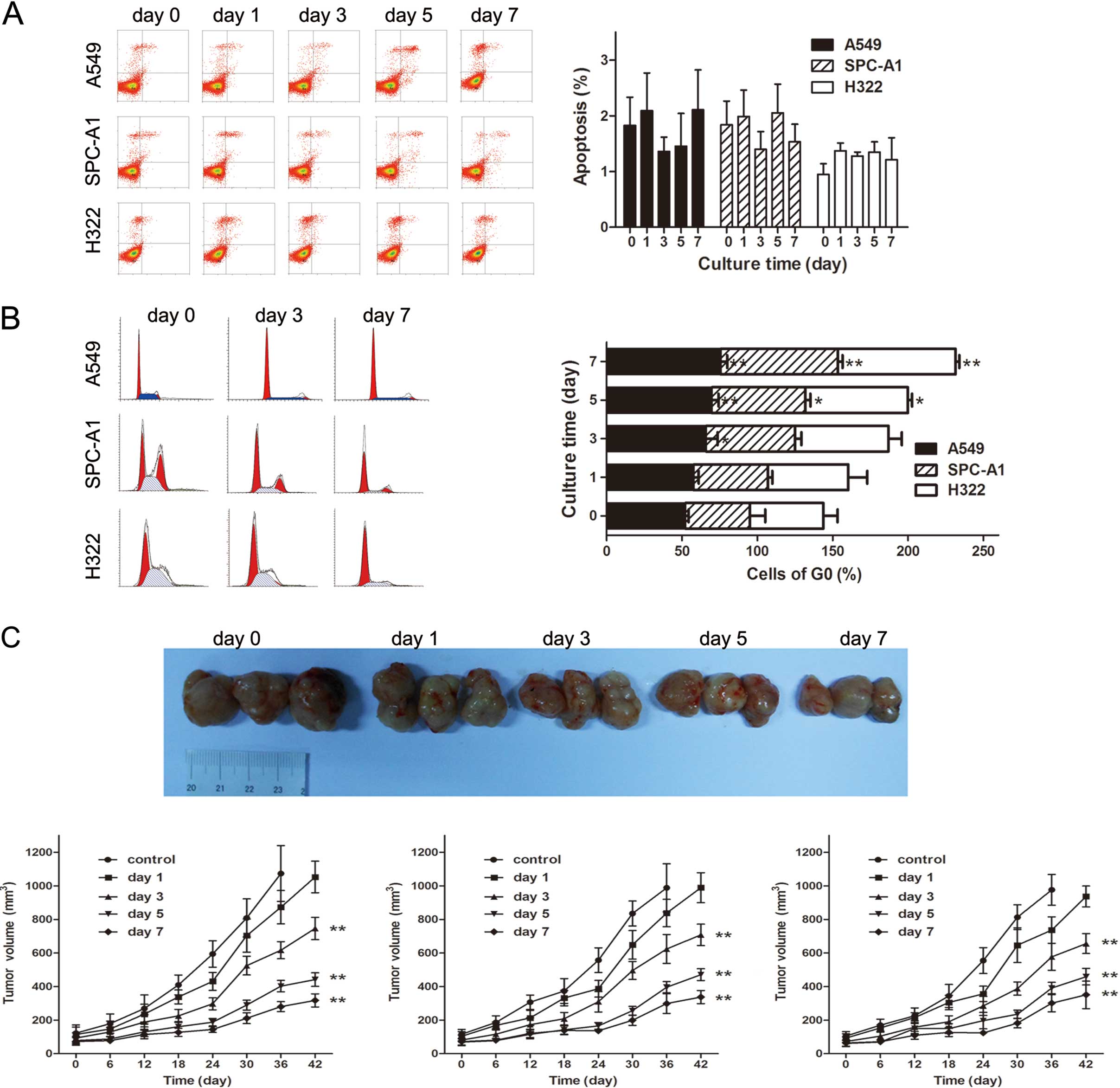
EtBr inhibits lung cancer cell growth in
vitro and in vivo without significantly inducing apoptosis
During the process of knocking out mitochondrial
genes, the biological behavior of EtBr-treated cells was assessed.
In vitro cell proliferation, clonogenic and migration assays
demonstrated that EtBr inhibited lung cancer cell growth and
migration in a time-dependent manner, but there was no significant
increase in apoptotic events in EtBr-treated cells (Fig. 1A). The PI staining results
demonstrated that EtBr-treated cells underwent cell cycle arrest
(Fig. 1B). In vivo,
EtBr-treated cells grew more slowly than untreated control cells in
the lung cancer xenograft models (Fig.
1C).
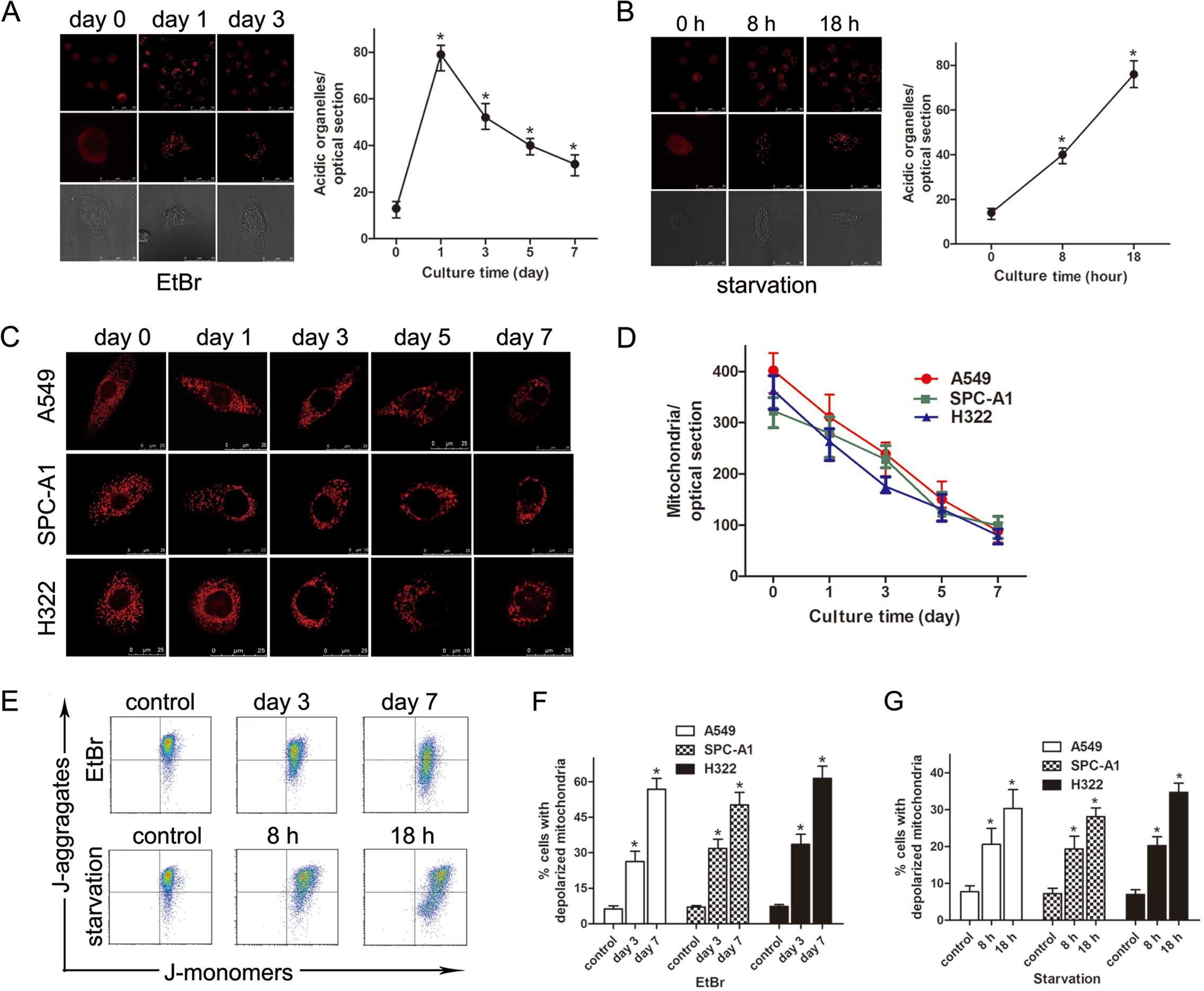
Increased LTR uptake and reduced TMRM
fluorescence in EtBr-treated cells
At different time points after exposure to 250 ng/ml
EtBr, A549, SPC-A1 and H322 cells were loaded with Lyso-Tracker Red
(LTR) to detect acidic organelles. Compared with the control cells,
the number of LTR-labeled organelles in EtBr-treated cells
significantly increased from 13±2.7 to 78±4.2 per optical section
by day 1 but decreased to 52±4.3 by day 3 (n=5 optical sections;
P<0.01) (Fig. 2A). The same
changes were observed in the nutrient-deprived cells (n=5 optical
sections; P<0.01) (Fig. 2B).
Subsequently, cells were loaded with TMRM to detect polarized
mitochondria. The results indicated that the mitochondrial membrane
potential (MMP) decreased in a time-dependent manner (n=5 optical
sections; P<0.05) (Fig. 2C and
D). The decreased MMP was confirmed by flow cytometry using
JC-1 staining (Fig. 2E–G).
Alterations in mitochondrial size and shape were identified by TMRM
microscopy. The mitochondria, typically homogeneous in size, became
large and branched after treatment with EtBr.
Since a significant depopulation of polarized
mitochondria was observed in EtBr-treated cells, the mtDNA content
and the mRNA expression of cytochrome c oxidase subunit II
(COX II) were measured by quantitative real-time PCR. The results
demonstrated that these two markers significantly decreased in a
time-dependent manner after EtBr treatment (data not shown).
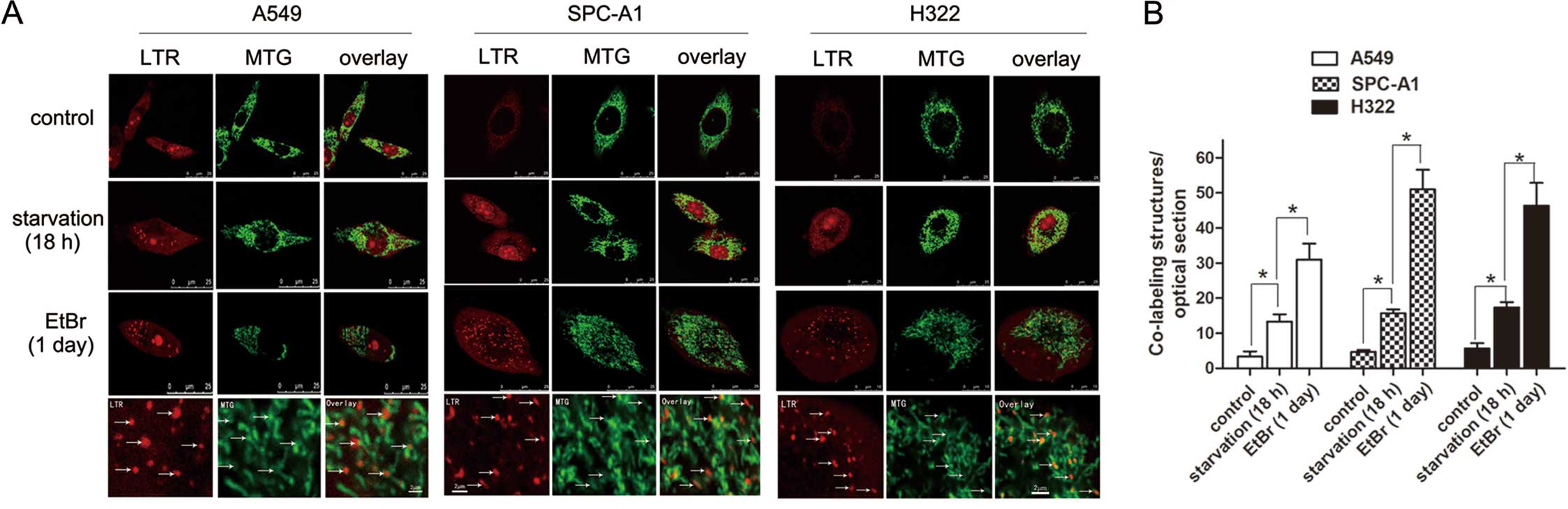
Mitochondrial degradation by LTR-labeled
organelles in cells treated with EtBr
To observe the movement of mitochondria into acidic
lysosomal structures in EtBr-treated cells, cells were co-loaded
with red-fluorescing LTR and green-fluorescing MTG. As illustrated
in Fig. 3, there was little
co-localization of LTR- and MTG-positive structures in the control
cells. After 1 day of treatment with EtBr, the number of
dual-labeled structures per section increased from 3.3±1.5, 4.7±0.6
and 5.7±1.5 to 31±4.6, 51±5.6 and 46.3±6.5 in the A549, SPC-A1 and
H322 cells, respectively (n=5 sections; P<0.01) (Fig. 3A). In cells starved for 18 h, a
significant increase in co-localization was observed (n=5 sections;
P<0.01) (Fig. 3A). Compared with
starvation, EtBr treatment more dramatically increased the
co-localization of MTG- and LTR-positive structures (Fig. 3B).
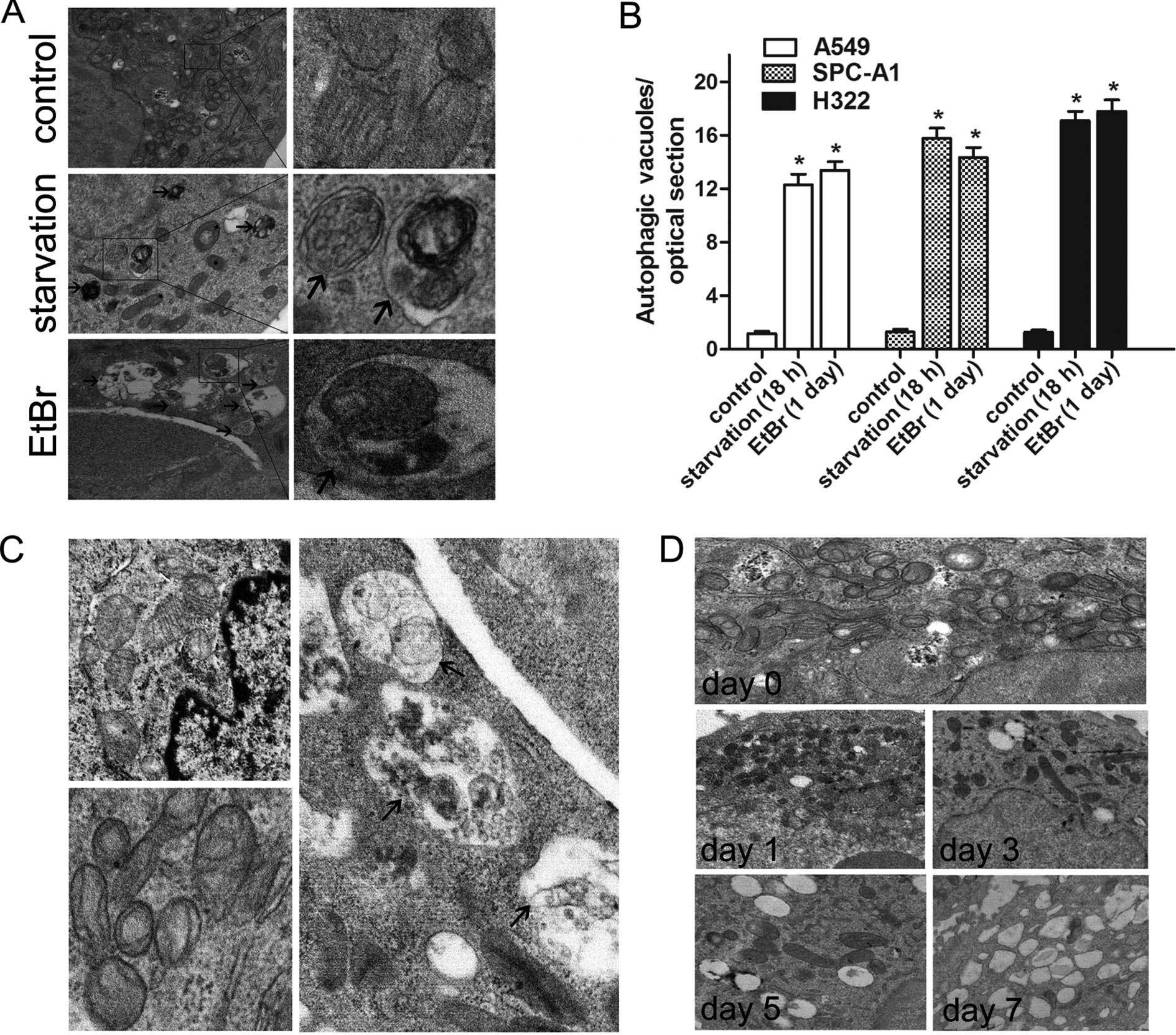
Transmission electron microscopy to
identify autophagic ultrastructures
To confirm the mitochondrial degradation via
autophagy, TEM was performed. In cells starved for 18 h, the
presence of autophagic vacuoles with cytoplasmic content (Fig. 4A, middle, arrows) increased
significantly when compared with the control cells (n=30 cells;
P<0.01) (Fig. 4A and B).
Similarly, more mitochondria encapsulated in double-membrane
vesicles (Fig. 4A, bottom, arrows)
were observed after EtBr treatment. Consistent with the TMRM
fluorescence microscopy results, there was an obvious depopulation
of mitochondria from day 1 to day 7. Morphological alterations in
the internal structures of the remaining mitochondria were
identified. After 7 days of EtBr treatment, the mitochondria
exhibited aberrant phenotypes with partial or complete loss of
regular crista patterns. In contrast, the untreated control cells
possessed typical elongated mitochondria with parallel cristae
(Fig. 4C). In addition, a
significant increase in cytoplasmic lipid droplets was observed in
EtBr-treated cells (Fig. 4D).
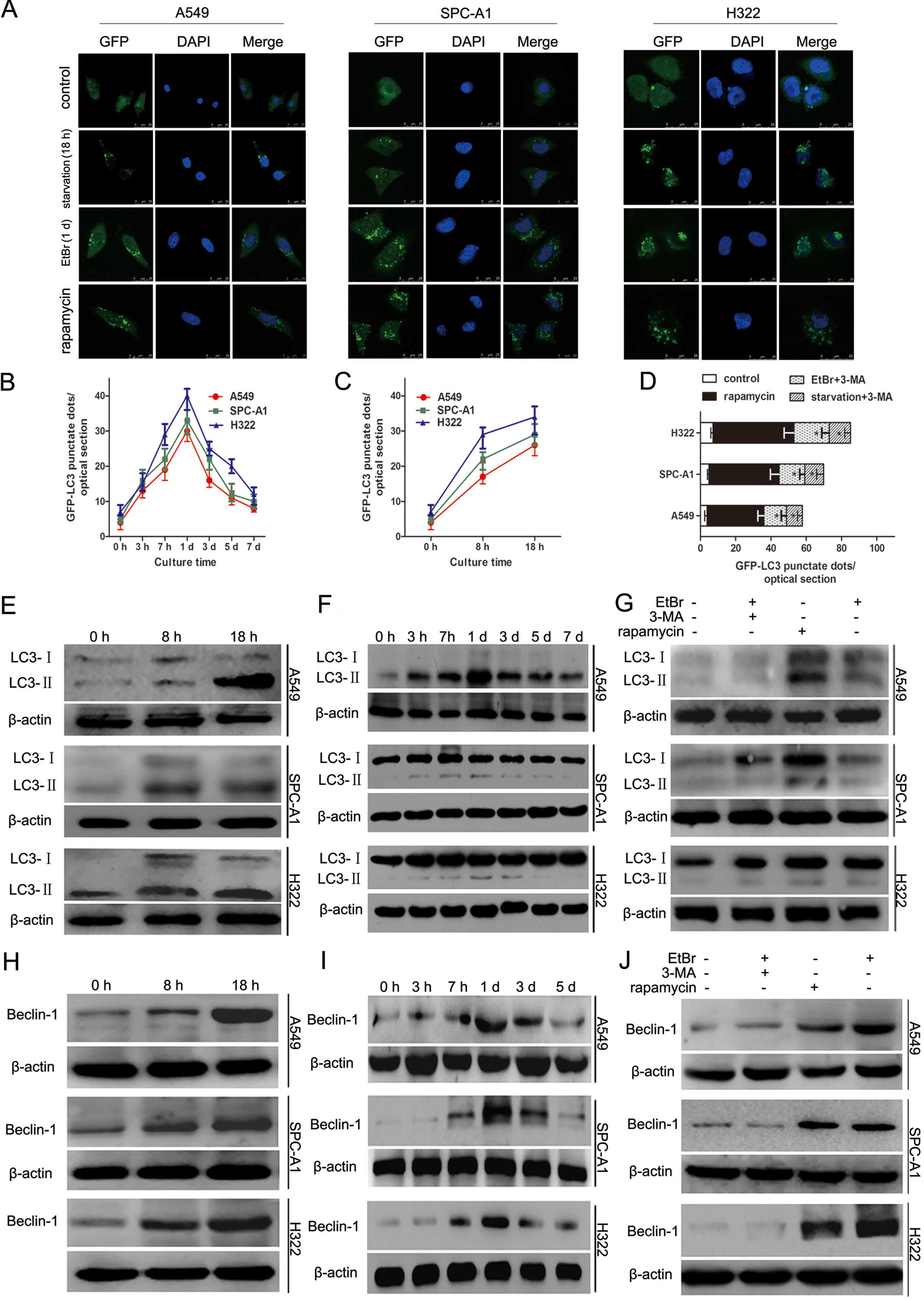
Recruitment of LC3-II to autophagosomes
during mitochondrial degradation via autophagy
To monitor autophagic activity, the conversion of
endogenous LC3-I to LC3-II was assessed. An expression vector
encoding pEGFP-C1-LC3 was utilized. In cells treated for 3 h with
EtBr, the expression level of GFP-LC3 began to increase. By day 1,
the number of GFP-LC3 punctae reached a maximum. After 24 h, the
number of green LC3 dots decreased but remained significantly
higher than that in the control cells (Fig. 5A and B). Similar patterns of LC3-II
conversion were identified by western blot analysis (Fig. 5F). We examined whether 3-MA, an
autophagy inhibitor, inhibits EtBr-induced autophagy. The results
indicated that EtBr-induced autophagy was inhibited by 3-MA
(P<0.05) (Fig. 5D and G). In
cells starved for 8 or 18 h, the number of GFP-LC3 dots increased
compared with the control cells (Fig.
5A and C). A significant conversion of LC3-I to LC3-II was
detected by western blotting (Fig.
5E), and starvation-induced LC3-II conversion was
time-dependent. As expected, 3-MA markedly decreased LC3-II
recruitment in the starved cells (P<0.05) (Fig. 5D).
The expression level of Beclin-1 was analyzed as it
has been reported to be required for the initial formation of
autophagosomes during autophagy (15). During culture in nutrient
deprivation medium, cancer cells exhibited increased Beclin-1
protein (Fig. 5H). A similar
increase in Beclin-1 protein was observed in cells treated with
EtBr (Fig. 5I); this increase was
inhibited by 3-MA, an autophagy inhibitor (Fig. 5J).
Discussion
Unlike nuclear DNA, mtDNA lacks protection by
histones, and mitochondria have a limited capacity for self-repair,
making mtDNA more susceptible to damage (16). Typically, a low concentration of
EtBr has been used to inhibit mtDNA replication and transcription
(17–19). During the establishment of lung
cancer cell lines without mtDNA, a progressive depopulation of
mitochondria was observed that was accompanied by increased LTR
uptake and co-localization of LTR- and MTG-positive structures.
Moreover, autophagosomal structures containing mitochondrial
remnants were directly observed with an electron microscope. The
expression of LC3-II and Beclin-1 significantly increased, but
these increases were inhibited by 3-MA, an autophagy inhibitor. Our
findings suggest that autophagy was responsible for mitochondrial
degradation after EtBr exposure, most likely through the
PI3K-Beclin-1 pathway.
TMRM is a cell-permeant, cationic, red-orange
fluorophore that localizes to mitochondria in response to a high
negative membrane potential (20).
In the present study, decreased TMRM red fluorescence was observed
by confocal microscopy, suggesting a significant loss of MMP.
Electron microscopy provided solid evidence of mitochondrial
depopulation. Moreover, the mtDNA copies and mRNA levels of COX
were significantly decreased in EtBr-treated cells. Taken together,
EtBr treatment led to mitochondrial degradation. Moreover,
alterations in mitochondrial ultrastructure were identified. In
EtBr-treated cells, the remaining mitochondria became large and
branched compared with the spherical, oval or short rod-like shape
of the control cells. The cristae inside the mitochondria were less
abundant and shorter.
During the loss of mitochondria, we observed a
significant increase in acidic organelles. This was verified by an
increased uptake of LTR, an acidotrophic fluorescent probe
(21). LTR labels all acidic
organelles, including lysosomes and late endosomes, not just
autophagosomes. However, Rodriguez-Enriquez et al(21) and Kim and Lemasters (22) reported that, in the context of
nutrient deprivation-induced autophagy, the increased number of LTR
fluorescent dots represented an increase in autophagosomes and
autolysosomes. Therefore, we believe that the increased LTR uptake
in EtBr-treated cells was due to an increase in autophagosomes and
autolysosomes. The movement of MTG-labeled mitochondria into
LTR-labeled acidic autolysosomal structures was visualized by
confocal microscopy. The autophagosomal structures containing
mitochondrial remnants were observed in the cytoplasm by electron
microscopy. These findings suggest that mitochondrial degradation
in cells treated with EtBr was accomplished via an autophagic
pathway. Well-established nutrient deprivation-induced autophagy
was employed as a control. As previously reported, mitochondria
occupy ~5–6% of the cytoplasmic volume (23,24),
thus mitochondria were a major target for autophagic digestion
after nutrient deprivation.
LC3-II conversion is usually studied as an indicator
of autophagic activity. LC3-tagged GFP has been utilized to monitor
autophagy through direct fluorescence microscopy (25). In complete growth medium, lung
cancer cells transfected with GFP-LC3 exhibited a diffuse
distribution of green fluorescence in the cytosol. In cells treated
with EtBr, a marked increase in GFP-LC3 punctae was observed. This
characteristic LC3-II conversion was verified by western blot
analysis. Similar changes in this autophagic marker were identified
in starved cells. Furthermore, GFP-LC3 patches containing
TMRM-labeled polarized mitochondria were observed in GFP-LC3
transgenic hepatocytes (22). This
convinced us that selective mitochondrial degradation via autophagy
was involved in nutrient deprivation-induced autophagy. It has been
reported that 3-MA can inhibit class III PI3K and subsequently
suppress LC3-II conversion under starvation conditions (26,27),
which is important in clarifying the effects of autophagy. Our data
demonstrated that 3-MA inhibited the formation of GFP-LC3 punctae
as well as the accumulation of LC3-II protein in EtBr-treated
cells. Therefore, since increased Beclin-1 expression following
EtBr treatment could be inhibited by 3-MA, EtBr-induced
mitochondrial autophagy was activated through the class III PI3K
pathway.
Currently, the role of autophagy in keeping cells
alive or inducing cell death is controversial (28–30).
We believe that excessive autophagy breaks the delicate balance
between cell survival and cell death. In our study, mitochondrial
autophagy was over-activated in lung cancer cells by continuous
exposure to EtBr, which resulted in a slower growth rate in
vitro and in vivo. As Kulawiec et al(31), Yu et al(32) and Singh (33) reported, mitochondrial damage can
result in cell cycle arrest and prevent the initiation of
apoptosis. In the present study, EtBr-induced mitochondrial damage
may activate the mito-checkpoint to maintain the mitochondrial
integrity of lung cancer cells. mtDNA dysfunction was reported to
cause chromosomal instability in the nucleus (34,35).
Therefore, interference with the normal function of nDNA was
thought to explain the decreased tumorigenicity of EtBr-treated
lung cancer cells. In the present study, our data demonstrated that
excessive mitochondrial degradation via autophagy was, at least in
part, responsible for inhibiting the cell growth and proliferation
of EtBr-treated lung cancer cells.
In conclusion, we demonstrated that autophagy is
responsible for mitochondrial degradation in lung cancer cell lines
exposed to a low concentration of EtBr. We also demonstrated that
the class III PI3K-Beclin-1 complex is involved in mediating
EtBr-induced mitochondrial autophagy. The present data indicate
that mitochondrial autophagy can inhibit cell proliferation,
migration, and tumorigenesis but cannot induce significant
apoptosis or cell death. Our findings provide new insight into the
effects of mitochondrial autophagy on lung cancer cells.
Acknowledgements
We would like to thank Jin Peng and Jie He (Central
Laboratory, The Second Affiliated Hospital of Third Military
Medical University, Chongqing, China) for their tremendous help
with laser confocal scanning microscopy and Halei Sheng for her
technical assistance with flow cytometry (Central Laboratory, The
Second Affiliated Hospital of Third Military Medical University,
Chongqing, China).
Abbreviations:
|
LTR
|
LysoTracker Red
|
|
MTG
|
MitoTracker Green
|
|
TMRM
|
tetramethylrhodamine methyl ester
|
|
TEM
|
transmission electron microscopy
|
|
LC3
|
microtubule-associated protein light
chain 3
|
|
mtDNA
|
mitochondrial DNA
|
|
EtBr
|
ethidium bromide
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
References
|
1
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eskelinen EL and Saftig P: Autophagy: a
lysosomal degradation pathway with a central role in health and
disease. Biochim Biophys Acta. 1793:664–673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang J and Klionsky DJ: Autophagy and
human disease. Cell Cycle. 6:1837–1849. 2007. View Article : Google Scholar
|
|
4
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jin S: Autophagy, mitochondrial quality
control, and oncogenesis. Autophagy. 2:80–84. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membrane after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malhotra JD and Kaufman RJ: ER stress and
its functional link to mitochondria: role in cell survival and
death. Cold Spring Harb Perspect Biol. 3:a0044242011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gupta KJ, Igamberdiev AU and Mur LA: NO
and ROS homeostasis in mitochondria: a central role for alternative
oxidase. New Phytol. 195:1–3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rambold AS and Lippincott-Schwartz J:
Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle.
10:4032–4038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhatia-Kiššová I and Camougrand N:
Mitophagy: a process that adapts to the cell physiology. Int J
Biochem Cell Biol. 45:30–33. 2013.PubMed/NCBI
|
|
13
|
Ashrafi G and Schwarz TL: The pathways of
mitophagy for quality control and clearance of mitochondria. Cell
Death Differ. 20:31–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hashiguchi K and Zhang-Akiyama QM:
Establishment of human cell lines lacking mitochondrial DNA.
Methods Mol Biol. 554:383–391. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alexeyev MF: Is there more to aging than
mitochondrial DNA and reactive oxygen species? FEBS J.
276:5768–5787. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Garcia N, Hernández-Esquivel L, Zazueta C,
Martínez-Abundis E, Pavón N and Chávez E: Induction of
mitochondrial permeability transition by the DNA-intercalating
cationic dye ethidium bromide. J Biochem. 146:887–894. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Armand R, Channon JY, Kintner J, White KA,
Miselis KA, Perez RP and Lewis LD: The effects of ethidium bromide
induced loss of mitochondrial DNA on mitochondrial phenotype and
ultrastructure in a human leukemia T-cell line (MOLT-4 cells).
Toxicol Appl Pharmacol. 196:68–79. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
von Wurmb-Schwark N, Cavelier L and
Cortopassi GA: A low dose of ethidium bromide leads to an increase
of total mitochondrial DNA while higher concentrations induce the
mtDNA 4997 deletion in a human neuronal cell line. Mutat Res.
596:57–63. 2006.PubMed/NCBI
|
|
20
|
Chazotte B: Labeling mitochondria with
TMRM or TMRE. Cold Spring Harb Protoc. 2011:895–897.
2011.PubMed/NCBI
|
|
21
|
Rodriguez-Enriquez S, Kim I, Currin RT and
Lemasters JJ: Tracker dyes to probe mitochondrial autophagy
(mitophagy) in rat hepatocytes. Autophagy. 2:39–46. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim I and Lemasters JJ: Mitochondrial
degradation by autophagy (mitophagy) in GFP-LC3 transgenic
hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol.
300:C308–C317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dewey WC and Fuhr MA: Quantification of
mitochondria during the cell cycle of Chinese hamster cells. Exp
Cell Res. 99:23–30. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Segui-Simarro JM, Coronado MJ and
Staehelin LA: The mitochondrial cycle of Arabidopsis shoot apical
meristem and leaf primordium meristematic cells is defined by a
perinuclear tentaculate/cage-like mitochondrion. Plant Physiol.
148:1380–1393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q,
Wenk MR, Ong CN, Codogno P and Shen HM: Dual role of
3-methyladenine in modulation of autophagy via different temporal
patterns of inhibition on class I and III phosphoinositide
3-kinase. J Biol Chem. 285:10850–10861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
|
|
28
|
Chen N and Karantza-Wadsworth V: Role and
regulation of autophagy in cancer. Biochim Biophys Acta.
1793:1516–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
White E, Karp C, Strohecker AM, Guo Y and
Mathew R: Role of autophagy in suppression of inflammation and
cancer. Curr Opin Cell Biol. 22:212–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gorski SM, Ries J and Lum JJ: Targeting
autophagy: the Achilles’ heel of cancer. Autophagy. 8:1279–1280.
2012.
|
|
31
|
Kulawiec M, Ayyasamy V and Singh KK: p53
regulates mtDNA copy number and mitocheckpoint pathway. J Carcinog.
8:82009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu M, Shi Y, Wei X, Yang Y, Zhou Y, Hao X,
Zhang N and Niu R: Depletion of mitochondrial DNA by ethidium
bromide treatment inhibits the proliferation and tumorigenesis of
T47D human breast cancer cells. Toxicol Lett. 170:83–93. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Singh KK: Mitochondria damage checkpoint,
aging, and cancer. Ann NY Acad Sci. 1067:182–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Amuthan G, Biswas G, Ananadatheerthavarada
HK, Vijayasarathy C, Shephard HM and Avadhani NG: Mitochondrial
stress-induced calcium signaling, phenotypic changes and invasive
behavior in human lung carcinoma A549 cells. Oncogene.
21:7839–7849. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen D, Xue W and Xiang J: The
intra-nucleus integration of mitochondrial DNA (mtDNA) in cervical
mucosa cells and its relation with c-myc expression. J Exp Clin
Cancer Res. 27:362008. View Article : Google Scholar : PubMed/NCBI
|