Introduction
In recent years, it has become apparent that
reactive oxygen species (ROS) play an important role during
induction of apoptotic cell death (1). ROS, such as H2O2
and O2−, are constantly produced during
metabolic processes in all living species. Under physiological
conditions, the maintenance of an appropriate level of
intracellular ROS is important in maintaining redox balance and
cell proliferation (2,3). However, excessive ROS accumulation
leads to cellular injury, including lipid peroxidation, protein
oxidation, enzyme inactivation (4)
and oxidative DNA damage (5,6).
An increase in ROS generation is a common feature of
cancer cells. Evidence suggests that most cancer cells are under
oxidative stress that is associated with increased metabolic
activity and production of ROS (7).
Several studies have provided evidence that intracellular
production of ROS can lead directly to activation of mitochondrial
permeability transition, loss of mitochondrial membrane potential
(ΔΨm), and cytochrome c release from mitochondria into the
cytoplasm, which is followed by activation of the caspase cascade
and, ultimately, apoptotic cell death (8).
Morphologically, apoptosis is characterized by
shrinkage of the cell, dramatic reorganization of the nucleus,
active membrane blebbing and fragmentation of the cell into
membrane-enclosed vesicles (apoptotic bodies) (9). In the early stage of apoptosis, the
membrane phospholipid phosphatidylserine (PS) is translocated from
the inner to the outer leaflet. Annexin V has a high affinity for
PS, which identifies apoptosis at this early stage (10).
Apoptosis has been characterized as a fundamental
cellular activity that maintains physiological balance within the
organism (11). It is involved in
immune defense mechanisms that play a necessary role in protecting
against carcinogenesis by eliminating damaged or abnormal excess
cells which have proliferated owing to the induction of various
chemical agents (12,13). The process of apoptosis is well
regulated, requiring extracellular (extrinsic) and intracellular
(intrinsic) inducers.
Upon intrinsic apoptotic stimulation, several
important events occur in the mitochondria, including the release
of cytochrome c from the mitochondria into the cytoplasm
(14,15). Cytochrome c binds to
apoptotic protease activating factor 1 (Apaf-1), which then
recruits procaspase-9 to form an apoptosome. This complex activates
caspase-9, which in turn cleaves and activates effector procaspases
to yield active effector caspases, such as caspase-3 (16). The Bcl-2 family of proteins plays an
important role in the mitochondrial pathway of apoptosis;
specifically, activation of mitochondria and release of
intermembrane contents of mitochondria are under regulatory control
of a number of Bcl-2 family proteins (17,18).
Anti-apoptotic proteins, including Bcl-2, prevent the release of
cytochrome c and pro-apoptotic proteins, such as Bax,
promote the release of cytochrome c. The extrinsic pathway
initiated by activation of the Fas receptor (also known as Apo-1 or
CD95) involves a series of death-associated molecules, including
the Fas-associated death domain-containing protein (FADD), an
adaptor protein that is recruited to the Fas receptor upon its
engagement (19,20). FADD binds to and activates
procaspase-8.
Marine invertebrates, such as sponges, tunicates and
jellyfish, exhibit complex association with diverse microorganisms
that are recognized as a prolific source of biologically active
molecules, some of which have effects on cell viability and
proliferation (21). One of these
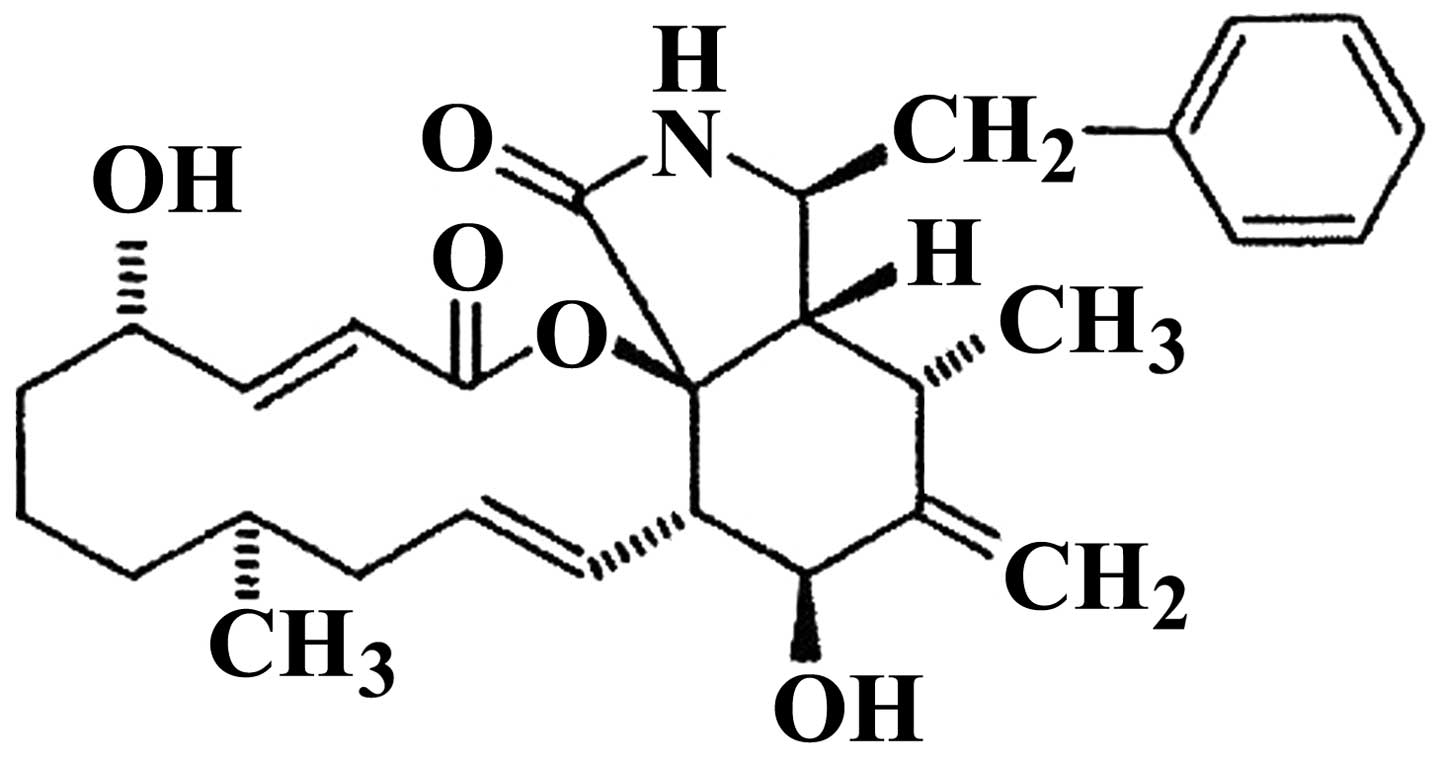
molecules, cytochalasin B (CB; Fig.
1) was isolated from Phoma sp. fungus obtained from the
giant jellyfish Nemopilema nomurai(22). This toxin interferes with
cytoskeleton functions by inhibiting actin polymerization (23,24).
At sufficiently high concentrations, cytochalasin poisoning of
cells leads to a number of morphological and functional effects,
including arborization, inhibition of endocytosis and secretion,
suppression of cytoplasmic division and enucleation (25,26).
Moreover, in a previous study by Kulms et al(27), CB was shown to cause apoptosis via
the extrinsic pathway. In the present study, we found that CB
exhibited a marked cytotoxic effect on HeLa cells via caspase
activation during apoptosis and we investigated the underlying
mechanism. Our results indicate that CB elicits apoptosis via both
the extrinsic and the intrinsic pathways.
Materials and methods
Chemicals
CB was a gift from Dr J.H. Jung. CB was dissolved in
dimethyl sulfoxide (DMSO). Eagle’s minimum essential medium (EMEM),
penicillin and trypsin-EDTA were purchased from HyClone (Logan, UT,
USA). Fetal bovine serum (FBS) was obtained from Gibco-BRL
(Carlsbad, CA, USA). Cell Counting Kit-8 (CCK-8) was obtained from
Dojindo (Japan). The propidium iodide (PI)/RNase staining buffer
and Annexin-FITC kit for apoptosis were from BD Pharmingen (San
Diego, CA, USA).
Cell culture
HeLa cells were obtained from the American Type
Culture Collection (ATCC, Manassas, VA, USA) and were cultured in
EMEM supplemented with 10% FBS 37°C in a humidified atmosphere with
5% CO2.
Cell viability and proliferation
assay
HeLa cells were plated at 5×103
cells/well in a 96-well microplate. After 24 h, media were
substituted by fresh media containing CB at various concentrations
(5, 10, 20 and 40 μM). The plate was incubated for a further 48 h
and the cell viability was then assessed using a WST-8 assay
according to the manufacturer’s recommendations. The optical
density for living cells was read at 450 nm in a multimicroplate
reader (Synergy HT; BioTek Instruments, Inc. Winooski, VT, USA)
(28). For the cell proliferation
assay, cells were seeded at 5×103 cells/ml media into
96-well plates and treated with or without CB (8 μM) for various
time periods.
Annexin V-FITC/PI apoptotic analysis
Cells (5×105 cells in a 60-mm petri
dish), treated with or without CB, were collected by trypsinization
and washed with ice-cold phosphate buffered saline (PBS) via
centrifugation. Then, 1×105 cells were resuspended in
100 μl of binding buffer and stained with 5 μl of Annexin V-FITC
and 10 μl of PI (50 μg/ml) for 15 min at room temperature, in the
dark. Analysis was performed by FACSCalibur flow cytometer
(Becton-Dickinson, San Jose, CA, USA) with 10,000 events/analysis.
The data were analyzed using CellQuest software (Becton-Dickinson
Instruments, Franklin Lakes, NJ, USA).
Measurement of apoptotic cell
morphology
HeLa cells were distributed (1×105
cells/well) into a 24-well plate and allowed to adhere overnight.
The cells were treated with CB (8 μM) for 24 and 48 h. Non-treated
wells received an equivalent volume of DMSO (<0.1%) as control.
Optic phase-contrast images were captured with a Phase Contrast-2,
ELWD 0.3 inverted microscope (Nikon, Japan).
DNA content analysis
Cells (3×105 cells in a 60-mm petri
dish), treated with or without CB, were collected by trypsinization
and washed with ice-cold PBS via centrifugation. The cells were
suspended in PBS and fixed with 70% ethanol (v/v). Samples were
washed with ice-cold PBS and stained with PI/RNase staining buffer
for 15 min at room temperature. The number of cells in different
phases of the cell cycle was analyzed using a FACScan flow
cytometer analysis system. The percentage of cells in the different
phases of cell cycle was determined using Modifit software
(Becton-Dickinson Instruments).
[3H]-thymidine incorporation
assay
The [3H]-dTTP incorporation assay was
performed as previously described (29). Briefly, HeLa cells were applied to
12-well plates in growth medium. After the cells had grown to
70–80% confluence, they were rendered quiescent by incubation for
24 h in EMEM containing 2% FBS. Cells were treated with or without
CB in EMEM supplemented with 10% FBS and the cultures were then
incubated for 21 or 45 h. [3H]-dTTP was added at 1
μCi/ml (1 μCi=37 kBq) and incubated for a further 3 h. Incorporated
[3H]-dTTP was extracted in cell lysis buffer and
measured in a liquid scintillation analyzer (Tris-Crab 2910TR;
Perkin-Elmer Inc., Waltham, MA, USA).
Measurement of intracellular ROS
Generation of ROS was assessed by using the
fluorescent indicator 2,7-dichlorodihydrofluorescein
(H2DCFDA), a cell-permeable indicator for ROS shown to
react with H2O2(30). As previously described,
H2DCFDA was oxidized to highly green fluorescent
2,7-dichlorofluorescein (DCF) by the generation of ROS. Cells
(3×105 cells in a 60-mm petri dish), treated with or
without CB, were collected by trypsinization and centrifugation.
Samples were washed with ice-cold PBS and stained with 2 μl
H2DCFDA for 30 min at 37°C in a dark room. Relative
fluorescence intensities were monitored using the FACSCalibur flow
cytometer and analyzed using CellQuest software with the FL-1
channel (green) set to 530 nm.
Measurement of ΔΨm
Depolarization of the mitochondrial membrane was
detected using a fluorescent probe, rhodamine 123. During
apoptosis, the integrity of the mitochondrial membrane is
disrupted, which leads to depolarization of the membrane and
opening of mitochondrial permeability transition pores and release
of sequestered rhodamine 123. Cells (3×105 cells in a
60-mm petri dish), treated with or without CB, were collected by
trypsinization and centrifugation. Samples were washed with
ice-cold PBS and stained with 5 μl rhodamine 123 for 30 min at 37°C
in a dark room. Relative fluorescence intensities were monitored
using a FACSCalibur flow cytometer and analyzed using CellQuest
software with the FL-1 channel.
Western blot analysis
After cells were treated with or without CB, total
cell lysates and cytosolic fractions were prepared as previously
described (31). Protein contents
of the lysates were determined by the Bradford protein assay
(Bio-Rad, Hercules, CA, USA). Proteins (30 μg) were resolved by
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto nitrocellulose membranes
(Schleicher & Schuell, BioScience, Inc., Keene, NH, USA) by
western blotting. The results were quantified using Image J v. 1.43
software (National Institutes of Health, Bethesda, MD, USA). The
following primary polyclonal antibodies were used: β-actin, Bcl-2,
caspase-9, caspase-8 (1:1,000 dilution; Cell Signaling Technology
Inc, Danvers, MA, USA), caspase-3, p53 (1:300; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) and Bax (1:1,000; BD
Pharmingen, San Diego, CA, USA).
Statistical analysis
All results reported were obtained from at least 3
independent experiments with similar results and the results are
expressed as means ± standard deviation (SD) in quantitative
experiments. Statistical analysis was performed by one-way analysis
of variance (ANOVA). P<0.05 was considered to indicate a
statistically significant difference. Microsoft Excel 2007 was used
for statistical and graphical evaluations.
Results
CB inhibits cell viability
In the present study, we investigated the
cytotoxicity of 48-h treatment with various concentrations (5, 10,
20 and 40 μM) of CB on the growth of HeLa human cervical carcinoma
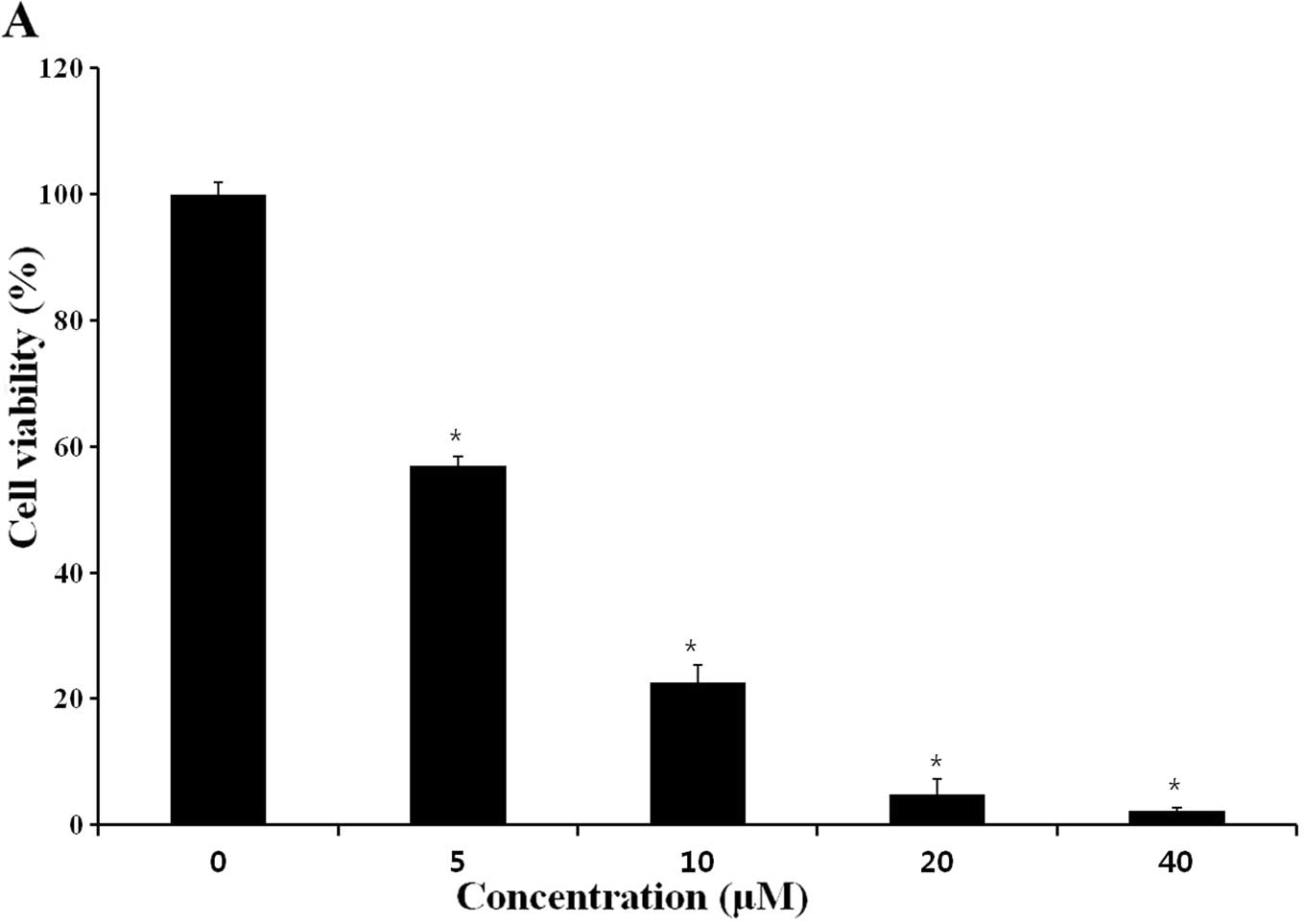
cells, using a WST-8 assay. As shown in Fig. 2A, CB inhibited cell viability, with
an IC50 of ~7.9 μM. Treatment of HeLa cells with 8 μM of
CB for 0, 12, 24, 36, 48 and 60 h, inhibited proliferation, whereas
untreated cells maintained exponential proliferation (Fig. 2B). Thus, CB treatment significantly
inhibited growth of HeLa cells in both a concentration- and a
time-dependent manner. These data indicate that CB may suppress
cancer cell proliferation or induce cancer cell apoptosis, leading
to growth-inhibition of cancer cells.
CB induces apoptosis
Light microscopy revealed morphological
characteristics of apoptosis in cells treated with an
IC50 concentration of CB at various time points. As
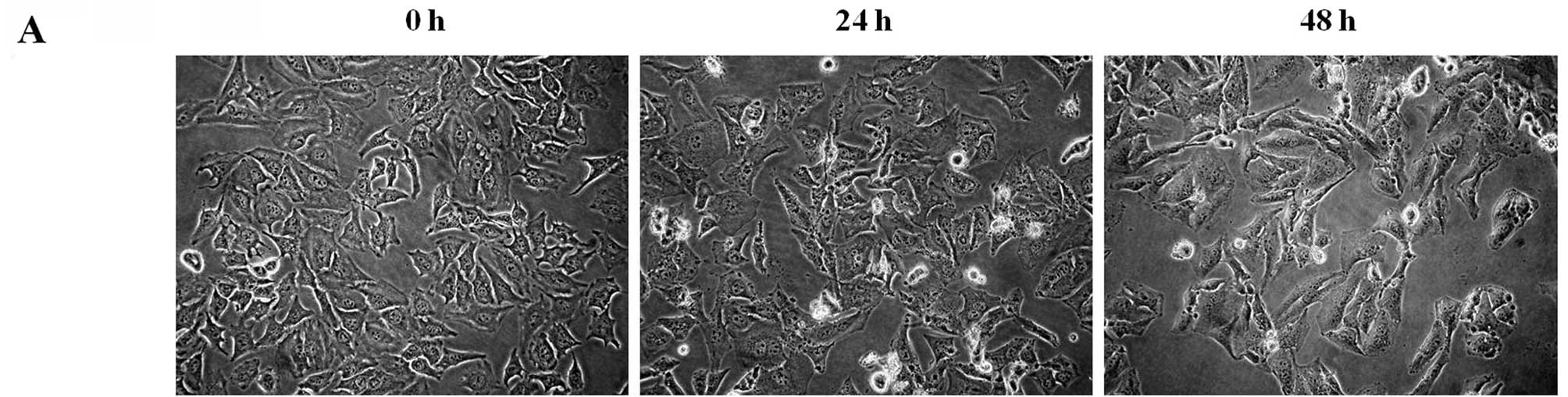
shown in Fig. 3A, untreated cells
spread regularly throughout the culture plates and grew to near
confluence. After 24 h of treatment, the cells showed a decrease in
size and rounding up of cells, with increased intercellular spaces,
but the majority of the attached cells retained a normal cellular
shape. After 48 h of treatment, the number of shrunk and floating
cells increased, as did the intercellular spaces and distinct
changes in morphology were observed.
Further evidence for the activation of apoptosis was
obtained by double-staining of cell cultures with PI and Annexin
V-FITC. Annexin V is a protein that binds to PS, which is
translocated from the inner to the outer membrane leaflet early in
the apoptotic process, with high affinity. Significant differences
were observed between the control and treated cells. As shown in
Fig. 3B, 93.58% of vehicle
alone-treated HeLa cells were viable (Annexin
V−/PI−) and 1.03% were early apoptotic cells
(Annexin V+/PI−). However, after 48 h of
treatment with CB, the percentage of early apoptotic cells
increased significantly from 3.15–16.87%, and late apoptotic (or
necrotic cells) cells increased from 9.84–17.21%. Taken together,
these findings provided evidence that CB induced apoptosis in HeLa
cells.
CB causes S-phase arrest
Cell growth and inhibition are both tightly
regulated through cell cycle control (32) and dysregulation of cell cycle
progression has been implicated in the initiation of apoptosis
(33,34). In order to further examine the
effects of CB on the cell cycle, cell cycle distribution of HeLa
cells treated by CB (8 μM) was studied by investigating DNA
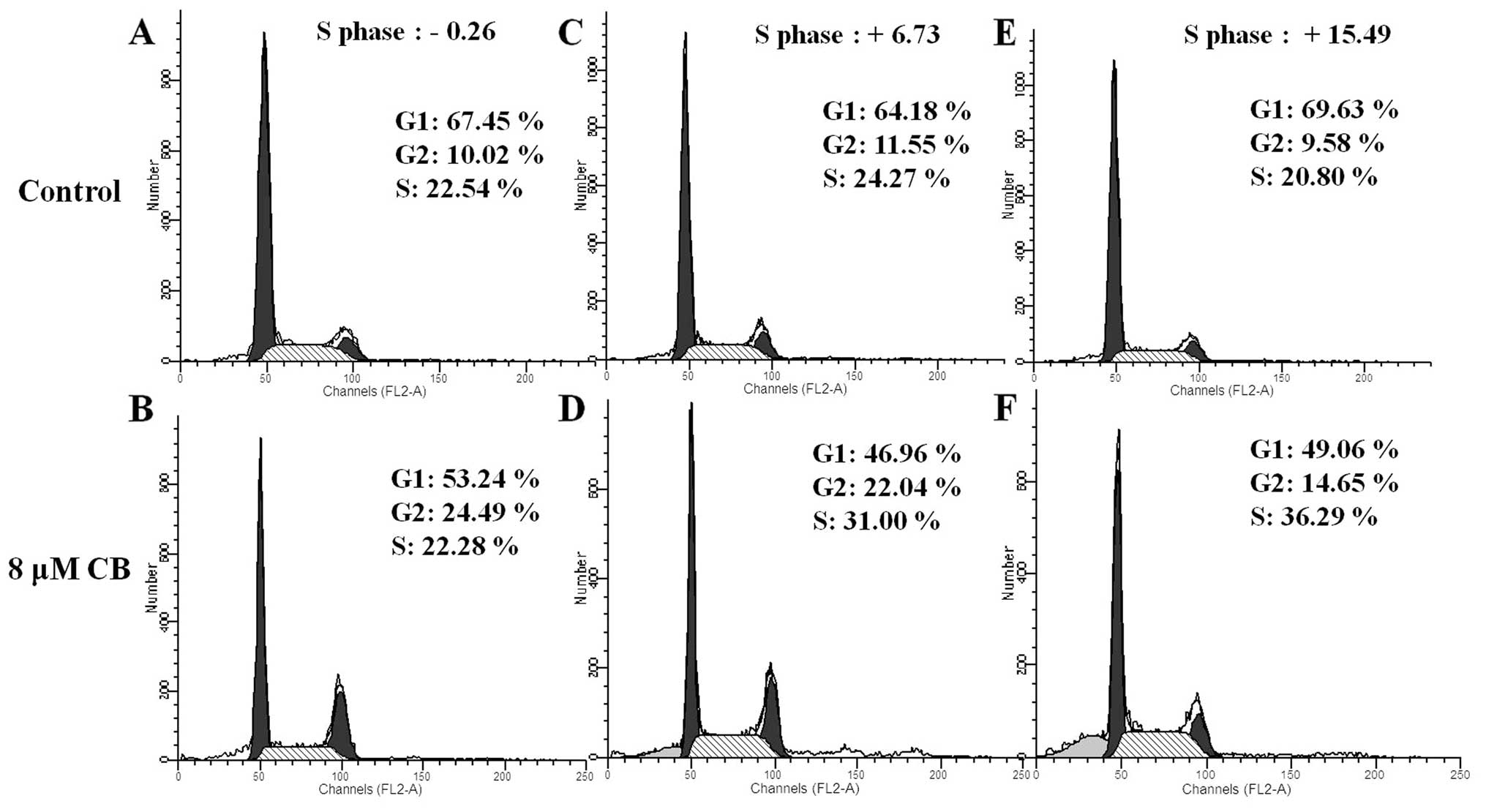
content. As shown in Fig. 4, for
all treatment durations, the percentage of cells in G2 phase was
increased. At 36 h, the percentage of cells in S phase was
increased. These results suggest that CB inhibited HeLa cell
proliferation, via S-phase arrest, in a time-dependent manner. We
hypothesized that CB induced cell cycle arrest in S phase due to
the inhibition of DNA replication. To confirm this hypothesis, we
analyzed DNA replication in cells treated with CB by determining
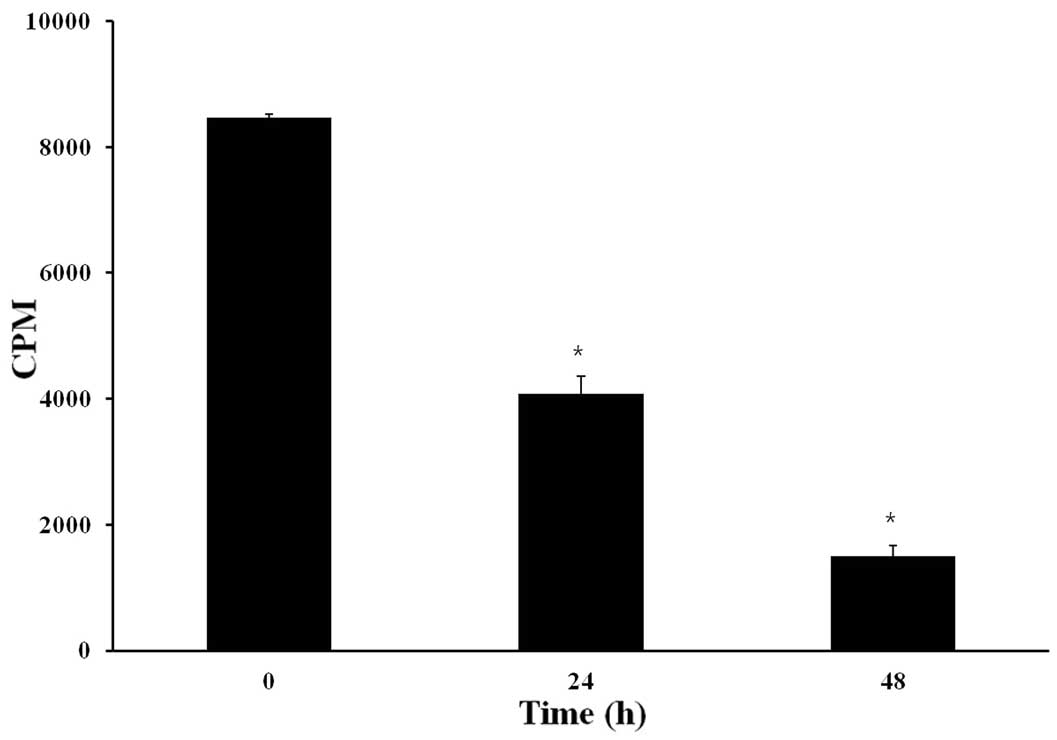
the incorporation of [3H]-dTTP. As shown in Fig. 5, CB inhibited [3H]-dTTP
incorporation after 24 and 48 h of treatment by ~50 and 75%,
respectively.
CB increases ROS and disrupts ΔΨm
Several studies have reported that generation of ROS
is associated with disruption of ΔΨm (35). CB was found to be capable of
generating intracellular ROS when examined by the cell permeable
dye H2DCF-DA. Treatment of cells for 3 h with 8 μM of CB
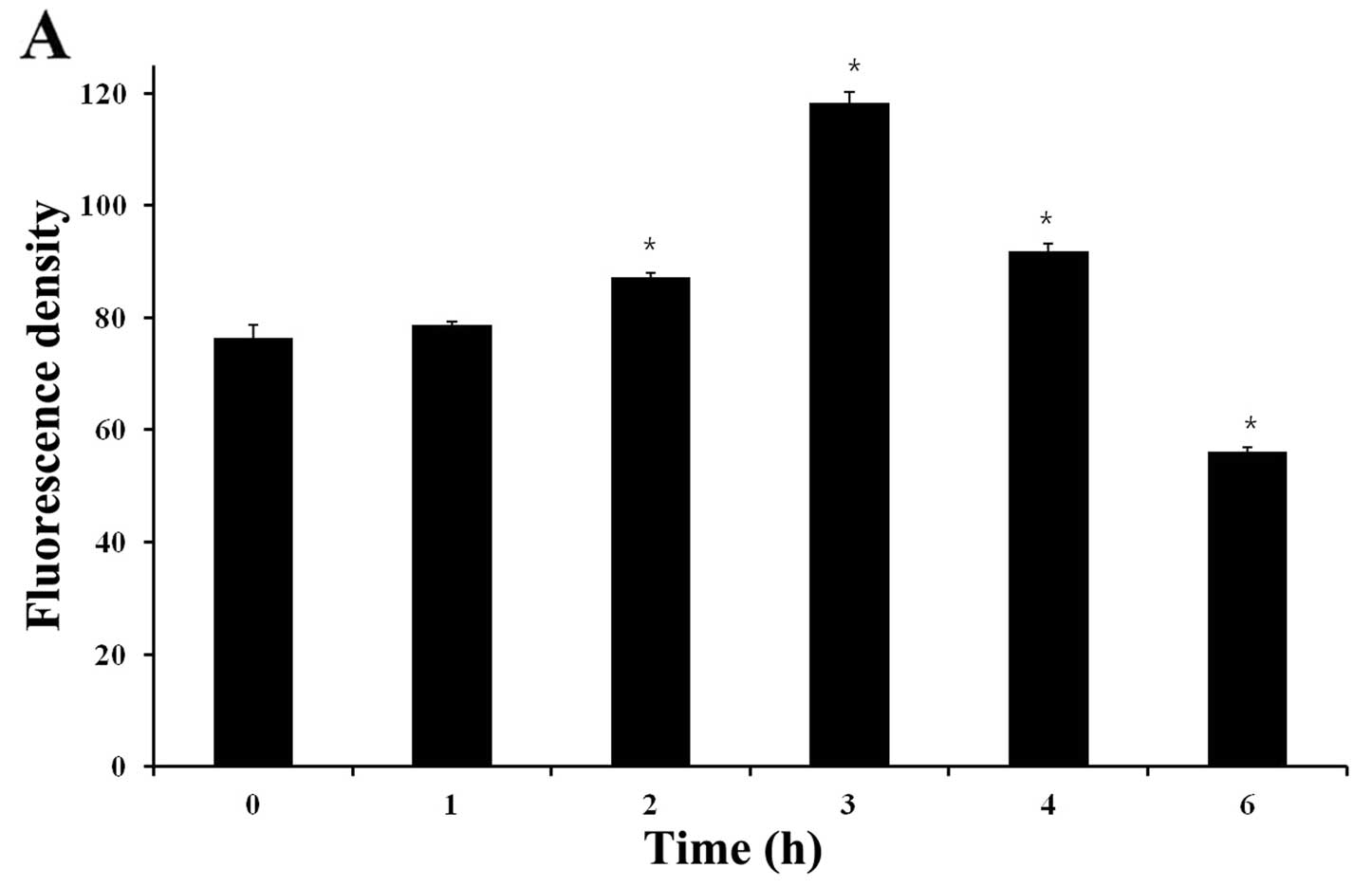
induced a change in ROS generation compared to control. Fig. 6A shows that the mean H2DCF-DA
fluorescence increased from ~76.48 (0 h) to 118.35 (3 h) after
treatment with 8 μM of CB.
Mitochondrial membrane depolarization (MMP) is a
known event associated with apoptosis. A decrease in
H2DCF-DA fluorescence indicates a loss of MMP. In this
study, we detected loss of MMP after treatment of cells with CB by
staining HeLa cells with rhodamine 123. Fig. 6B showed that the mean rhodamine 123
fluorescence decreased from ~89.99 (0 h) to 73.55 (6 h) and 43.63
(12 h) by treatment with 8 μM of CB, suggesting that mitochondria
was involved in CB-induced apoptosis.
CB-induced apoptosis is mediated via the
mitochondrial pathway
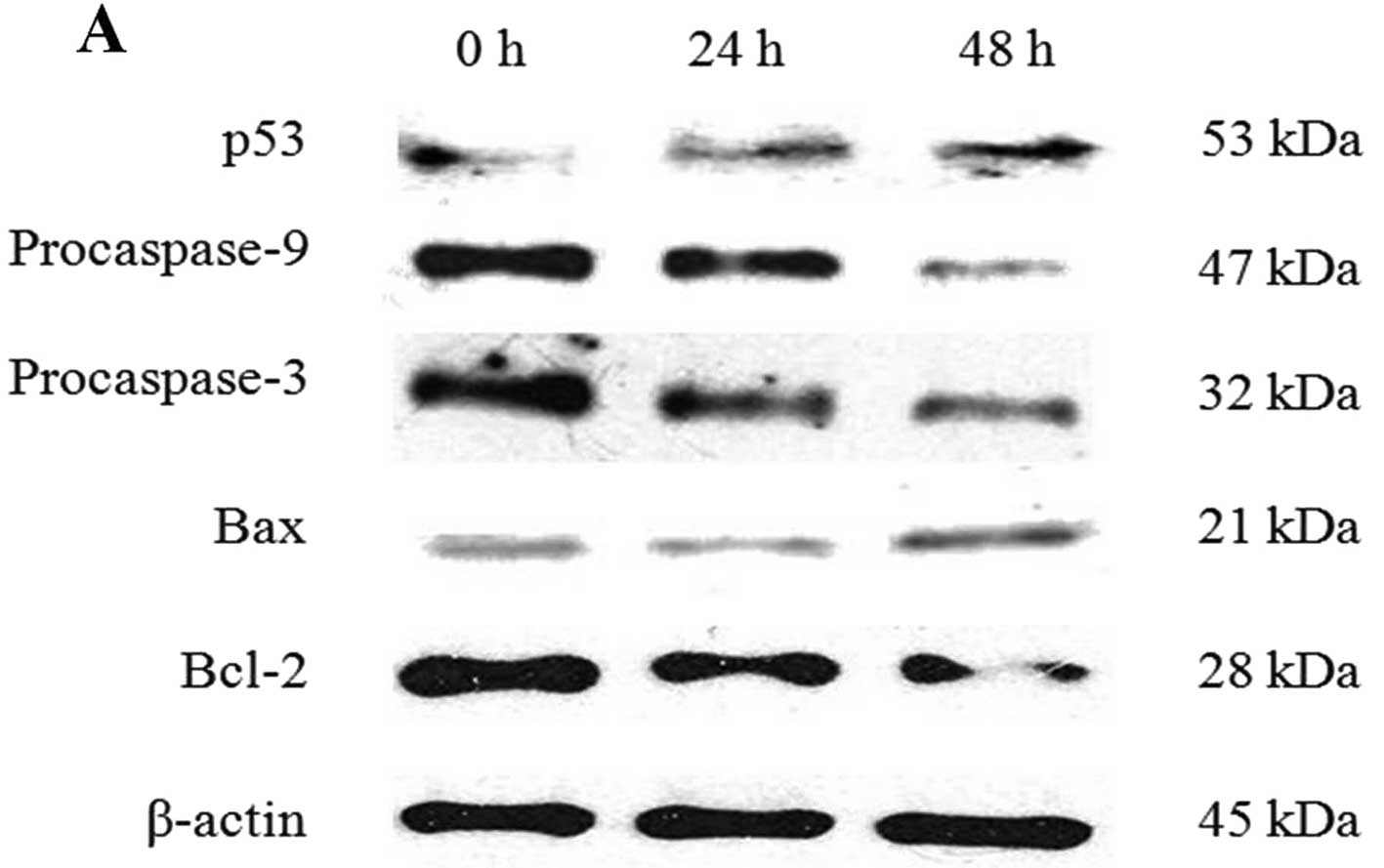
The expression of apoptosis-related proteins was
analyzed by western blotting. Fig.
7B shows that the expression of procaspase-8 was decreased
after CB treatment of HeLa cells. This is in agreement with a
previous report by Kulms et al(27), who found that CB caused apoptosis
via activation of CD95, the Fas receptor. To further characterize
CB-induced apoptosis, we examined whether CB also activates the
intrinsic apoptotic pathway in HeLa cells.
CB induced activation of caspases, increased p53
levels, and regulated the expression of the pro-apoptotic protein
Bax and the anti-apoptotic protein Bcl-2 (Fig. 7A). The Bcl-2 family proteins, Bax
and Bcl-2, play important roles in initiating the mitochondrial
death cascade (36). In particular,
protein expression of Bax was increased and that of Bcl-2 was
decreased, resulting in an increase in the Bax/Bcl-2 ratio
(Fig. 7C).
To determine which apoptotic pathway is activated by
CB, we examined the activity of procaspase-8 and -9, the important
proteins in the extrinsic and intrinsic pathway, respectively.
Collectively, these findings demonstrate that CB-induced apoptosis
in HeLa cells is mediated via the mitochondrial pathway.
Discussion
Cytochalasin B (CB) is a cell-permeable mycotoxin.
It inhibits cytoplasmic division by blocking the formation of
contractile microfilaments. It inhibits cell movement and induces
nuclear extrusion. In this study, we investigated the effects of CB
on human cervical carcinoma HeLa cells. We found that CB inhibited
HeLa cell proliferation, and induced S-phase arrest and apoptosis
through the generation of ROS and the disruption of MMP. CB
treatment significantly inhibited the growth of HeLa cells in both
a concentration- and a time-dependent manner. CB exhibited
inhibition on cell viability with an IC50 of
approximately 7.9 μM after 48 h treatment and inhibited
proliferation, whereas untreated cells maintained an exponential
proliferation state.
We analyzed DNA content using flow cytometry and
found that CB inhibited HeLa cell proliferation via S-phase arrest
in a time-dependent manner. During the S phase of the cell division
cycle, cells replicate their DNA. If DNA replication is blocked by
an inhibitor or if the template is damaged by radiation or other
factors, signals are generated that can induce cell-cycle arrest or
apoptosis (37). Therefore, we
hypothesized that cell cycle arrest in S phase was induced due to
the inhibition of DNA replication; this hypothesis was supported by
our finding that [3H]thymidine incorporation was
inhibited by CB treatment after 24 and 48 h. Morphological changes
of cell apoptosis were clearly observed, and the result of Annexin
V-FITC/PI double-staining indicated that CB induced early apoptosis
in HeLa cells.
Recent studies have reported that generation of ROS
is associated with disruption of MMP, thereby triggering a series
of mitochondrial-associated events, including apoptosis (35). In the present study, we found that
CB induced HeLa cell apoptosis, which was associated with a
significant increase in the levels of intracellular ROS and
disruption of MMP. Based on these results, we performed western
blot analysis to further investigate the apoptotic pathway
activated in CB-treated HeLa cells. A direct link between ROS and
apoptosis is possible as ROS production causes dimerization of Bax
in the cytosol. The Bcl-2 family proteins Bax and Bcl-2 play
important roles in initiating the mitochondria-mediated apoptotic
pathway (36). Anti-apoptotic
protein Bcl-2 prevents this process by preserving mitochondrial
integrity. Thus, the ratio of Bax to Bcl-2 is crucial to sustaining
drug-induced apoptosis in the mitochondria-mediated apoptotic
pathway (38). The present study
showed that CB upregulated the expression level of Bax and
downregulated the expression level of Bcl-2, eventually leading to
an increase in the ratio of Bax/Bcl-2 protein levels. The release
of mitochondrial cytochrome c facilitates the formation of
the apoptosome complex, consisting of Apaf-1 and caspase-9, which
subsequently activates effector caspases, such as caspase-3 and
leads to apoptosis (16). In the
present study, activation of both caspase-9 and -3 were detected.
Moreover, the level of p53, a well-known tumor suppressor protein,
increased. These results suggest that CB induced apoptosis via the
mitochondria-dependent pathway.
In conclusion, the results of the present study
revealed that CB inhibited proliferation of HeLa cells, by inducing
arrest in the S phase of the cell cycle and by inducing apoptosis
through the generation of ROS and disruption of MMP. Although a
previous study by Kulms et al(27) suggested that CB causes apoptosis via
the extrinsic pathway, our findings suggest that CB activates the
intrinsic apoptotic pathway. Therefore, CB triggers apoptosis via
both the intrinsic pathway as well as the extrinsic pathway. The
results of our study suggest that CB may suppress cancer cell
proliferation or induce cancer cell apoptosis, inhibiting the
growth of cancer cells.
Acknowledgements
This study was supported by the 2012 Inje University
research grant.
References
|
1
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Preeta R and Nair RR: Stimulation of
cardiac fibroblast proliferation by cerium: a superoxide
anion-mediated response. J Mol Cell Cardiol. 31:1573–1580. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nicotera TM, Privalle C, Wang TC, et al:
Differential proliferative responses of Syrian hamster embryo
fibroblasts to paraquat-generated superoxide radicals depending on
tumor suppressor gene function. Cancer Res. 54:3884–3888. 1994.
|
|
4
|
Morel I, Lescoat G, Cillard J, et al:
Kinetic evaluation of freemalondialdehyde and enzyme leakage as
indices of iron damage in rat hepatocyte cultures. Involvement of
free radicals. Biochem Pharmacol. 39:1647–1655. 1990. View Article : Google Scholar
|
|
5
|
Randerath K, Randerath E, Smith CV and
Chang J: Structural origins of bulky oxidative DNA adducts (type II
I-compounds) as deduced by oxidation of oligonucleotides of known
sequence. Chem Res Toxicol. 9:247–254. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ames BN: Endogenous DNA damage as related
to cancer and aging. Mutat Res. 214:41–46. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Szatrowski TP and Nathan CF: Production of
large amounts of hydrogen peroxide by human tumor cells. Cancer
Res. 51:794–798. 1991.PubMed/NCBI
|
|
8
|
Chan WH, Wu CC and Yu JS: Curcumin
inhibits UV irradiation-induced oxidative stress and apoptotic
biochemical changes in human epidermoid carcinoma A431 cells. J
Cell Biochem. 90:327–338. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Earnshaw WC: Nuclear changes in apoptosis.
Curr Opin Cell Biol. 7:337–343. 1995. View Article : Google Scholar
|
|
10
|
Miao C, Du J, Dang HT, et al: Apoptotic
activity of fatty acid derivatives may correlate with their
inhibition of DNA replication. Int J Oncol. 33:1291–1298.
2008.PubMed/NCBI
|
|
11
|
Zhang X, Zhao J, Kang S, et al: A novel
cromakalim analogue induces cell cycle arrest and apoptosis in
human cervical carcinoma HeLa cells through the caspase- and
mitochondria-dependent pathway. Int J Oncol. 39:1609–1617.
2011.
|
|
12
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown JM and Wouters BG: Apoptosis, p53,
and tumor cell sensitivity to anticancer agents. Cancer Res.
59:1391–1399. 1999.PubMed/NCBI
|
|
14
|
Reed JC and Green DR: Remodeling for
demolition: changes in mitochondrial ultrastructure during
apoptosis. Mol Cell. 9:1–3. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang C and Youle RJ: The role of
mitochondria in apoptosis. Ann Rev Genet. 43:95–118. 2009.
View Article : Google Scholar
|
|
16
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marzo I, Brenner C, Zamzami N, et al: The
permeability transition pore complex: a target for apoptosis
regulation by caspases and Bcl-2 related proteins. J Exp Med.
187:1261–1271. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuwana T and Newmeyer DD: Bcl-2-family
proteins and the role of mitochondria in apoptosis. Curr Opin Cell
Biol. 15:691–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boldin MP, Varfolomeev EE, Pancer Z, et
al: A novel protein that interacts with the death domain of
Fas/APO1 contains a sequence motif related to the death domain. J
Biol Chem. 270:7796–7798. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chinnaiyan AM, O’Rourke KO, Tewari M and
Dixit VM: FADD, a novel death domain-containing protein, interacts
with death domain of Fas and initiates apoptosis. Cell. 81:505–512.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wilkinson CR: Symbiotic interactions
between marine sponges and algae. Algae and Symbioses. Reisser W:
Biopress; Bristol: pp. 112–151. 1992
|
|
22
|
Kim EL, Li JL, Dang HT, et al: Cytotoxic
cytochalasins from the endozoic fungus Phoma sp of the giant
jellyfish Nemopilema nomurai. Bioorg Med Chem Lett.
22:3126–3129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Spooner BS: Cytochalasins as probes in
selected morphogenetic processes. Cytochalasins. Biochemical and
Cell Biological Aspects. Tanenbaum SW: Elsevier/North Holland
Biomedical Press; Amsterdam: pp. 161–189. 1978, PubMed/NCBI
|
|
24
|
Bonder EM and Mooseker MS: Cytochalasin B
slows but does not prevent monomer addition at the barbed end of
the actin filament. J Cell Biol. 102:282–288. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Foissner I and Wasteneys GO: Wide-ranging
effects of eight cytochalasins and latrunculin A and B on
intracellular motility and actin filament reorganization in
characean internodal cells. Plant Cell Physiol. 48:585–587. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ohmori H and Toyama S and Toyama S: Direct
proof that the primary site of action of cytochalasin on cell
motility processes is actin. J Cell Biol. 116:933–941. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kulms D, Düssmann H, Pöppelmann B, et al:
Apoptosis induced by disruption of the actin cytoskeleton is
mediated via activation of CD95 (Fas/APO-1). Cell Death Differ.
9:598–608. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tominaga H, Ishiyama M, Ohseto F, et al: A
water-soluble tetrazolium salt useful for colorimetric cell
viability assay. Anal Commun. 36:47–50. 1999. View Article : Google Scholar
|
|
29
|
Lin SY, Liu JD, Chang HC, et al: Magnolol
suppresses proliferation of cultured human colon and liver cancer
cells by inhibiting DNA synthesis and activating apoptosis. J Cell
Biochem. 84:532–544. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bobyleva V, Pazienza TL, Maseroli R, et
al: Decrease in mitochondrial energy coupling by thyroid hormones:
a physiological effect rather than a pathological hyperthyroidism
consequence. FEBS Lett. 430:409–413. 1998. View Article : Google Scholar
|
|
31
|
Qi F, Li A, Zhao L, et al: Cinobufacini,
an aqueous extract from Bufo bufo gargarizans Cantor,
induces apoptosis through a mitochondria-mediated pathway in human
hepatocellular carcinoma cells. J Ethnopharmacol. 128:654–661.
2010.PubMed/NCBI
|
|
32
|
Sánchez I and Dynlacht BD: New insights
into cyclins, CDKs, and cell cycle control. Semin Cell Dev Biol.
16:311–321. 2005.PubMed/NCBI
|
|
33
|
Lee S, Christakos S and Small MB:
Apoptosis and signal transduction: clues to a molecular mechanism.
Curr Opin Cell Biol. 5:286–291. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Smith DM, Gao G, Zhang X, et al:
Regulation of tumor cell apoptotic sensitivity during the cell
cycle (Review). Int J Mol Med. 6:503–507. 2000.PubMed/NCBI
|
|
35
|
Park MT, Kim MJ, Kang YH, et al:
Phytosphingosine in combination with ionizing radiation enhances
apoptotic cell death in radiation-resistant cancer cells through
ROS-dependent and -independent AIF release. Blood. 105:1724–1733.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang YH, Ahn EY, Ryu SH, et al:
Cytotoxicity of psammaplin A from a two-sponge association may
correlate with the inhibition of DNA replication. BMC Cancer.
4:702004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qi F, Inagaki Y, Gao B, et al: Bufalin and
cinobufagin induce apoptosis of human hepatocellular carcinoma
cells via Fas- and mitochondria-mediated pathways. Cancer Sci.
102:951–958. 2011. View Article : Google Scholar : PubMed/NCBI
|