Introduction
Although the incidence of gastric cancer has been
declining globally since World War II, and it is one of the least
common cancers in North America, the incidence of gastric cancer is
still high in many countries around the world. In 2012, an
estimated 21,320 new cases were diagnosed, and 10,540 cases will
eventually die of the disease in the United States (1). Gastric cancer is estimated to be the
fourth most common cancer worldwide (2).
Despite a major decline in the incidence of gastric
cancer and substantial breakthroughs in our understanding of
gastric cancer both from a clinical as well as a preclinical
perspective over the past decades, gastric cancer remains a
significant public health burden worldwide, particularly in
developing countries. Hence, it is urgent to develop novel
therapeutics for gastric cancer. The pathogenesis of gastric
carcinoma is still unclear, and increasing evidence shows that many
factors contribute to this process.
In the past three decades, utilization of the
estrogen receptor (ER) in breast carcinoma is well established.
Women with ER-positive breast cancer benefit by substantial
improvements in outcomes due to current endocrine therapies
(3,4). Unfortunately, the role of the ER in
other types of cancers is largely unknown. Estrogen receptor (ER)α
expression in human gastric cancer was first reported by Tokunaga
et al(5). Since that time,
the relationship between ERα status and tumor progression was
reported in a series of studies. Our previous study demonstrated
that ERα status strongly influences patient survival in gastric
cancer (37). It is tempting to
postulate that ERα may play an essential role in the carcinogenesis
of gastric cancer; however, its definitive role in the cell
biological characteristics and related involved mechanisms have not
yet been fully elucidated. Better understanding of the role of ERα
as well as the related pathway will lead to more effective
targeting of this pathway for cancer prevention and therapy.
The present study aimed to investigate the
involvement of ERα in cell growth and progression in gastric
cancer. To accomplish this, we constructed an eukaryotic expression
vector with the ERα gene to determine the effects of ERα
overexpression on the cell biological characteristics of the
gastric cancer cell line MKN28. The long-term goals of our research
are to ascertain whether ERα may serve as a potential diagnostic
and prognostic marker of gastric cancer and as a target for the
development of therapeutic approaches to treat this disease.
Materials and methods
Cell culture
Human gastric adenocarcinoma cell lines, BGC823,
KATOIII, MKN45, MKN28, AGS, N87 and SGC7901, were purchased from
the Cell Bank, Chinese Academy of Science, Shanghai, China. They
were cultured in an incubator at 37°C under a humidified atmosphere
of 5% CO2 and 95% air in Dulbecco’s modified Eagle’s
medium (DMEM) (Invitrogen, Life Technologies, Gaithersburg, MD,
USA). All media were supplemented with 10% heat-inactivated fetal
bovine serum (FBS) (Invitrogen Life Technologies), penicillin (100
U/ml) and streptomycin (100 μg/ml). Cells were incubated for 24 h
in Phenol red-free minimum essential medium (MEM; Invitrogen Life
Technologies, Carlsbad, CA, USA) without FBS prior to all
experiments (termed cell cycle synchronization).
Construction and transfection of the ERα
plasmid expression vector
We used plasmid pcDNA3.1+ (Shanghai Cancer
Institute, China) to construct the ERα expression vector. The
methods of pcDNA3.1+ERα and transfection of MKN28 gastric cancer
cells with pcDNA3.1+ (vector) or pcDNA3.1+ERα were conducted as
follows. Briefly, an ERα cDNA PCR product and pcDNA3.1+ (vector)
were digested with EcoRI. The digested PCR product was
electrophoresed through and isolated from an agarose gel. After
purification, it was ligated into the cut vector to form
pcDNA3.1+ERα. After the ligation, the plasmid was transformed into
Escherichia coli TOP10 cells, and then planted on solid LB
medium. Ampicillin-resistant colonies were cultured at 37°C
overnight in a rocking bed. The recombinant plasmid was prepared,
and the sequences were verified by electrophoresis of the digested
product. MKN28 cells (1×105) were inoculated into a
6-well plate and transfected with pcDNA3.1+ or pcDNA3.1+ERα
recombination plasmids when the confluency achieved 90%.
Forty-eight hours after transfection, cells were diluted to 1:10
for passage, and cultured for at least 2 weeks in medium containing
G418 (Geneticin® selection agent; Invitrogen Life
Technologies, Carlsbad, CA, USA).
MTT assay
MKN28 cells with or without ERα overexpression
(1×103/well) were seeded into a 96-well plate and
incubated in an incubator at 37°C under a humidified atmosphere of
5% CO2 and 95% air. Growth was measured by adding 20 μl
of 5 mg/ml methyl-thiazolyl-tetrazolium (MTT) to each well, and the
plates were incubated at 37°C for 4 h. Then, 200 μl dimethyl
sulfoxide was added to each well after removal of the old medium,
and absorbance was measured at 570 nm using a multi-well
spectrophotometer (Bio-Rad, Hercules, CA, USA).
Colony formation assay
Cell suspensions from each group were diluted in
DMEM supplemented with 10% FBS, and immediately re-plated (1,000
cells/well) in 6-well plates. The plates were incubated until the
cells had formed sufficiently large colonies. The colonies were
fixed with dehydrated ethanol and stained with 0.5% crystal violet.
The plates were photographed and their digital images were manually
analyzed to determine the colony number.
Flow cytometric analysis
For cell cycle analysis, cells (1×106)
were washed twice with ice-cold PBS, treated with trypsin, and
fixed in cold 70% ethanol at 4°C for at least 24 h, washed twice in
PBS, and incubated in 25 μg/ml of RNase for 30 min at 37°C. Before
analysis, cells were stained with 50 μg/ml of propidium iodide (PI)
(Cell Apoptosis PI Detection kit; Keygentec, China) at room
temperature for 30 min. Analyses were performed using a FACScan
flow cytometer (Becton-Dickinson, Sunnyvale, CA, USA). Data
obtained from the cell cycle distributions were analyzed using
FlowJo v7.6 (Tree Star, Inc., Ashland, OR, USA).
Analysis of apoptosis
Enumeration of apoptotic cells was carried out using
the Cell Apoptosis PI Detection kit (Keygentec). Cells were washed
twice in cold PBS and re-suspended in 1X buffer A at a
concentration of 100×106 cells/ml. This suspension (95
μl) was stained with 5 μl of PI. The cells were gently vortexed and
incubated for 5 min at room temperature in the dark. Cells were
observed under a fluorescence microscope according to the protocol.
The number of cells undergoing apoptotic cell death was analyzed by
an inverted fluorescence microscope. Images were captured randomly
from 5 fields of vision with ×200 magnification. Independent
experiments were performed in triplicate.
Transwell migration and invasion
assays
For the migration studies, cells with or without ERα
overexpression were dispersed using trypsin and adjusted to a
density of 1×106 cells/ml with serum-free DMEM. Then,
100 μl of the solution (1×105 cells/ml) was placed in
the upper chambers of Transwell plates (Millipore, Billerica, MA,
USA), and 500 μl of DMEM with 10% FBS was added to the lower
chambers. The plates were then placed in an incubator at 37°C with
5% CO2 for 24 h. After incubation, the cells remaining
in the upper chamber were carefully removed, and the Transwell
membrane was fixed with dehydrated ethanol and stained with 0.5%
crystal violet. To count the fixed cells, images were captured
randomly from 5 fields of vision with ×200 magnification.
Independent experiments were performed in triplicate.
For the cell invasion assay, Matrigel (BD
Biosciences, Franklin Lakes, NJ, USA) was thawed on ice at 4°C
overnight and diluted with serum-free medium at a ratio of 1:3.
Then, the Transwell chambers were coated with 30 μl of diluted
Matrigel in a 24-well plate and incubated at 37°C for 2 h.
Afterward, 1×105 cells in serum-free DMEM were seeded
into the prepared Transwell chambers. Then, 500 μl of DMEM with 10%
FBS was added to the basal chamber. The 24-well plate was then
incubated at 37°C with 5% CO2 for 24 h. Cells were
stained and counted as in the migration assay.
Western blot analysis
Whole-cell proteins were isolated using a protein
extraction buffer containing 150 mmol/l NaCl, 10 mmol/ml Tris (pH
7.2), 5 mmol/l ethylenediaminetetraacetic acid, 0.1% Triton X-100,
5% glycerol and 1% sodium dodecyl sulfate. Equal amounts (40
μg/lane) of proteins were fractionated on 10% sodium dodecyl
sulfate polyacrylamide gels and transferred to polyvinylidene
difluoride membranes. The membranes were probed with anti-ERα
(Epitomics, Inc., Burlingame, CA, USA), -GAPDH (Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA) and -β-catenin (Epitomics,
Inc.) primary antibodies. After being washed with TBS Tween-20
(0.1%), the membranes were incubated with peroxidase-conjugated
rabbit anti-mouse or goat anti-rabbit secondary antibody (Santa
Cruz Biotechnology, Inc.) for 2 h at room temperature and subjected
to enhanced chemiluminescent staining using an ECL detection system
(Bio-Rad). All experiments were conducted in triplicate.
Statistical analysis
Data are presented as means ± standard error of the
mean (SEM) of 3 independent experiments. The Student’s t-test was
used to determine the statistical differences between various
experimental and control groups, and one-way ANOVA test was used to
determine the difference between three or more groups. P-values
<0.05 were considered to indicate statistically significant
differences.
Results
ERα expression in gastric cancer cell
lines
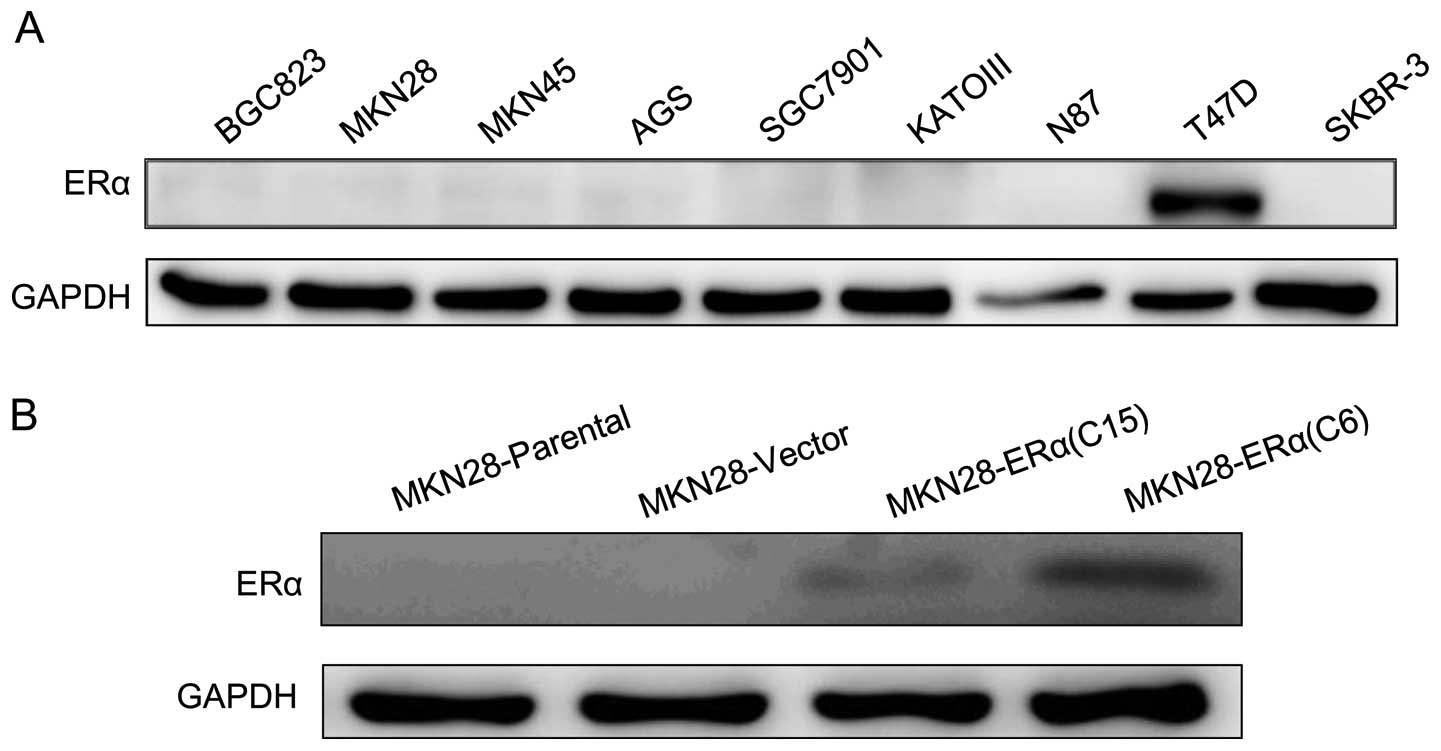
Western blotting was performed using 7 gastric
cancer cell lines. The protein level of ERα was not detectable in
all 7 gastric cancer cell lines based on western blot analysis
(Fig. 1A).
Construction and transfection with the
pcDNA3.1+ERα recombinant vector
To examine the effect of ERα on gastric tumor cell
progression in vitro, the plasmid pcDNA3.1 was used to
construct the ERα expression vector, pcDNA3.1+ERα. The MKN28 cell
line was engineered to stably express increased levels of ERα
protein, and the engineered cell lines are referred to as
MKN28-ERα(C6) and MKN28-ERα(C15), respectively. A control cell line
was transfected with the empty vector and is referred to as
MKN28-Vector. ERα protein was detectable in the MKN28-ERα(C6) and
MKN28-ERα(C15) cells. In contrast, no ERα expression was observed
in the MKN28-Vector cells (Fig.
1B). The data suggest that the pcDNA3.1+ERα recombinant vector
was successfully constructed, and ERα was stably overexpressed in
the MKN28-ERα(C6) and MKN28-ERα(C15) cells. Furthermore among the
two cell lines expressing ERα, MKN28-ERα(C6) cells exhibited much
higher expression than the other, which enabled us to examine how
different degrees of ERα expression influence the progression of
MKN28 cell.
ERα overexpression inhibits cell growth
and proliferation in gastric cancer MKN28 cells
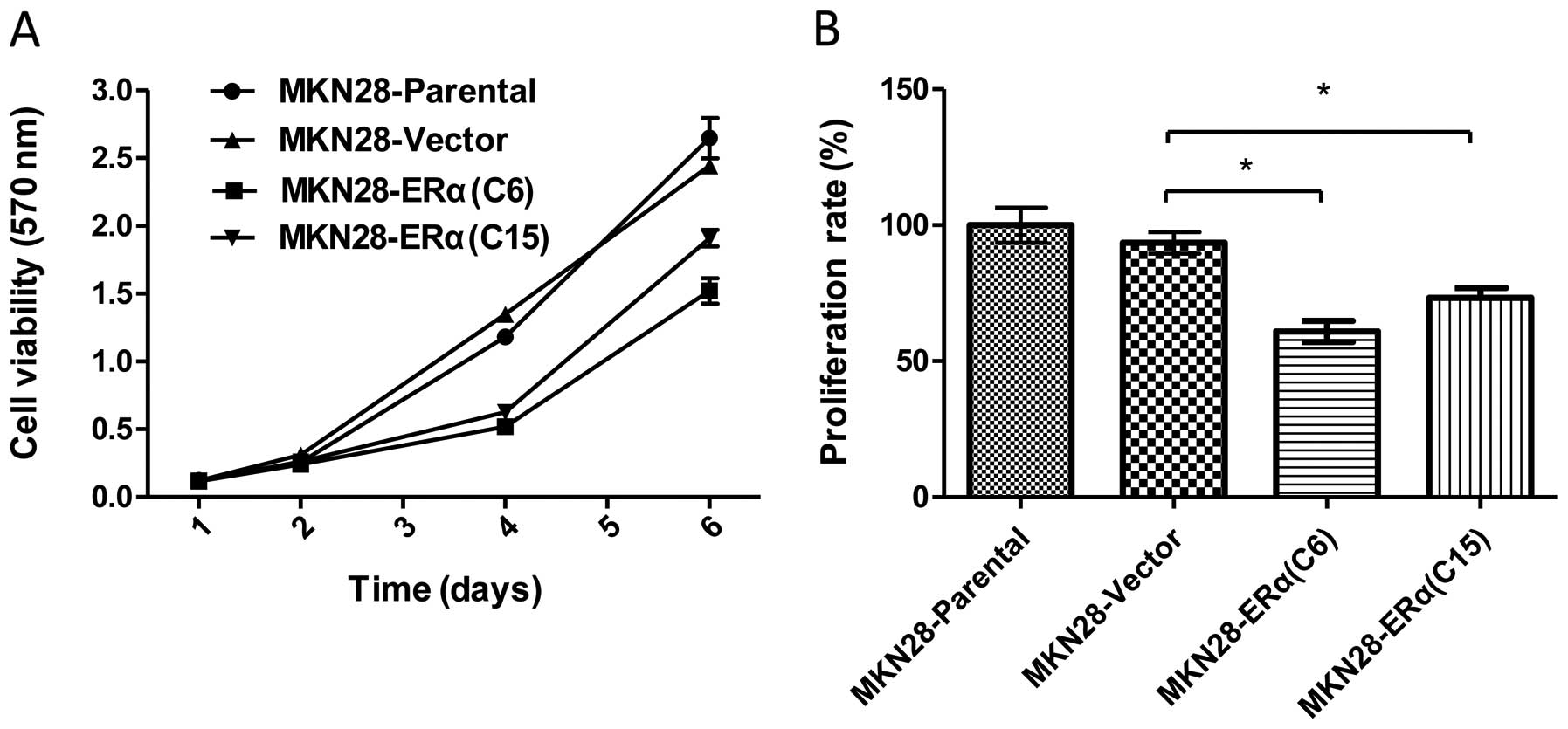
To determine the effect of ERα expression on the
growth and proliferation of MKN28, we determined the in
vitro survival rates of the MKN28-ERα(C6) and MKN28-ERα(C15)
cells. MKN28-ERα(C6) and MKN28-ERα(C15) cells exhibited
significantly reduced cell survival, as assessed by the MTT assay
(Fig. 2A). The mean proliferation
rate was 1.2- to 1.5-fold higher in the MKN28-Parental and
MKN28-Vector cells when compared to the rate in the MKN28-ERα(C6)
and MKN28-ERα(C15) cells (P<0.05) (Fig. 2B). In addition, MKN28-ERα(C6) cells
had a slower rate of growth than the MKN28-ERα(C15) cells, which
was consistent with the elevated levels of ERα in MKN28-ERα(C6)
cells.
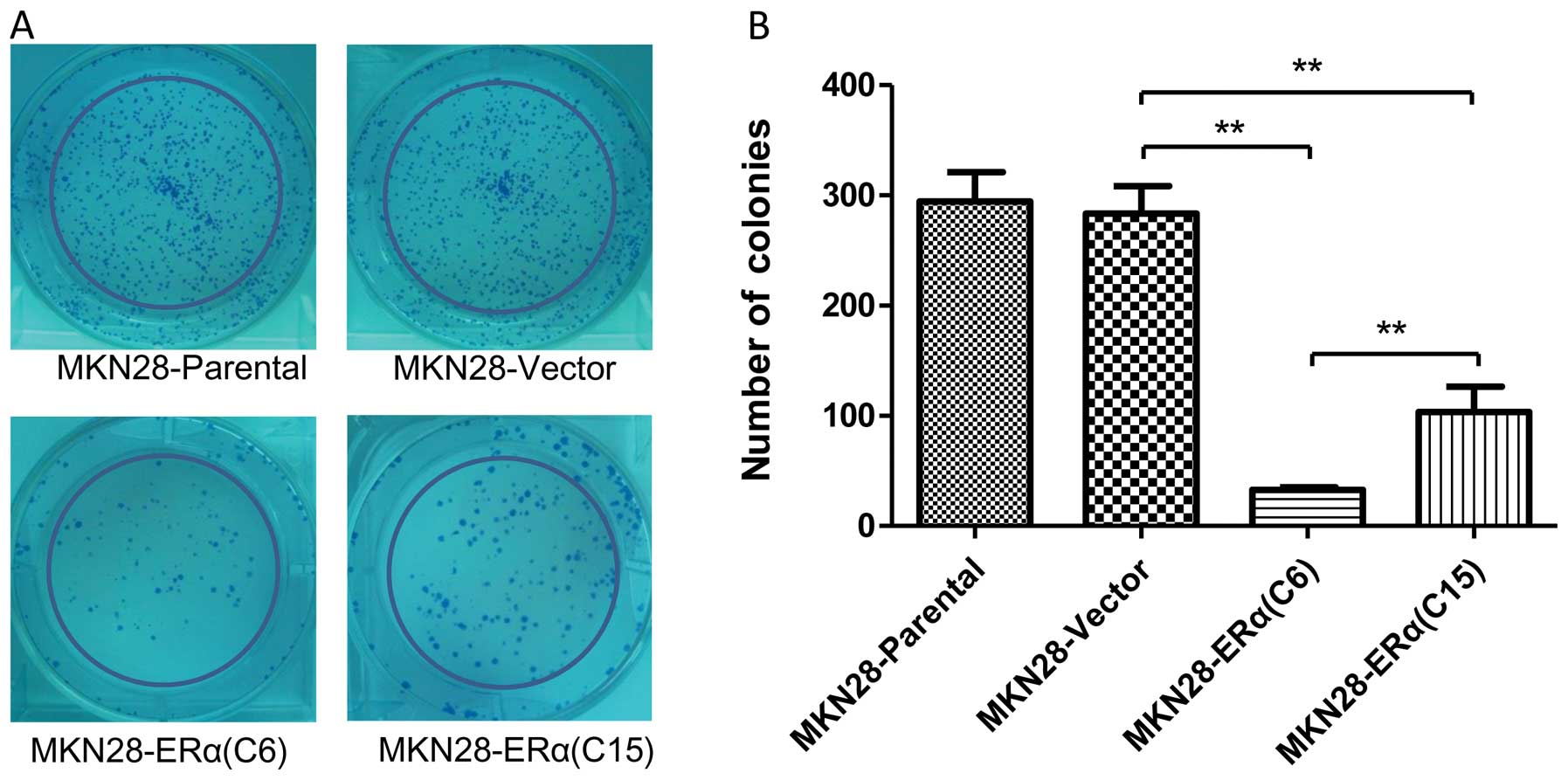
We utilized a colony formation assay to further
confirm the suppressive effect of ERα on the growth of MKN28 cells.
The mean number of colonies formed by MKN28-ERα(C6) cells after 10
days of culture was 32.67±4.16, and it was 59.7 and 62.1%
decreased, respectively, when compared with that of the
MKN28-Vector and MKN28 cells (P<0.001) (Fig. 3). Furthermore, more colonies were
observed in the MKN28-ERα(C15) cells when compared to the number of
colonies in the MKN28-ERα(C6) cells. Taken together, these data
suggest that ERα inhibits cell growth and proliferation in gastric
cancer MKN28 cells.
Effect of ERα overexpression on cell
cycle control in gastric cancer MKN28 cells
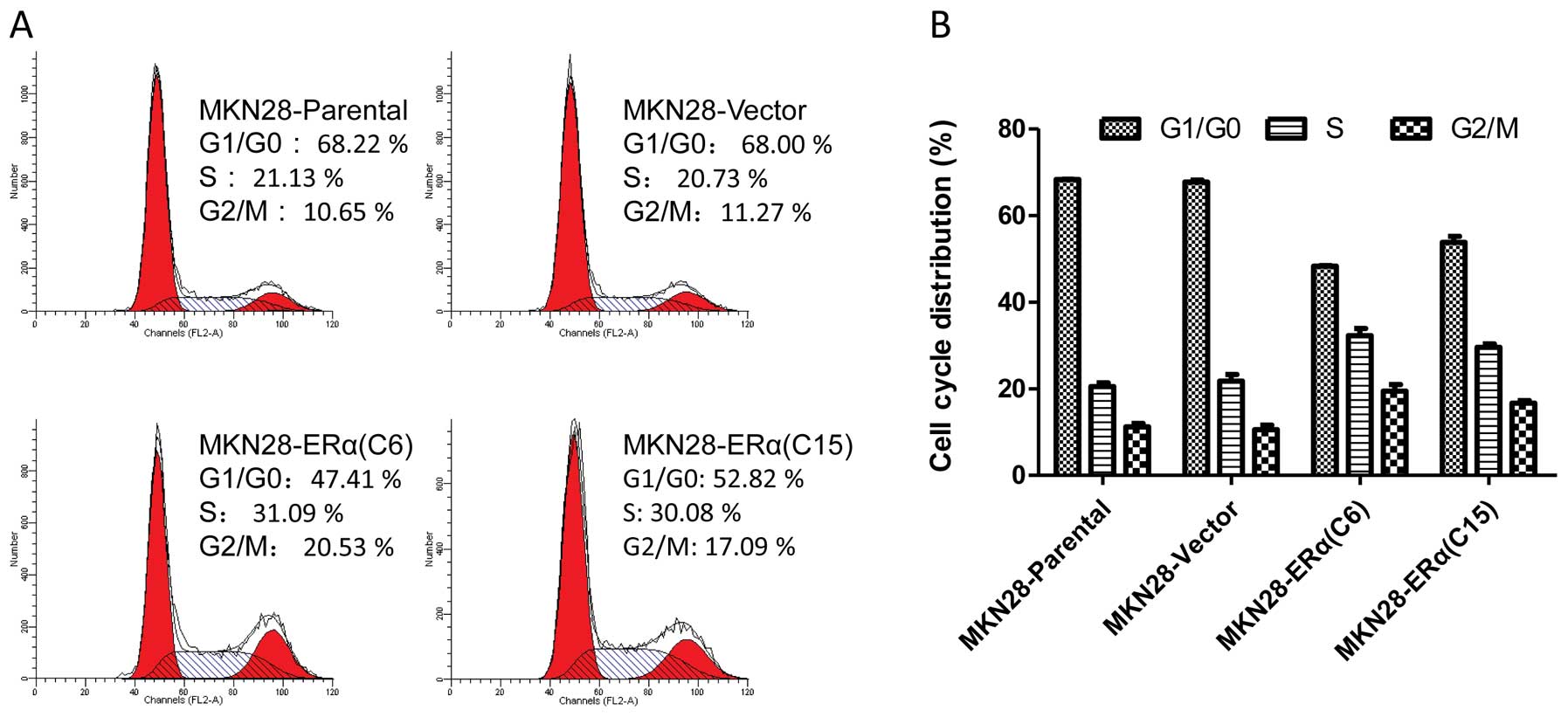
Flow cytometry was used to determine whether the
inhibitory effect of ERα on MKN28 cell proliferation was mediated,
at least partly, by the cell cycle. Both MKN28-ERα(C6) and
MKN28-ERα(C15) cells showed an increase in the number of
G2-M-arrested cells, when compared to this number in the parental
cells (Fig. 4). Cell growth
inhibition by ERα was associated with significant cell cycle arrest
at the G2/M phase, implicating that ERα suppresses cell
proliferation by controlling the G2 and M checkpoints and induces
specific blockage of cell cycle progression.
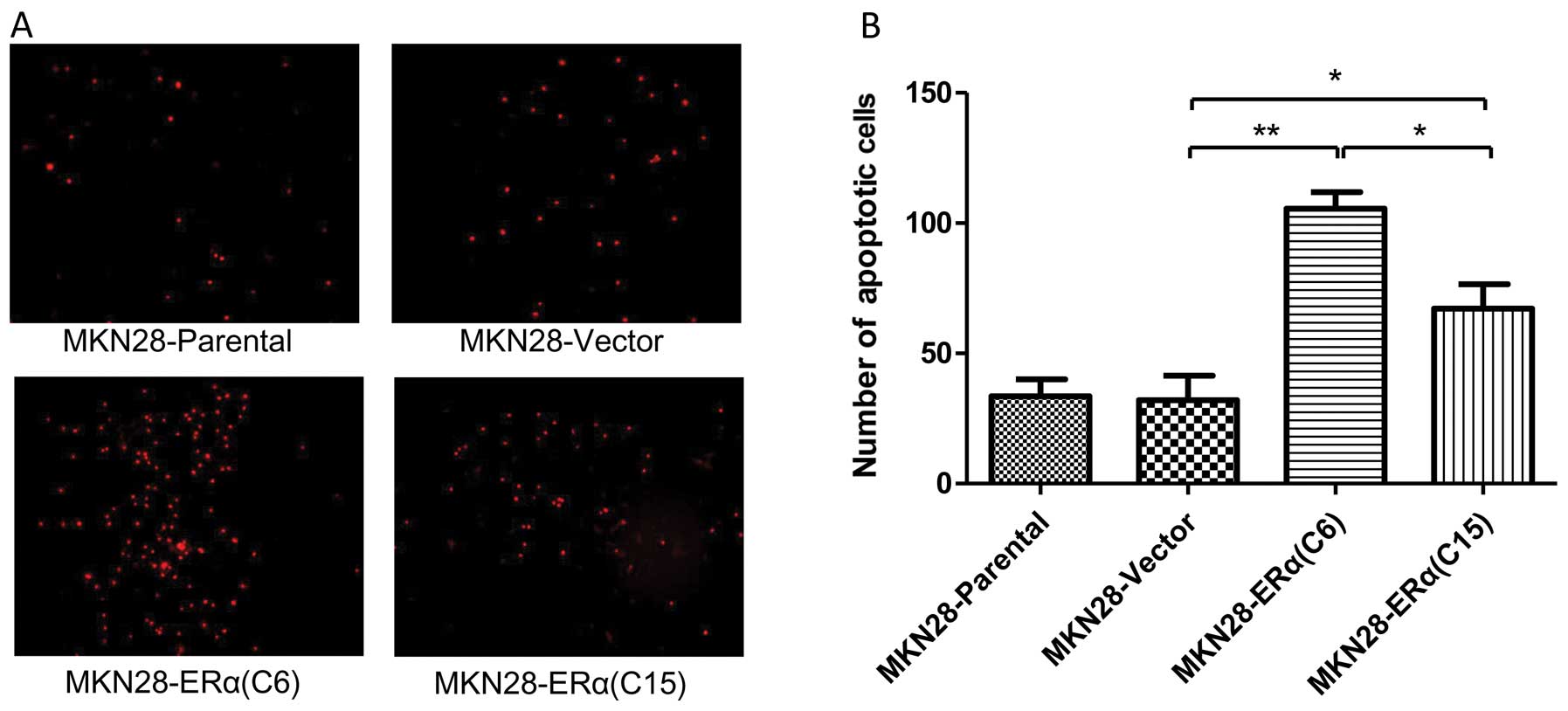
ERα overexpression induces apoptosis of
gastric cancer cells
PI staining was used to evaluate the degree of
apoptosis in the different cell lines. There was a significantly
increased number of apoptotic cells noted in the MKN28-ERα(C6) and
MKN28-ERα(C15) cells, as compared with the MKN28-Parental and
MKN28-Vector cells. Notably, the proportion of apoptotic cells in
the MKN28-ERα cells was in line with the level of ERα expression
(Fig. 5).
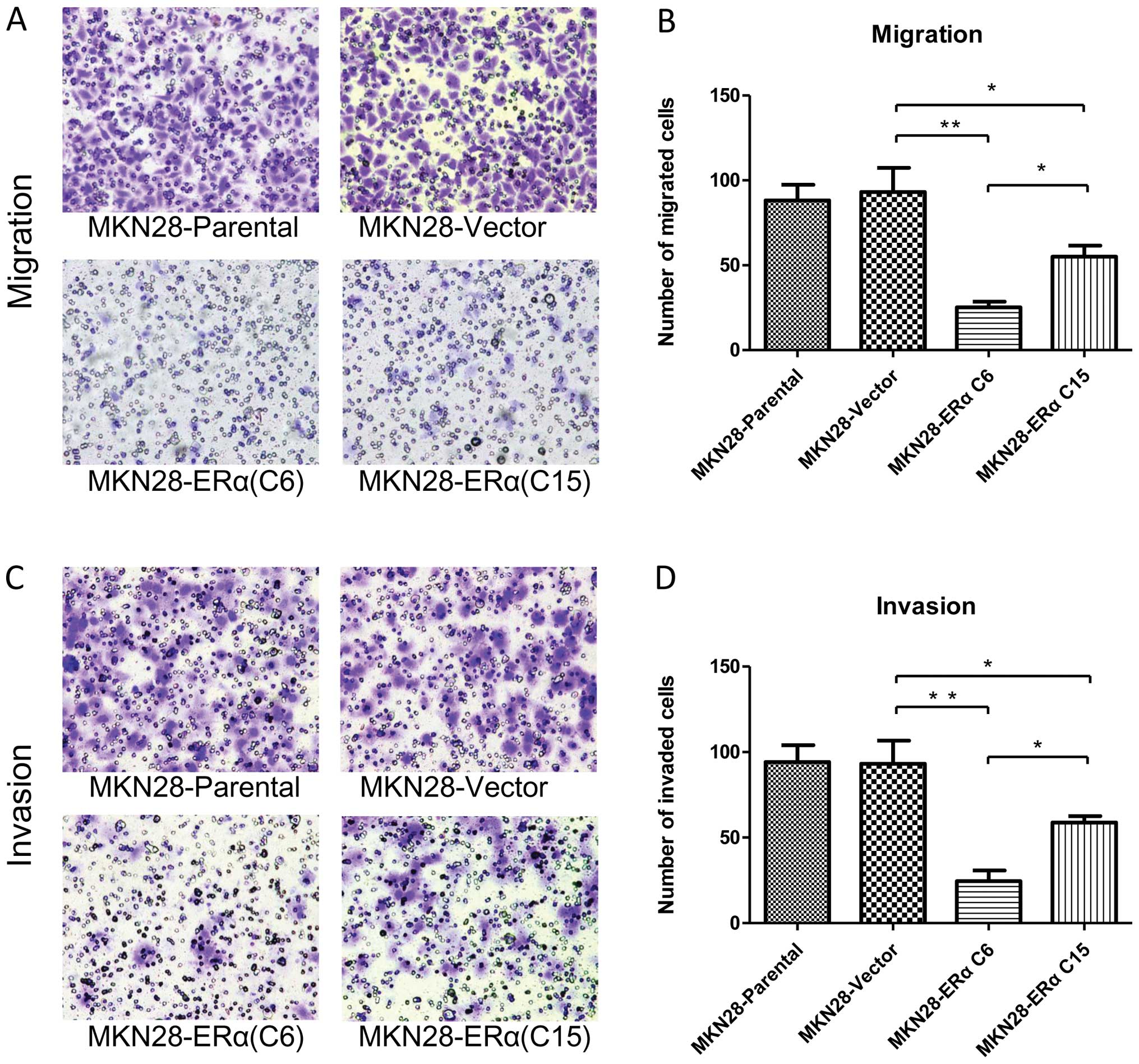
ERα overexpression inhibits the migration
and invasion of gastric cancer cells
To determine whether ERα is involved in mediating
the migration and invasion of gastric cancer cells, we performed
in vitro migration and invasion assays using Transwell
chambers. After the cells were incubated for 24 h in the Transwell
assay system, the number of MKN28-Vector cells that had moved
through the membrane of the chamber was ~3.7 and 1.7-fold higher
than the number of the MKN28-ERα(C6) (P<0.001) and
MKN28-ERα(C15) cells (P<0.05) (Fig.
6A and B), respectively. MKN28-ERα cells migrated at a
significantly lower rate than the control cells after 24 h. The
results indicate that ERα reduces the migratory ability of MKN28
cells.
Similarly, MKN28-ERα cells were observed to be less
invasive. After the cells were incubated for 24 h in the Transwell
assay system, the number of MKN28-Vector cells that had invaded
through the membrane of the Matrigel chamber was ~3.8-fold higher
than that of the MKN28-ERα(C6) cells (P<0.001) and 1.6-fold
higher than that of the MKN28-ERα(C15) cells (P<0.05) (Fig. 6C and D).
The migration and invasion ability was 2.2-fold
reduced in the MKN28-ERα(C6) cells when compared to the
MKN28-ERα(C15) cells (Fig. 6B and
D). Taken together, these results showed that overexpression of
ERα may suppress the migration and invasion of MKN28 cells.
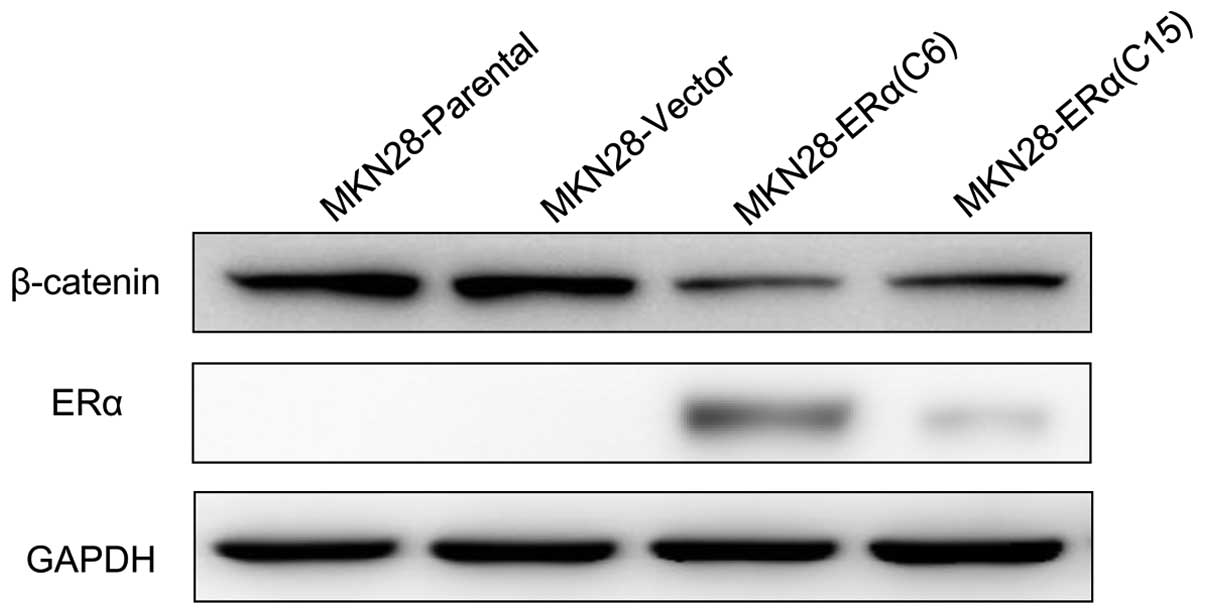
Overexpression of ERα inhibits
progression by suppressing β-catenin in MKN28 cells
Next, to determine the potential molecular mechanism
of the phenotypes gained by ERα overexpression in MKN28 cells, we
examined the protein level of β-catenin, as β-catenin has been
implicated in the initiation and progression of gastric cancer
(6,7); interaction between ERα and β-catenin
has been delineated (8). Our
results showed that β-catenin was significantly reduced in
MKN28-ERα(C6) and MKN28-ERα(C15) cells, compared with the control
cells (Fig. 7).
Discussion
In the present study, we investigated the role of
ERα expression on the cell growth and metastasis of gastric cancer
cell line MKN28. We observed that ERα protein was not expressed in
the 7 gastric cell lines studied. ERα transfection inhibited cell
growth by G2/M arrest, induced apoptosis, and suppressed cell
migration and invasion in gastric cancer cell line MKN28. In
addition, these inhibitory effects by ERα were in line with the
level of ERα expression in gastric cancer. Our study also observed
that the biological changes induced by ERα may possibly be through
the suppression of β-catenin expression.
Estrogen protects against gastric cancer
through ERs
Gastric cancer has an unexplained strong and
enigmatic male predominance (2:1) (9,10),
which cannot entirely be explained on the basis of gender
differences in the prevalence of known risk factors (11). Accumulative evidence suggests that
the differences are rooted in basic biological differences between
men and women, and this phenomenon would be explained by the
hypothesis that estrogens are protective in this respect (reviewed
in ref. 12).
This hypothesis has gained support from a number of
studies based on different aspects. Epidemiological studies have
confirmed that estrogen exposure is associated with a decreased
risk of developing gastric cancer (13–16).
Women with a longer fertility life and those on hormone replacement
therapy appear to have a decreased risk of gastric cancer.
Furthermore, men who have been treated with estrogen for prostate
cancer also have a decreased risk. In a meta-analyses, risks for
ever vs. never use of hormone treatment (HT) were significantly
reduced for gastric cancer (RR 0.78, confidence interval (CI)
0.65–0.94; P=0.008) (17). A nested
case-control study of hormone replacement therapy (HRT)
demonstrated that a greater than 50% reduced risk of gastric
adenocarcinoma was found among users of HRT compared to nonusers
[odds ratio (OR), 0.48, 95% CI 0.29–0.79] (18). Furthermore, tamoxifen (TAM) an
anti-estrogen, may increase the risk of gastric cancer and
accelerate tumor progression (19,20).
On the contrary, Harrison et al(21) demonstrated that estradiol caused
significant stimulation of gastric cell lines at physiologic
concentrations, meanwhile, addition of the active metabolite of the
estrogen-receptor blocker/partial-agonist 4-hydroxytamoxifen had a
stimulating effect on the growth rate of the gastric cell lines. On
the other hand, another study reported that estrogen did not affect
the proliferation of gastric cancer cell lines (22). Despite these contradictory results,
in animal studies, female and castrated rats have a lower incidence
of chemically induced gastric cancer (23). This hypothesis was further validated
by animal studies (23–26). These preclinical studies indicate
that estrogen may offer protection against the development of
gastric cancer, as for example, ovariectomized mice are at an
increased risk, while administration of female sex hormones
decreases the incidence of gastric cancer.
The biological means behind this hypothesis is still
inconclusive but various mechanisms have been suggested. Estrogen
exerts its biological actions through the activation of two nuclear
receptors, ERα and ERβ, with distinctive tissue distribution and a
counteracting function (27–29).
In addition, ERα has been proven to have a critical role in gastric
cancer, which will be subsequently discussed. It is therefore
reasonable to hypothesize that estrogen may protect women against
gastric cancer through the ERα pathway.
ERα is involved in the development and
progression of gastric cancer
The discovery of the ERα provided us not only with a
powerful predictive and prognostic marker, but also an efficient
target for the treatment of hormone-dependent breast cancer with
anti-estrogens. The important role of ERα in the development,
progression and treatment of breast cancer are well established,
but the role of such an evaluation in other types of cancers is
largely unknown.
ERα expression in human gastric cancer was first
reported by Tokunaga et al(5) as far back as in 1986. Nonetheless, the
role of ERα in human gastric cancer is not yet fully elucidated. It
has been suggested that the ERα pathway may have a role in the
progression of gastric cancer (5,22,30–32).
Contradictory findings have emerged on the basis of publicly
accessible in vivo and in vitro studies. We found
that numerous investigators have reported the relationship of ERα
status to carcinogenesis and tumor progression; even though, their
findings are still controversial (33–36),
including our previous study (37).
Most recently, another study indicated that sex hormone receptors
(including ERα) may be partly involved in gastric carcinogenesis
yet their clinicopathological and prognostic significance in
gastric cancer appears to be limited (38). The possibility exists that these
discrepancies result from small numbers and inconsistencies in
methodologies.
Based on the evidence that ERα is expressed in
poorly differentiated adenocarcinomas more frequently than in well
differentiated gastric cancer (39–43),
several clinical trials using a partial estrogen antagonist,
tamoxifen, have been conducted for the management of ERα-positive
gastric cancer patients. However, the results have not been
consistent, and the utility of ERα for the treatment of gastric
cancer is still inconclusive (21,39,44,45).
Even more, the expression of ERα in gastric cancer
has shown marked variability (0–62.5%) (12,46).
Consistent with our result, several studies also found that ERα
cannot be detected in gastric cancer cell lines (46,47).
Based on the currently available evidence, the
clinical significance and implication of ERα expression in human
gastric carcinoma are still not fully elucidated. Elucidation of
the precise roles of estrogen and/or its receptors in gastric
cancer will provide new insights that will contribute to diagnosis
and treatment.
Role of β-catenin in gastric cancer
Wnt and estrogen signaling represent important
regulatory pathways, each controlling a wide range of biological
processes. Crosstalk between Wnt and estrogen signaling pathways
via functional interaction between β-catenin and ERα (8), can provide fine-tuned regulation of
many cellular processes. In the present study, we aimed to
ascertain whether ERα plays a suppressive role in the proliferation
and metastasis of MKN28 cells through altering β-catenin
expression.
The roles of β-catenin in mediating intercellular
adhesion and regulation of cell growth, differentiation, invasion
and metastasis have been well characterized (48,49).
The β-catenin-TCF/LEF complex regulates and activates its
downstream target transcription genes which are involved in the
development and progression of cancer (50–52).
The abnormal activation of β-catenin frequently occurs in gastric
cancer and has been proven to promote tumor growth, invasion and
metastasis (6,7). Furthermore, previous studies have
confirmed that high β-catenin expression is an independent
indicator of poor prognosis for these carcinomas and is closely
correlated with enhanced tumor progression (53,54).
In the present study, expression of β-catenin was
found to be notably decreased in ERα-overexpressing MKN28 cells
(Fig. 7). Importantly, the degree
of decrease was in line with the level of ERα expression in the two
different MKN28-ERα cells.
Limitation of this present study and
future perspectives
These preliminary findings will require further
replication and in-depth investigation. In our present study, only
one gastric cancer cell line MKN28 was studied. Thus, it would be
sensible to reanalyze our findings using another gastric cancer
cell line to reconfirm our results and to exclude a cell-specific
phenomenon.
In addition, cell lines do not always accurately
represent the phenotype of the tumors from which they were derived.
Therefore, in vivo studies using xenografts should be
embarked on in the near future.
It should also be noted that this study was
primarily concerned with gain-of-function analysis of the
biological effect of overexpression of ERα in MKN28 cells.
Unfortunately as mentioned above, none of the 7 gastric cancer cell
lines in our study had an inherent ERα protein level expression.
Because of this reason, we could not provide evidence whether
inhibition of ERα promotes the aggressive phenotype of gastric
cancer cells.
Additionally, based on the present study, whether or
not the effect of ERα is estrogen mediated was not directly
determined. Nonetheless, estrogen-free Dulbecco’s modified Eagle’s
medium (DMEM) was used in this study, which can exclude the
influence of estrogen. An estrogen-containing condition should be
further investigated, to delineate whether ERα can exhibit a
further suppressive effect on the malignant phenotype of MKN28
cells in the context of the presence of estrogen.
Finally, the specific mechanism between ERα and
β-catenin interaction needs to be further clarified.
In conclusion, despite its preliminary character,
the research reported here indicates that overexpression of ERα
inhibits tumor cell proliferation, migration and invasion. To the
best of our knowledge, this is the first finding that demonstrates
that ERα expression is a possible protective mechanism against the
progression of gastric cancer and suggests that ERα may be a
potential target for utilization in gastric cancer treatment.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81101659/H1609,
81101659), the Natural Science Foundation of Zhejiang Province
(grant no. Y2110073), the Science and Health Care Foundation of
Zhejiang Province (grant no. 2011KYA086), and the Program for
Innovative Research Team in Zhejiang Province (grant no.
2010R50046).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar
|
|
3
|
Clarke M, Collins R, Davies C, et al:
Tamoxifen for early breast cancer: an overview of the randomised
trials. Early Breast Cancer Trialists’ Collaborative Group
(EBCTCG). Lancet. 351:1451–1467. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abe O, Abe R, Enomoto K, et al: Effects of
chemotherapy and hormonal therapy for early breast cancer on
recurrence and 15-year survival: an overview of the randomised
trials. Early Breast Cancer Trialists’ Collaborative Group
(EBCTCG). Lancet. 365:1687–1717. 2005.PubMed/NCBI
|
|
5
|
Tokunaga A, Nishi K, Matsukura N, et al:
Estrogen and progesterone receptors in gastric cancer. Cancer.
57:1376–1379. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bianchi F, Hu J, Pelosi G, et al: Lung
cancers detected by screening with spiral computed tomography have
a malignant phenotype when analyzed by cDNA microarray. Clin Cancer
Res. 10:6023–6028. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang N, Zhang J, Shuai L, et al:
Krüppel-like factor 4 negatively regulates β-catenin expression and
inhibits the proliferation, invasion and metastasis of gastric
cancer. Int J Oncol. 40:2038–2048. 2012.
|
|
8
|
Kouzmenko AP, Takeyama K, Ito S, et al:
Wnt/β-catenin and estrogen signaling converge in vivo. J Biol Chem.
279:40255–40258. 2004.
|
|
9
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.
|
|
10
|
Brenner H, Rothenbacher D and Arndt V:
Epidemiology of stomach cancer. Methods Mol Biol. 472:467–477.
2009. View Article : Google Scholar
|
|
11
|
Parkin D, Whelan S, Ferlay J, Teppo L and
Thomas D: Cancer incidence in five continents. Volume VIII. IARC
Sci Publ. 155:1–781. 2002.PubMed/NCBI
|
|
12
|
Chandanos E and Lagergren J: Oestrogen and
the enigmatic male predominance of gastric cancer. Eur J Cancer.
44:2397–2403. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
La Vecchia C, D’Avanzo B, Franceschi S,
Negri E, Parazzini F and Decarli A: Menstrual and reproductive
factors and gastric-cancer risk in women. Int J Cancer. 59:761–764.
1994.
|
|
14
|
Frise S, Kreiger N, Gallinger S, Tomlinson
G and Cotterchio M: Menstrual and reproductive risk factors and
risk for gastric adenocarcinoma in women: findings from the
Canadian National Enhanced Cancer Surveillance system. Ann
Epidemiol. 16:908–916. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lindblad M, Ye W, Rubio C and Lagergren J:
Estrogen and risk of gastric cancer: a protective effect in a
nationwide cohort study of patients with prostate cancer in Sweden.
Cancer Epidemiol Biomarkers Prev. 13:2203–2207. 2004.PubMed/NCBI
|
|
16
|
Freedman ND, Chow WH, Gao YT, et al:
Menstrual and reproductive factors and gastric cancer risk in a
large prospective study of women. Gut. 56:1671–1677. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Green J, Czanner G, Reeves G, et al:
Menopausal hormone therapy and risk of gastrointestinal cancer:
nested case-control study within a prospective cohort, and
meta-analysis. Int J Cancer. 130:2387–2396. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lindblad M, García Rodríguez LA, Chandanos
E and Lagergren J: Hormone replacement therapy and risks of
oesophageal and gastric adenocarcinomas. Br J Cancer. 94:136–141.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chandanos E, Lindblad M, Jia C, Rubio CA,
Ye W and Lagergren J: Tamoxifen exposure and risk of oesophageal
and gastric adenocarcinoma: a population-based cohort study of
breast cancer patients in Sweden. Br J Cancer. 95:118–122. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chandanos E, Lindblad M, Rubio CA, et al:
Tamoxifen exposure in relation to gastric adenocarcinoma
development. Eur J Cancer. 44:1007–1014. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harrison JD, Watson S and Morris DL: The
effect of sex hormones and tamoxifen on the growth of human gastric
and colorectal cancer cell lines. Cancer. 63:2148–2151. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takano N, Iizuka N, Hazama S, Yoshino S,
Tangoku A and Oka M: Expression of estrogen receptor-α and -β mRNAs
in human gastric cancer. Cancer Lett. 176:129–135. 2002.
|
|
23
|
Furukawa H, Iwanaga T, Koyama H and
Taniguchi H: Effect of sex hormones on carcinogenesis in the
stomachs of rats. Cancer Res. 42:5181–5182. 1982.PubMed/NCBI
|
|
24
|
Campbell-Thompson M, Lauwers GY, Reyher
KK, Cromwell J and Shiverick KT: 17β-estradiol modulates
gastroduodenal preneoplastic alterations in rats exposed to the
carcinogen N-methyl-N′-nitro-nitrosoguanidine.
Endocrinology. 140:4886–4894. 1999.
|
|
25
|
Furukawa H, Iwanaga T, Hiratsuka M, et al:
Suppressive effect of sex hormones on spreading of stomach cancer.
Gan To Kagaku Ryoho. 16:3691–3695. 1989.(In Japanese).
|
|
26
|
Furukawa H, Iwanaga T, Koyama H and
Taniguchi H: Effect of sex hormones on the experimental induction
of cancer in rat stomach - a preliminary study. Digestion.
23:151–155. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Greene GL, Gilna P, Waterfield M, Baker A,
Hort Y and Shine J: Sequence and expression of human estrogen
receptor complementary DNA. Science. 231:1150–1154. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kuiper GG, Enmark E, Pelto-Huikko M,
Nilsson S and Gustafsson JA: Cloning of a novel estrogen receptor
expressed in rat prostate and ovary. Proc Natl Acad Sci USA.
93:5925–5930. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Green S, Walter P, Kumar V, et al: Human
oestrogen receptor cDNA: sequence, expression and homology to
v-erb-A. Nature. 320:134–139. 1986. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu CW, Chang YF, Yeh TH, et al: Steroid
hormone receptors in three human gastric cancer cell lines. Dig Dis
Sci. 39:2689–2694. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karat D, Brotherick I, Shenton BK, Scott
D, Raimes SA and Griffin SM: Expression of oestrogen and
progesterone receptors in gastric cancer: a flow cytometric study.
Br J Cancer. 80:1271–1274. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chandanos E, Rubio CA, Lindblad M, et al:
Endogenous estrogen exposure in relation to distribution of
histological type and estrogen receptors in gastric adenocarcinoma.
Gastric Cancer. 11:168–174. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matsui M, Kojima O, Uehara Y and Takahashi
T: Characterization of estrogen receptor in human gastric cancer.
Cancer. 68:305–308. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu CW, Lui WY, P’eng FK and Chi CW:
Hormonal therapy for stomach cancer. Med Hypotheses. 39:137–139.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Machado JC, Carneiro F, Gärtner F, Ribeiro
P and Sobrinho-Simões M: Female sex hormone receptors are not
involved in gastric carcinogenesis. A biochemical and
immunohistochemical study. Eur J Cancer Prev. 3:31–37. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Singh S, Poulsom R, Wright NA, Sheppard MC
and Langman MJ: Differential expression of oestrogen receptor and
oestrogen inducible genes in gastric mucosa and cancer. Gut.
40:516–520. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu CY, Guo JL, Jiang ZN, et al: Prognostic
role of estrogen receptor α and estrogen receptor β in gastric
cancer. Ann Surg Oncol. 17:2503–2509. 2010.
|
|
38
|
Gan L, He J, Zhang X, et al: Expression
profile and prognostic role of sex hormone receptors in gastric
cancer. BMC Cancer. 12:5662012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kitaoka H: Sex hormone dependency and
endocrine therapy in diffuse carcinoma of the stomach]. Gan To
Kagaku Ryoho. 10:2453–2460. 1983.(In Japanese).
|
|
40
|
Matsui M, Kojima O, Kawakami S, Uehara Y
and Takahashi T: The prognosis of patients with gastric cancer
possessing sex hormone receptors. Surg Today. 22:421–425. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhao XH, Gu SZ, Liu SX and Pan BR:
Expression of estrogen receptor and estrogen receptor messenger RNA
in gastric carcinoma tissues. World J Gastroenterol. 9:665–669.
2003.PubMed/NCBI
|
|
42
|
Yokozaki H, Takekura N, Takanashi A,
Tabuchi J, Haruta R and Tahara E: Estrogen receptors in gastric
adenocarcinoma: a retrospective immunohistochemical analysis.
Virchows Arch A Pathol Anat Histopathol. 413:297–302. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang M, Pan JY, Song GR, Chen HB, An LJ
and Qu SX: Altered expression of estrogen receptor α and β in
advanced gastric adenocarcinoma: correlation with prothymosin α and
clinicopathological parameters. Eur J Surg Oncol. 33:195–201.
2007.
|
|
44
|
Kitaoka H: Chemo-endocrine therapy of
diffuse carcinoma of the stomach and its clinical evaluation. Gan
No Rinsho. 31:1189–1194. 1985.(In Japanese).
|
|
45
|
Koullias GJ, Pratsinis CI, Korkolis DP, et
al: Brief tamoxifen pretreatment enhances the chemosensitivity of
gastric carcinoma cells to 5-fluorouracil in vitro. Anticancer Res.
23:1575–1580. 2003.PubMed/NCBI
|
|
46
|
Matsuyama S, Ohkura Y, Eguchi H, et al:
Estrogen receptor β is expressed in human stomach adenocarcinoma. J
Cancer Res Clin Oncol. 128:319–324. 2002.
|
|
47
|
Ryu WS, Kim JH, Jang YJ, et al: Expression
of estrogen receptors in gastric cancer and their clinical
significance. J Surg Oncol. 106:456–461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ozawa M, Ringwald M and Kemler R:
Uvomorulin-catenin complex formation is regulated by a specific
domain in the cytoplasmic region of the cell adhesion molecule.
Proc Nat Acad Sci USA. 87:4246–4250. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Conacci-Sorrell M, Simcha I, Ben-Yedidia
T, Blechman J, Savagner P and Ben-Ze’ev A: Autoregulation of
E-cadherin expression by cadherin-cadherin interactions: the roles
of β-catenin signaling, Slug, and MAPK. J Cell Biol. 163:847–857.
2003.PubMed/NCBI
|
|
50
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kanwar SS, Yu Y, Nautiyal J, Patel BB and
Majumdar AP: The Wnt/β-catenin pathway regulates growth and
maintenance of colonospheres. Mol Cancer. 9:2122010.
|
|
52
|
Hervieu V, Lepinasse F, Gouysse G, et al:
Expression of β-catenin in gastroenteropancreatic endocrine
tumours: a study of 229 cases. J Clin Pathol. 59:1300–1304.
2006.
|
|
53
|
Lin SY, Xia W, Wang JC, et al: β-catenin,
a novel prognostic marker for breast cancer: its roles in cyclin D1
expression and cancer progression. Proc Nat Acad Sci USA.
97:4262–4266. 2000.
|
|
54
|
Wong SC, Lo ES, Lee KC, Chan JK and Hsiao
WL: Prognostic and diagnostic significance of β-catenin nuclear
immunostaining in colorectal cancer. Clin Cancer Res. 10:1401–1408.
2004.
|