Introduction
Doxorubicin (DOX, Adriamycin), an anthracycline
isolated from Streptomyces strains, is one of the most
efficacious anticancer drugs for the treatment of hematological
malignancies and a broad range of solid tumors (1,2).
However, the clinical application of DOX has long been limited by
its dose-dependent toxicities to the heart, kidney, liver and bone
marrow. To reduce these side effects, significant efforts have been
made to develop DOX-prodrugs, such as PK1 and PK2, which remain
inactive in blood and normal tissues but release DOX at the tumor
site (3), and which have entered
phase II/III clinical studies (4,5).
Cathepsin B (Cat B), a lysosomal cysteine protease
in normal cells and tissues, is highly upregulated in malignant
tumors and premalignant lesions at the mRNA, protein and activity
levels (6). The active cleavage
sites of Cat B cover a range of oligopeptides, including Arg-Arg,
Ala-Leu, Phe-Arg, Phe-Lys, Ala-Phe-Lys, Gly-Leu-Phe-Gly,
Gly-Phe-Leu-Gly and Ala-Leu-Ala-Leu (7–11).
Therefore, several low- and high-molecular-weight DOX prodrugs
through Cat B-cleavable oligopeptides, have been designed (4,5,12) and
have demonstrated rapid and nearly quantitative DOX release in the
presence of Cat B.
Based on the characteristics of Cat B, we designed
and developed a smart DOX prodrug, Ac-Phe-Lys-PABC-DOX (PDOX), in
which a Cat B-specific dipeptide (Phe-Lys) is introduced,
containing a self-immolative spacer para-aminobenzyloxycarbonyl
(PABC) to increase the distance between the dipeptide and DOX
(13–16), so that the dipeptide can be directly
accessable to the Cat B′ active site. PDOX remains inactive and
stable in blood circulation and normal tissues. When PDOX reaches
Cat B-enriched areas such as tumor sites, the dipeptide Phe-Lys is
cleaved by Cat B, exposing the PABC spacer that is hydrolyzed
spontaneously, releasing free DOX.
An in vivo study in a nude mouse model of
gastric cancer peritoneal carcinomatosis showed that, compared with
free DOX, PDOX produced superior anti-metastasis effects in terms
of the experimental peritoneal carcinomatosis index (ePCI) and body
weight, and reduced toxicities to the liver, kidney and
particularly the heart (16).
However, the underlying mechanism remains unclear.
It has been reported that apoptosis plays a key role
in the anticancer effects and toxicities of DOX (17), mainly via the mitochondria-centered
intrinsic pathway (18,19), which involves the release of
cytochrome c from the mitochondria (20,21).
DOX promotes the generation of reactive oxygen species (ROS) in
various cell types (22,23) and mitochondria are the major source
of ROS production (24). The
present study was aimed to investigate the in vitro
mechanisms of action of PDOX with special focus on the
mitochondria-centered intrinsic pathway.
Materials and methods
Drug preparation and cell culture
DOX (Pharmacia, Milan, Italy) and PDOX (synthesized
by Y.-P.H.) were used. MGC-803 gastric cancer cells were cultured
in 25 cm2 tissue culture flasks at 37°C in 5%
CO2, 95% air and 100% humidity. Cells were grown in
RPMI-1640 medium containing 10% newborn calf serum and 1%
penicillin and streptomycin. Throughout the study, the medium was
replaced every 2 to 3 days. The cells were passaged when grown to
90% confluence.
Immunocytochemistry
Cells were transferred onto a coverslip in 6-well
culture plates, grown for 2 days to reach 90% confluence and then
fixed by ice-cold 4% paraformaldehyde for 30 min.
Immunocytochemistry for Cat B was then performed following a
standard method (25). Briefly,
cells were first blocked with 2% bovine serum albumin (BSA) at 37°C
for 30 min. Next, cells were incubated with a primary rabbit
anti-human Cat B antibody (cat no. 3190-100, dilution 1:200;
BioVision, Mountain View, CA, USA) at 4°C overnight. Cells were
washed with Tris-buffered saline-Tween (TBST; 0.05% Tween, 0.1 M
Tris-base, 0.9% NaCl, pH 7.6) 3 times and then incubated with a
peroxidase-labled goat anti-rabbit IgG at 37°C for 30 min. The
antibody reaction products were visualized with diaminobenzidine
(Dako, Denmark). The images were captured using an Olympus BX51
fluorescence microscope equipped with an Olympus Micro DP 72
camera.
Cell viability study by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
assay
Cells were passaged and 1×104 cells in
100 μl medium were transferred into 96-well culture plates. After
24 h, the MGC-803 cells were treated with 0, 0.86, 1.72, 3.44, 6.88
and 13.76 μM of PDOX, or 0, 0.86, 1.72, 3.44, 6.88 and 13.76 μM DOX
in medium for 24, 48, 72 and 96 h, respectively; then immediately
incubated with 100 μl 0.5 mg/ml MTT at 37°C in 5%CO2,
95% air and 100% humidity for 4 h. After medium removal, 150 μl
DMSO was added and incubated for 10 min. OD560 was
obtained by the ELISA (modulus microplate), and IC50
values were calculated.
Cell cycle analysis by flow
cytometry
MGC-803 cells (2×105) in 2 ml of medium
were transferred into 6-well culture plates and cultured for 24 h.
The cells were treated with 0, 14.9 μM (IC50 of PDOX
treated for 48 h), 4.9 μM PDOX and 14.9 and 4.9 μM (IC50
of DOX treated for 48 h) DOX for 48 h. The harvested cells were
subjected to flow cytometry assay by Coulter® DNA PREP™
reagents kit according to the manufacturer’s protocol. Briefly,
cells were harvested, washed twice with phosphate-buffered saline
(PBS) and re-suspended in serum-free medium, then 50 μl DNA PREP™
LPR buffer was added and incubated in a light-protected moist
chamber for 20 min. Next, 500 μl DNA PREP™ stain was added and
incubated in the dark for 20 min. Finally, cell cycle analysis was
performed by flow cytometry (FC 500; Beckman Coulter, USA).
ROS generation and mitochondrial membrane
potential (ΔΨm) measurement
ROS generation was assayed using dichlorofluorescein
diacetate (DCF-DA; Sigma, USA). The polar derivative from DCF-DA by
intracellular esterases rapidly reacts with ROS to form a highly
fluorescent compound (26). MGC-803
cells (2×105) in 2 ml of medium were transferred into
6-well culture plates for 24 h. The medium was replaced with
serum-free medium the following day. Twelve hours later, cells were
treated with 0, 4.9 and 14.9 μM PDOX or 4.9 and 14.9 μM DOX,
respectively, for 48 h. For ROS generation, cells were immediately
incubated with 100 μM DCF-DA at 37°C for 1 h in the dark. Images
were captured using a laser confocal scanning microscope (Leica,
Wetzlar, Germany) at 488 nm excitation and 525 nm emission.
For ΔΨm assay, MGC-803 cells were treated in the
same way as above and then immediately incubated with 100 nM
MitoTracker® Red CMXRos at 37°C for 30 min in the dark.
Images were captured by a laser confocal scanning microscope at 579
nm excitation and 599 nm emission. For semi-quantitative analysis
of ROS production and ΔΨm, total fluorescence intensity was
analyzed with the LCS lite (Leica).
Mitochondrial morphology as determined by
transmission electron microscopy
MGC-803 cells (2×106) in 8 ml of medium
were transferred into 10-cm culture dishes. The cells were treated
in the same way as above and harvested cells were initially fixed
in 2.5% glutaraldehyde, then post-fixed in 1% osmic acid and
embedded in epoxide resin. Ultrathin sections were cut with an
ultramicrotome (LKB-V; Bromma, Sweden), stained with uranyl acetate
and lead citrate, and examined using a transmission electron
microscope (H-600; Hitachi, Japan) and photographed.
Western blotting
MGC-803 cells (2×105) in 2 ml of medium
were transferred into 6-well culture plates and treated in the same
way as above. The harvested cells were homogenated, centrifuged at
12,000 × g at 4°C for 30 min, and the protein concentration in
supernatants was determined using the BCA assay.
A total of 100 μg proteins were separated by
SDS-polyacrylamide gel electrophoresis (4% stacking and 10%
separating gels) and then transferred overnight onto PVDF
membranes. The membranes were blocked with 5% skimmed milk in 0.01
M PBS containing 0.05% (v/v) Tween. Next, they were immunoblotted
with rabbit anti-human p-ERK1/2 antibody (cat no. 4370, dilution
1:2,000), rabbit anti-human ERK1/2 (cat no. 4695, di1ution 1:2,000;
both from Cell Signaling Technology, Danvers, MA, USA), rabbit
anti-human cytochrome c (cat no. ab133504, di1ution 1:1,000;
Abcam, USA) for 2 h. Blots were then incubated with a
peroxidase-conjugated sheep anti-rabbit IgG (dilution 1:8,000;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 1 h and
developed using chemiluminescent detection with a Supersignal West
Pico Assay kit (Thermo, USA) and autoradiography film.
Results
Cat B expression in MGC-803 cells
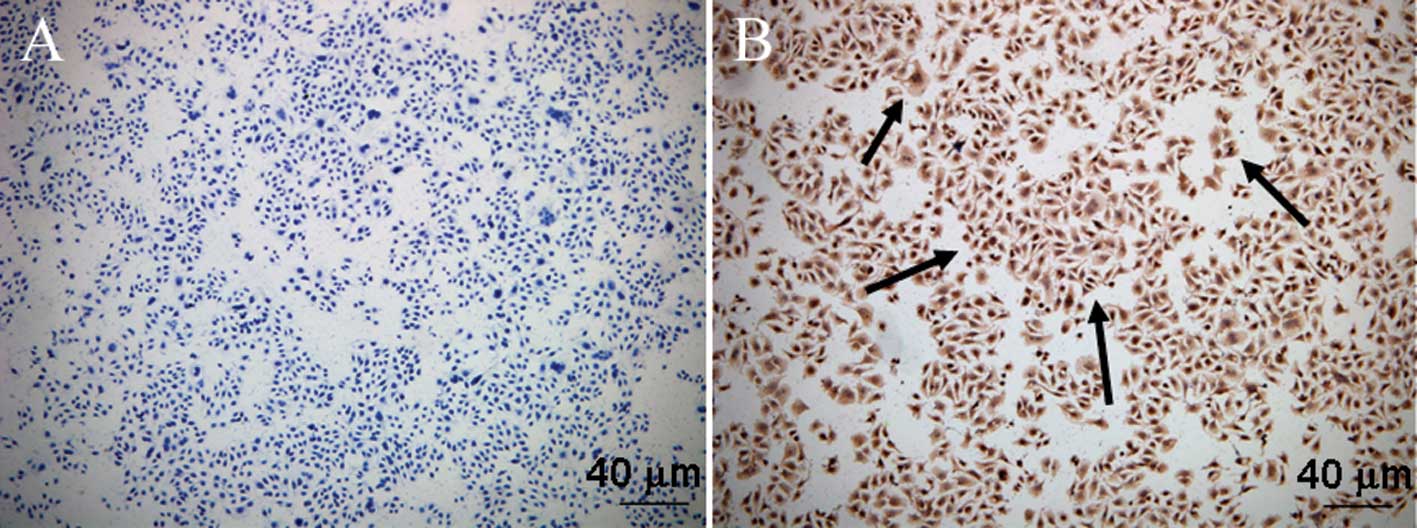
As shown in Fig. 1,
immunocytochemical analysis revealed abundant Cat B expression in
the cytoplasm of the MGC-803 cells.
Cytotoxic effects of DOX and PDOX on
MGC-803 cells
To investigate the cytotoxic effects of DOX and
PDOX, MGC-803 cells were treated with the indicated concentrations
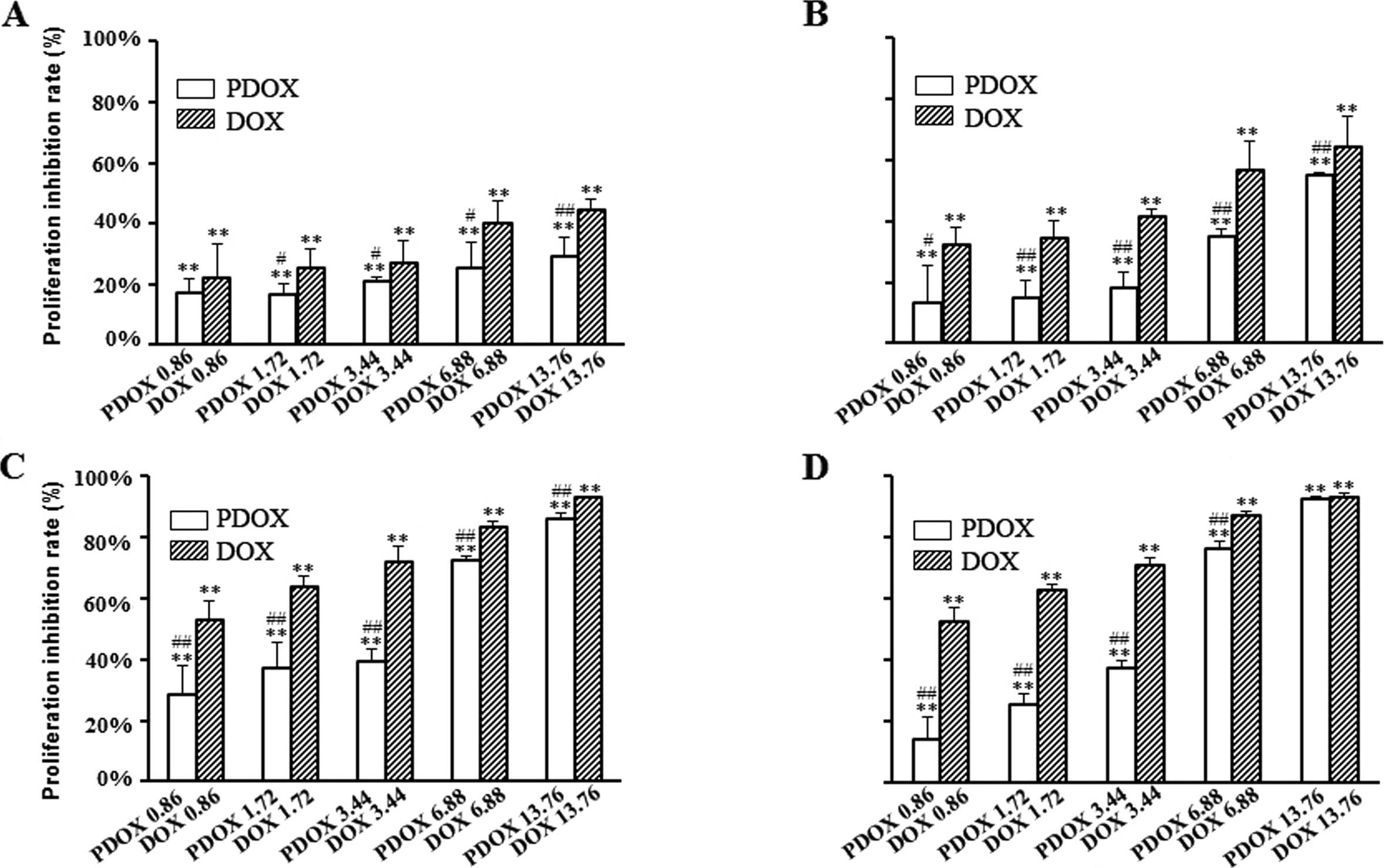
of DOX and PDOX for 24, 48, 72 and 96 h, respectively. As confirmed
by the MTT assay, DOX and PDOX triggered dose-dependent
cytotoxicity and resulted in a significant reduction in cell
viability (Fig. 2). At 48 h, the
rates of proliferation inhibition following treatment with 0.86,
1.72, 3.44, 6.88 and 13.76 μM of PDOX were 13.17±12.12, 15.10±5.46,
18.33±4.85, 34.99±2.27 and 54.86±0.93%, respectively; and following
treatment with the same concentrations of DOX were 32.30±5.47,
34.39±5.81, 41.50±2.58, 52.35±9.25 and 59.97±10.29%, respectively
(Fig 2B). At 48 h of treatment, the
IC50 concentration of PDOX (14.9 μM) was 3.04 times that
of DOX (4.9 μM).
Effects of DOX and PDOX on the cell cycle
distribution of MGC-803 cells
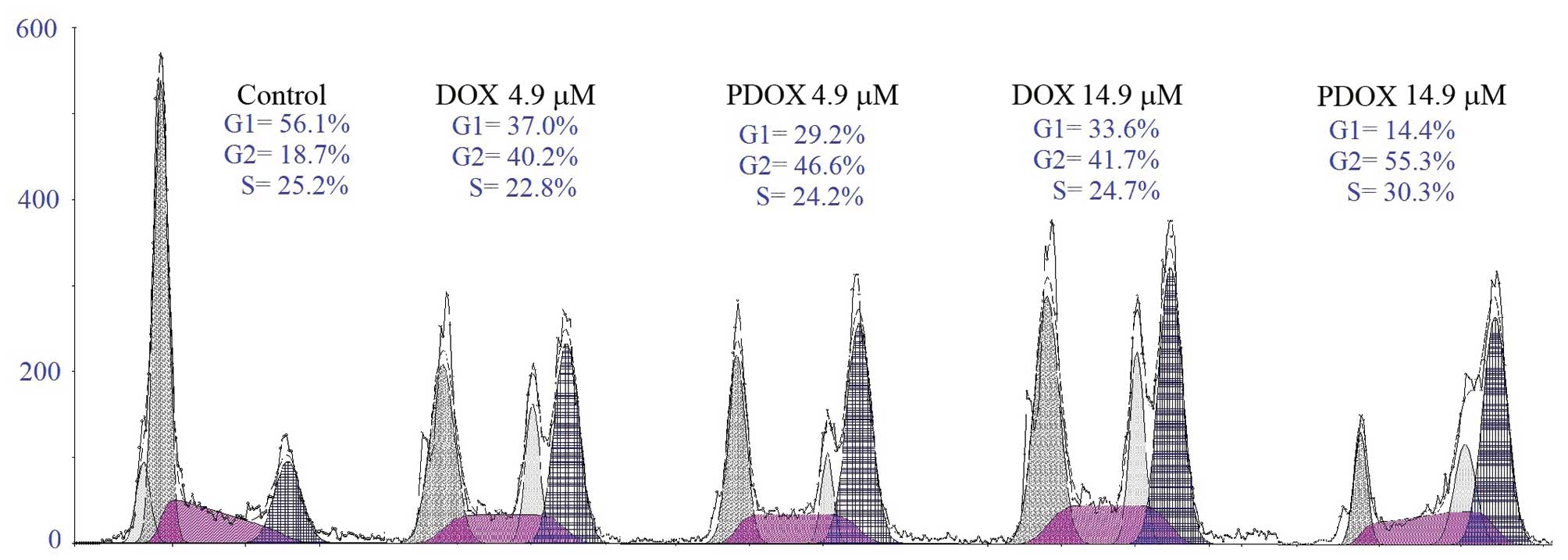
To study the effects of DOX and PDOX on the cell
cycle, MGC-803 cells were treated with 4.9 and 14.9 μM PDOX or 4.9
and 14.9 μM DOX for 48 h, respectively. The percentages of cells at
phases G1, G2 and S were 56.1, 18.7 and 25.5% in the control group;
37.0, 40.2 and 22.8% in the 4.9 μM DOX group; 29.2, 46.6 and 24.2%
in the 4.9 μM PDOX group; 33.6, 41.7 and 24.7% in the 14.9 μM DOX
group; and 14.4, 55.3 and 30.3% in the 14.9 μM PDOX group (Fig. 3). These results suggest that both
PDOX and DOX arrested the cell cycle at the G2/S phase. Moreover,
PDOX at IC50 arrested more cells at the G2/S phase
compared with DOX at IC50.
Effects of DOX and PDOX on ROS generation
of MGC-803 cells
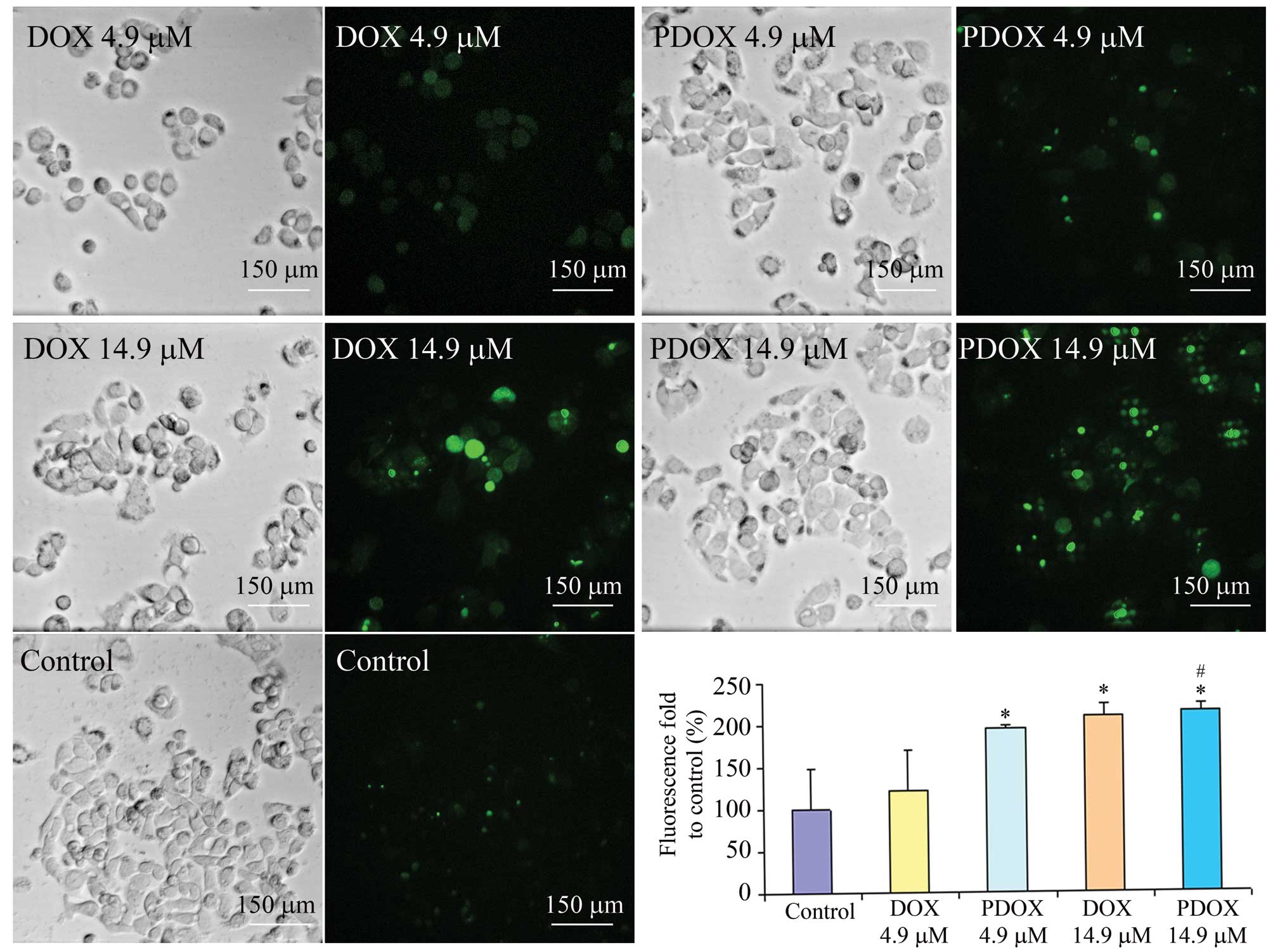
ROS generation of MGC-803 cells was significantly
increased by PDOX when compared with that by DOX (Fig. 4). Compared with the control, ROS
levels were increased by 1.20-fold (P>0.05), 1.95-fold
(P<0.05), 2.09-fold (P<0.05) and 2.15-fold (P<0.05)
following treatment of 4.9 μM DOX or PDOX, 14.9 μM DOX or PDOX,
respectively. Meanwhile, compared with DOX at IC50, PDOX
at IC50 significantly increased ROS generation
(1.53-fold, P<0.05).
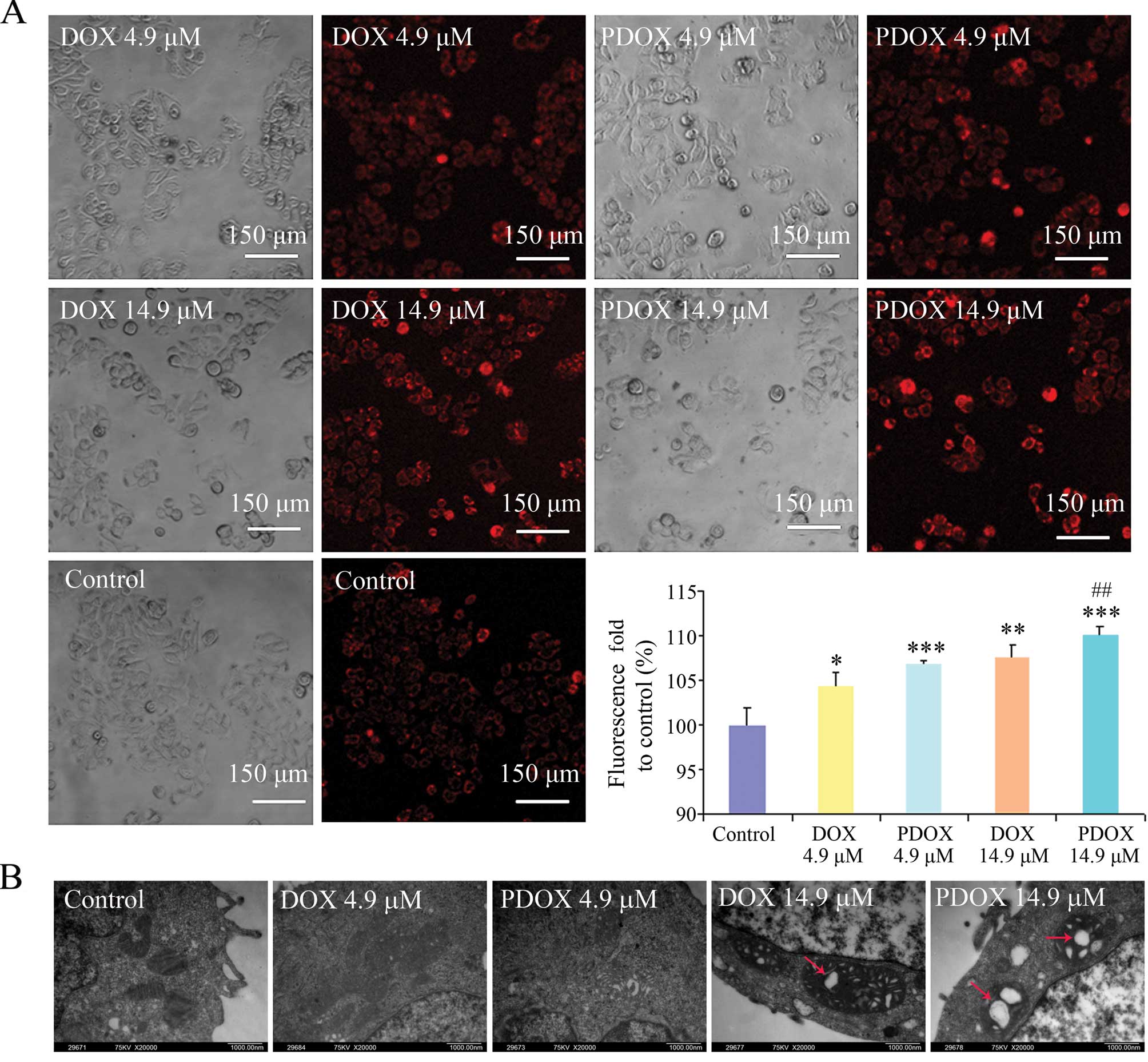
Effects of DOX and PDOX on the
mitochondria in MGC-803 cells
ΔΨm of MGC-803 cells was significantly decreased
following treatment with both DOX and PDOX (Fig. 5A). Compared with the control,
fluorescence intensity was increased by 1.04-fold (P<0.05),
1.07-fold (P<0.001), 1.08-fold (P<0.01) and 1.10-fold
(P<0.001) after treatment with 4.9 μM DOX or PDOX and 14.9 μM
DOX or PDOX, respectively. Meanwhile, compared with DOX at
IC50, PDOX at IC50 significantly increased
fluorescence values (1.53-fold, P<0.01).
Results of transmission electron microscopy revealed
that PDOX and DOX induced mitochondrial swelling, particularly in
cells treated with PDOX at IC50 (Fig. 5B).
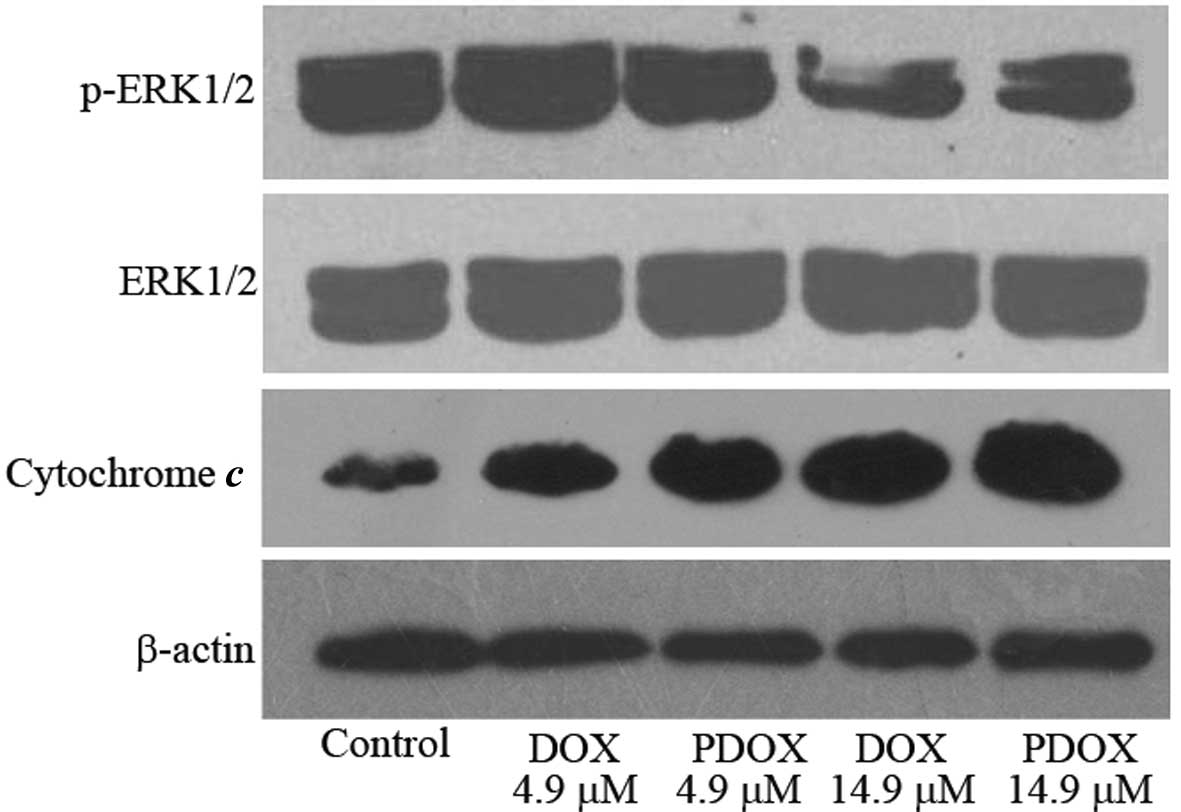
Effects of DOX and PDOX on ERK1/2
phosphorylation and cytoplasmic cytochrome c release in MGC-803
cells
Phosphorylation of ERK1/2 was significantly reduced
by both PDOX and DOX treatments, and the effect of PDOX was much
more marked. Similarly, cytoplasmic cytochrome c was
increased following both PDOX and DOX treatments, and the effect of
PDOX was more obvious (Fig. 6).
Discussion
DOX is one of the most commonly used
chemotherapeutic drugs with proven efficacy, but is associated with
serious side effects. To resolve this issue, the ‘smart’
DOX-prodrug PDOX was designed with increased in vivo
anti-metastasis effects and reduced toxicities in gastric cancer
peritoneal carcinomatosis (16).
The present study aimed to elucidate the molecular
mechanisms of action of PDOX using MGC-803 cells as a model, since
immunocytochemical analysis demonstrated that this cell line is
rich in Cat B. Our results showed that: i) the direct cytotoxicity
of PDOX on cancer cells did not exceed that of DOX since the
IC50 of PDOX was significantly higher than that of DOX;
ii) PDOX induced a greater degree of mitochondria-centered
intrinsic apoptosis than DOX, since more cytochrome c was
released from the mitochondria into the cytoplasm following PDOX
treatment; iii) PDOX promoted more ROS production and significantly
inhibited p-ERK1/2 to a greater degree than DOX. Therefore,
treatment with both PDOX and DOX caused mitochondrial swelling and
arrested the cell cycle at the G2/S phase; moreover, the effects of
PDOX on mitochondria and the cell cycle were more marked than
DOX.
Previous studies found that the two
N-(2-hydroxypropyl) methacrylamide copolymer-bound DOX-prodrugs
(PK1 and HYD) and free DOX greatly differ in regards to their
antiproliferative effect and cell death signals in EL-4 cancer
cells; treatment with free DOX greatly increased p38
phosphorylation, while PK1 increased it slightly; PK1 also
significantly increased ERK phosphorylation, while the free DOX
slightly decreased it (27). Based
on our study and on the results from the PK1 study, we conclude
that antitumor mechanisms of action of a prodrug may be different
from the original drug itself. Molecular modifications could bring
new active groups, structural changes, drug metabolic alterations,
which together may account for the different mechanisms of action
from free DOX. This also suggests that the prodrugs may have a
different antitumor spectrum. Therefore, more studies are needed to
investigate the molecular mechanisms of action and the antitumor
spectrum.
In conclusion, the present study found that PDOX has
different mechanisms of action, particularly the
mitochondria-centered intrinsic pathway involving reactive
oxidative stress and the ERK1/2 signaling pathway. The new
knowledge gained from this study may aid in the development of PDOX
as a ‘smart’ molecular targeting drug against cancer
metastasis.
Acknowledgements
The present study was supported by the State Key
Research Project on Infectious Diseases (2012ZX10002012-012) and
the National Natural Science Foundation of China (no. 81171396) and
National University Students Innovation Training Project of China
(101048639).
References
|
1
|
Gianni L, Grasselli G, Cresta S, Locatelli
A, Viganò L and Minotti G: Anthracyclines. Cancer Chemother Biol
Response Modif. 21:29–40. 2003. View Article : Google Scholar
|
|
2
|
Abu Ajaj K, Graeser R, Fichtner I and
Kratz F: In vitro and in vivo study of an albumin-binding prodrug
of doxorubicin that is cleaved by cathepsin B. Cancer Chemother
Pharmacol. 64:413–418. 2009.PubMed/NCBI
|
|
3
|
Kratz F, Warnecke A, Schmid B, Chung DE
and Gitzel M: Prodrugs of anthracyclines in cancer chemotherapy.
Curr Med Chem. 13:477–523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seymour LW, Ferry DR, Kerr DJ, Rea D,
Whitlock M, Poyner R, et al: Phase II studies of
polymer-doxorubicin (PK1, FCE28068) in the treatment of breast,
lung and colorectal cancer. Int J Oncol. 34:1629–1636. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seymour LW, Ferry DR, Anderson D,
Hesslewood S, Julyan PJ, Poyner R, et al: Hepatic drug targeting:
Phase I evaluation of polymer-bound doxorubicin. J Clin Oncol.
20:1668–1676. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Podgorski I and Sloane BF: Cathepsin B and
its role(s) in cancer progression. Biochem Soc Symp. 70:263–276.
2003.PubMed/NCBI
|
|
7
|
Calderón M, Graeser R, Kratz F and Haag R:
Development of enzymatically cleavable prodrugs derived from
dendritic polyglycerol. Bioorg Med Chem Lett. 19:3725–3728.
2009.PubMed/NCBI
|
|
8
|
Kovár M, Strohalm J, Etrych T, Ulbrich K
and Ríhová B: Star structure of antibody-targeted HPMA
copolymer-bound doxorubicin: a novel type of polymeric conjugate
for targeted drug delivery with potent antitumor effect. Bioconjug
Chem. 13:206–215. 2002.PubMed/NCBI
|
|
9
|
Thanou M and Duncan R: Polymer-protein and
polymer-drug conjugates in cancer therapy. Curr Opin Investig
Drugs. 4:701–709. 2003.PubMed/NCBI
|
|
10
|
Mai J, Waisman DM and Sloane BF: Cell
surface complex of cathepsin B/annexin II tetramer in malignant
progression. Biochim Biophys Acta. 1477:215–230. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kratz F, Müller IA, Ryppa C and Warnecke
A: Prodrug strategies in anticancer chemotherapy. Chem Med Chem.
3:20–53. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vasey PA, Kaye SB, Morrison R, Twelves C,
Wilson P, Duncan R, et al: Phase I clinical and pharmacokinetic
study of PK1 (N-(2-hydroxypropyl)methacrylamide copolymer
doxorubicin): first member of a new class of chemotherapeutic
agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II
Committee. Clin Cancer Res. 5:83–94. 1999.
|
|
13
|
Dubowchik GM and Firestone RA: Cathepsin
B-sensitive dipeptide prodrugs. 1. A model study of structural
requirements for efficient release of doxorubicin. Bioorg Med Chem
Lett. 8:3341–3346. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dubowchik GM, Firestone RA, Padilla L,
Willner D, Hofstead SJ, Mosure K, et al: Cathepsin B-labile
dipeptide linkers for lysosomal release of doxorubicin from
internalizing immunoconjugates: model studies of enzymatic drug
release and antigen-specific in vitro anticancer activity.
Bioconjug Chem. 13:855–869. 2002. View Article : Google Scholar
|
|
15
|
Dubowchik GM, Mosure K, Knipe JO and
Firestone RA: Cathepsin B-sensitive dipeptide prodrugs. 2. Models
of anticancer drugs paclitaxel (Taxol), mitomycin C and
doxorubicin. Bioorg Med Chem Lett. 8:3347–3352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shao LH, Liu SP, Hou JX, Zhang YH, Peng
CW, Zhong YJ, et al: Cathepsin B cleavable novel prodrug
Ac-Phe-Lys-PABC-ADM enhances efficacy at reduced toxicity in
treating gastric cancer peritoneal carcinomatosis: an experimental
study. Cancer. 118:2986–2996. 2012. View Article : Google Scholar
|
|
17
|
Liu LL, Li QX, Xia L, Li J and Shao L:
Differential effects of dihydropyridine calcium antagonists on
doxorubicin-induced nephrotoxicity in rats. Toxicology. 231:81–90.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hancock JT, Desikan R and Neill SJ: Role
of reactive oxygen species in cell signalling pathways. Biochem Soc
Trans. 29:345–350. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fruehauf JP and Meyskens FL Jr: Reactive
oxygen species: a breath of life or death? Clin Cancer Res.
13:789–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Korsmeyer SJ, Yin XM, Oltvai ZN,
Veis-Novack DJ and Linette GP: Reactive oxygen species and the
regulation of cell death by the Bcl-2 gene family. Biochim Biophys
Acta. 1271:63–66. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gupta S: Molecular signaling in death
receptor and mitochondrial pathways of apoptosis (Review). Int J
Oncol. 22:15–20. 2003.PubMed/NCBI
|
|
22
|
Tsang WP, Chau SP, Kong SK, Fung KP and
Kwok TT: Reactive oxygen species mediate doxorubicin induced
p53-independent apoptosis. Life Sci. 73:2047–2058. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang S, Konorev EA, Kotamraju S, Joseph J,
Kalivendi S and Kalyanaraman B: Doxorubicin induces apoptosis in
normal and tumor cells via distinctly different mechanisms.
intermediacy of H(2)O(2)- and p53-dependent pathways. J Biol Chem.
279:25535–25543. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moungjaroen J, Nimmannit U, Callery PS,
Wang L, Azad N, Lipipun V, et al: Reactive oxygen species mediate
caspase activation and apoptosis induced by lipoic acid in human
lung epithelial cancer cells through Bcl-2 down-regulation. J
Pharmacol Exp Ther. 319:1062–1069. 2006. View Article : Google Scholar
|
|
25
|
Peng CW, Liu XL, Chen C, Liu X, Yang XQ,
Pang DW, et al: Patterns of cancer invasion revealed by QDs-based
quantitative multiplexed imaging of tumor microenvironment.
Biomaterials. 32:2907–2917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shinomol GK and Muralidhara: Effect of
Centella asiatica leaf powder on oxidative markers in brain
regions of prepubertal mice in vivo and its in vitro efficacy to
ameliorate 3-NPA-induced oxidative stress in mitochondria.
Phytomedicine. 15:971–984. 2008.
|
|
27
|
Kovár L, Strohalm J, Chytil P, Mrkvan T,
Kovár M, Hovorka O, et al: The same drug but a different mechanism
of action: comparison of free doxorubicin with two different
N-(2-hydroxypropyl)methacrylamide copolymer-bound doxorubicin
conjugates in EL-4 cancer cell line. Bioconjug Chem. 18:894–902.
2007.
|