Introduction
The Jun kinase (JNK) pathway is a member of the
mitogen-activated protein kinase (MAPK) pathways involved in signal
transduction, and is an important cellular pathway associated with
oncogenic transformation triggered in response to DNA damage
(1,2). JNK phosphorylates c-Jun (a component
of the AP-1 transcription complex, consisting of heterodimers of
the Fos, Jun and ATF-2 family of proteins) on serine 63 and 73 in
the N-terminal domain, thereby greatly enhancing transcriptional
transactivation by c-Jun (3–5). Thus,
phosphorylation of c-Jun at serines 63 and 73 leads to increased
formation of the AP-1 complex consisting of c-Jun and a member of
the Fos family of transcription factors or c-Jun in complex with
NH2-terminal phosphorylated ATF-2. JNK activation has
been correlated with apoptosis induced by death receptor activation
(Fas and TNF-α) and cellular stress (6–8).
However, JNK is also activated by non-apoptotic stimuli (8,9). In
fact, JNK is strongly induced in response to a variety of DNA
damaging treatments such as UV irradiation (10), DNA damaging chemotherapeutics such
as cisplatin (11,12), camptothecin (13) and etoposide (14), as well as epidermal growth factor
(EGF) which causes rapid induction of both JNK and ERK signaling
pathways (15,16). The downmodulation of the EGF
signaling pathway by EGF pre-treatment was found to inhibit
UVB-induced JNK-1 activation (17).
Members of these pathways have been the subject of intense interest
in recent years owing to their role in mediating numerous growth
factors and in responding to numerous agents and cytokines, in
inflammatory stimuli and DNA damaging agents. Activation of the JNK
pathway by any of the various upstream mechanisms leads to
activation of central kinase, which acts directly on 3 major
regulatory proteins, c-Jun, ATF-2 and Elk-1 by phosphorylating
serine residues of activation domains which greatly increases their
transcriptional (c-Jun, ATF-2) or DNA binding (Elk-1) potential. In
fact, activation of the JNK pathway potentially influences the
regulation of gene promoters bearing 7-bp AP-1 sites (18,19).
Several DNA repair genes that are known to be involved in cisplatin
DNA adduct repair contain ATF/CREB regulatory elements in their
promoters (12,20). These genes are known to be activated
by DNA damage and are of the type known to be activated by JNK.
Indeed, several studies have shown that the JNK pathway is
activated by DNA damaging chemotherapeutic agents such as cisplatin
and that this pathway is required for repair of cisplatin DNA
adducts (12,21,22).
It has been previously observed that the NH2-terminal
JNK pathway is required for formation of a variety of human tumors
both in vivo and in vitro(2,23–25).
It is known that the chemotherapeutic agent cisplatin, which
damages DNA through the formation of bi-functional platinum adducts
(25), activates JNK/SAPK up to
10-fold in a dose-dependent manner (12,26).
Nevertheless, activation of the JNK/SAPK pathway by genotoxic
stress appears to be general. Several other well characterized DNA
damaging agents have been shown to activate JNK/SAPK (12,16,27–29).
Thus, JNK is activated by cisplatin-induced DNA damage and is
required for DNA repair and survival following cisplatin treatment
(12,30). Further evidence for the generality
of the role of the JNK pathway in response to cisplatin comes from
studies using a dominant-negative c-Jun (3,4,31,32).
To analyze the mechanisms of sensitivity of tumor cells to
cisplatin by inhibition of the JNK-1/JNK-2 pathway, we tested
whether small interfering RNA (siRNA) against Jnk-1 and Jnk-2 was
able to induce loss of viability thereby sensitizing tumor cells to
cisplatin. In the present study, we provide data showing that
specific inhibition by siRNA led to a block in JNK-1/JNK-2
expression leading to loss of viability and cell growth arrest
thereby sensitizing PC-3 and LNCaP prostate tumor cells to
cisplatin treatment.
Materials and methods
The protease inhibitors, phenylmethylsulfonyl
fluoride (PMSF), leupeptin, pepstatin, aprotinin and bestatin, were
purchased from Roche (San Francisco, CA, USA).
[γ-32P]ATP was purchased from Amersham Pharmacia Biotech
(Piscataway, NJ, USA). T4 polynucleotide kinase and
poly(dI-dC)20 were obtained from Amersham.
Tris-borate-EDTA buffer and acrylamide-bisacrylamide (29:1) were
obtained from Bio-Rad (Richmond, CA, USA). Luciferase assay
reagent, lysis buffer and the pGL2 luciferase vector were obtained
from Promega Corporation (Madison, WI, USA). Phorbol 12-myristate
13-acetate (TPA) was purchased from Strategene Inc. (La Jolla, CA,
USA). Anti-c-Jun, -JNK, -JNK-1, -JNK-2 and -β-actin antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). TPA luciferase assay reagent, lysis buffer and the pGL2/pGL3
luciferase vector were obtained from Promega. GST-c-Jun (1–79) was
a gift from Dr Roger Davis (Howard Hughes Medical Institute, Chevy
Chase, MA, USA). Cisplatin and transplatin were purchased from
Sigma Chemical Co. (St. Louis, MO, USA).
Cell lines and culture
Human prostate carcinoma cell lines PC-3 and LNCaP
were a gift from Dr Dan Mercola (The SKCC, La Jolla, CA, USA). The
cells were cultured in RPMI-1640 medium supplemented with 100 ml/l
fetal bovine serum (FBS), 8×105 U/l penicillin and 0.1
g/l streptomycin in a humidified incubator containing 50 ml/l
CO2 at 37°C (14,15).
The antibodies used for western blotting included those against
protein kinase JNK-1, JNK-2, (Santa Cruz Biotechnology, Inc) and
β-actin (Sigma Chemical Co.). Western blot analysis was performed
as previously described (33).
siRNA preparation and transfection of
small interfering RNA
siRNA oligonucleotides with two thymidine residues
(tt) at the 3′-end of the sequence were designed for JNK-1 (sense,
5′-AAG CCCAGTAATATAGTAGTA and antisense, 5′-TACTACTA
TATTACTGGGC-3′) and JNK-2 (sense, 5′-CATGATGTT ATCATATCTTAT-3′ and
antisense, 5′-ATAAGATATG ATAACATCATG-3′). Cells were treated in
parallel with a non-silencing siRNA (sense, 5′-AATTCTCCGAACGTGT
CACGT-3′ and antisense, 5′-ACGTGACACGTTCGGAGA ATT-3′) as control.
Oligonucleotides were synthesized by Shanghai Genechem Co. Ltd. The
cells were cultured in medium without antibiotics, and 24 h before
transfection resulting in a confluence of the cell monolayer by
60–80%. Jnk-1-siRNA and Jnk-2-siRNA or non-silencing siRNA (70
nmol) were mixed with Lipofectamine™ 2000 (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer’s
recommendation and added to the cells. After 6 h at 37°C, the
medium was replaced and the cells were cultivated in RPMI-1640
supplemented with 10% heat-inactivated FBS (33,34).
Western immunoblot analysis
PC-3 and LNCaP prostate carcinoma cells lines
(5×105) were seeded onto 6-well plates. Forty-eight
hours after transfection, cells were collected and washed twice by
cold PBS, and each well was treated with 50 ml lysis buffer (2
mmol/l Tris-HCl, pH 7.4, 50 mmol/l NaCl, 25 mmol/l EDTA, 50 mmol/l
NaF, 1.5 mmol/l Na3VO4, 1% Triton X-100, 0.1%
SDS, supplemented with protease inhibitors 1 mmol/l
phenylmethylsulfonyl fluoride, 10 mg/l pepstatin, 10 mg/l aprotinin
and 5 mg/l leupeptin) (all from Sigma-Aldrich, St. Louis, MO, USA).
Protein concentrations were determined using the Bradford protein
assay. Equal amounts of protein (50 μg) were separated on a 15% SDS
polyacrylamide gel and transferred to nitrocellulose membranes
(Hybond C; Amersham, Freiburg, Germany). Membranes were blocked in
5% non-fat dry milk in TBS for 1 h at room temperature and probed
with rabbit anti-JNK-1 (sc-1648) and anti-JNK-2 (sc-571) antibodies
(dilution, 1:500) overnight at 4°C. After 3 washings with TBS
containing 0.1% Tween-20, membranes were incubated with anti-rabbit
IgG-horseradish peroxidase (1:5,000; Santa Cruz Biotechnology,
Inc.), and developed by luminal mediated chemiluminescence
(Appylgen Technologies, Inc., China). To confirm equal protein
loading, membranes were reprobed with a 1:1,000 dilution of an
anti-actin antibody (Santa Cruz Biotechnology, Inc.). Densitometric
analyses were performed using Scion Image software (35).
Reverse transcription-polymerase chain
reaction (RT-PCR)
PC-3 and LNCaP prostate carcinoma cell lines
(5×105) were seeded onto 6-well plates. Total RNA was
extracted 48 h after transfection using TRIzol reagent. Reverse
transcription was performed using One Step RT-PCR kit. The primers
included Jnk-1, 5′-CGTCTGGTGGAAGGAGAGAG-3′ (forward primer) and
5′-TAATAACGGGGGTGGAGGAT-3′ (reverse primer); Jnk-2,
5′-TCTGACGTCCTGGGCTGGAC-3′ (forward primer) and
5′-GCAGCAGCCCTCAGGATCCT-3′ (reverse primer); human β-actin,
5′-TCACCAACTGGGACGA CAT-3′ (forward primer) and 5′-GAAGTCCAGGGCGACG
TAG-3′ (reverse primer). Thermal cycling conditions were as
follows: 42°C for 30 min, 94°C for 2 min, followed by 28 cycles of
94°C for 15 sec, 55°C for 30 sec, 72°C for 1 min, with a final
extension at 72°C for 10 min. RT-PCR products were visualized by
ethidium bromide-stained agarose gels and the images were scanned
using a UV light.
Kinase assays (JNK-1 activity)
Cells were incubated in the absence of serum for 16
h and then treated with various agents. They were then washed twice
with PBS and lysed in ice-cold lysis buffer (20 mM HEPES, pH 7.4,
150 mM NaCl, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EDTA, 1
mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerol phosphate, 1
mM sodium orthovanadate, 1 μg/ml leupeptin and 1 mM
phenylmethylsulfonyl fluoride). The extracts were centrifuged to
remove cellular debris, and the protein content of the supernatants
was determined using the Bio-Rad protein assay reagent. Protein
(100 μg) from the lysate samples was incubated at 4°C overnight
with the N-terminal c-Jun (1–79) and ATF-2-glutathione
S-transferase fusion protein bound to glutathione-Sepharose beads
in order to selectively precipitate JNK-1 and p38 from the cell
lysates. Next, the beads were washed to remove non-specifically
bound proteins. Then, the kinase reaction was carried out in the
presence of cold ATP, and samples were resolved on 12% SDS-gel
electrophoresis followed by western blotting with the
phospho-specific c-Jun antibody. This antibody specifically
recognizes JNK-1-induced phosphorylation of c-Jun at Ser63, a site
important for c-Jun-dependent transcriptional activity (36).
Cell toxicity assay (MTT)
The effect of cisplatin on antitumor activity in
human LNCaP and PC-3 prostate carcinoma cells was determined by the
MTT survival assay, or using a commercial MTT assay kit (CellTiter
96® AQueous One Solution cell proliferation assay;
Promega Corporation) according to the manufacturer’s instructions.
The MTT survival assay was performed as described previously
(37). The MTT assay is a commonly
used method to evaluate cell survival, based on the ability of
viable cells to convert 3-(4,5-dimethylthiazole-2-yl)-2,5
diphenyltetrazolium bromide (MTT), a soluble tetrazolium salt, into
an insoluble formazan precipitate, which is quantitated by
spectrophotometry following solubilization in dimethyl sulfoxide
(DMSO). Briefly, LNCaP and PC-3 cells untreated and treated with
cisplatin alone, or the combination of siRNA and cisplatin in
96-well tissue culture dishes were incubated with MTT (2 mg/ml) for
4 h. The cells were then solubilized in 125 ml of DMSO, and
absorbance readings were taken using a 96-well Opsys MR™ microplate
reader (Thermo/Labsystems, Chantilly, VA, USA). The amount of MTT
dye reduction was calculated based on the difference between
absorbance at 570 and 630 nm. Cell viability in treated cells was
expressed as the amount of dye reduction relative to that of the
untreated control cells. The wells which contained only medium and
10 ml of MTT were used as blanks for the plate reader. Three sets
of experiments were performed in 8–12 wells for each treatment.
Results
The ability to turn off individual genes at will in
growing cells provides a powerful tool for elucidating the role of
a particular gene and for therapeutic intervention when that gene
is overexpressed or mutated. Since the discovery of siRNAs as the
key mediators of RNA-induced gene silencing, they have been applied
to inhibit the expression of a wide variety of target genes. Two of
them, the Jnk-1 and Jnk-2 genes expressing the respective kinases
with important functions in the regulation of growth and
differentiation, are involved in the transformed phenotype of
several types of cancers.
TPA and cisplatin activate the JNK-1
pathway in PC-3 and LNCaP prostate carcinoma cell lines
c-Jun-NH2 kinase (JNK) is among the
protein kinases that play an important role in cellular stress
response via the phosphorylation of c-Jun, ATF-2 and p53.
Activation of JNK-1 by UV irradiation requires cooperation between
membrane and nuclear components, including DNA lesions per
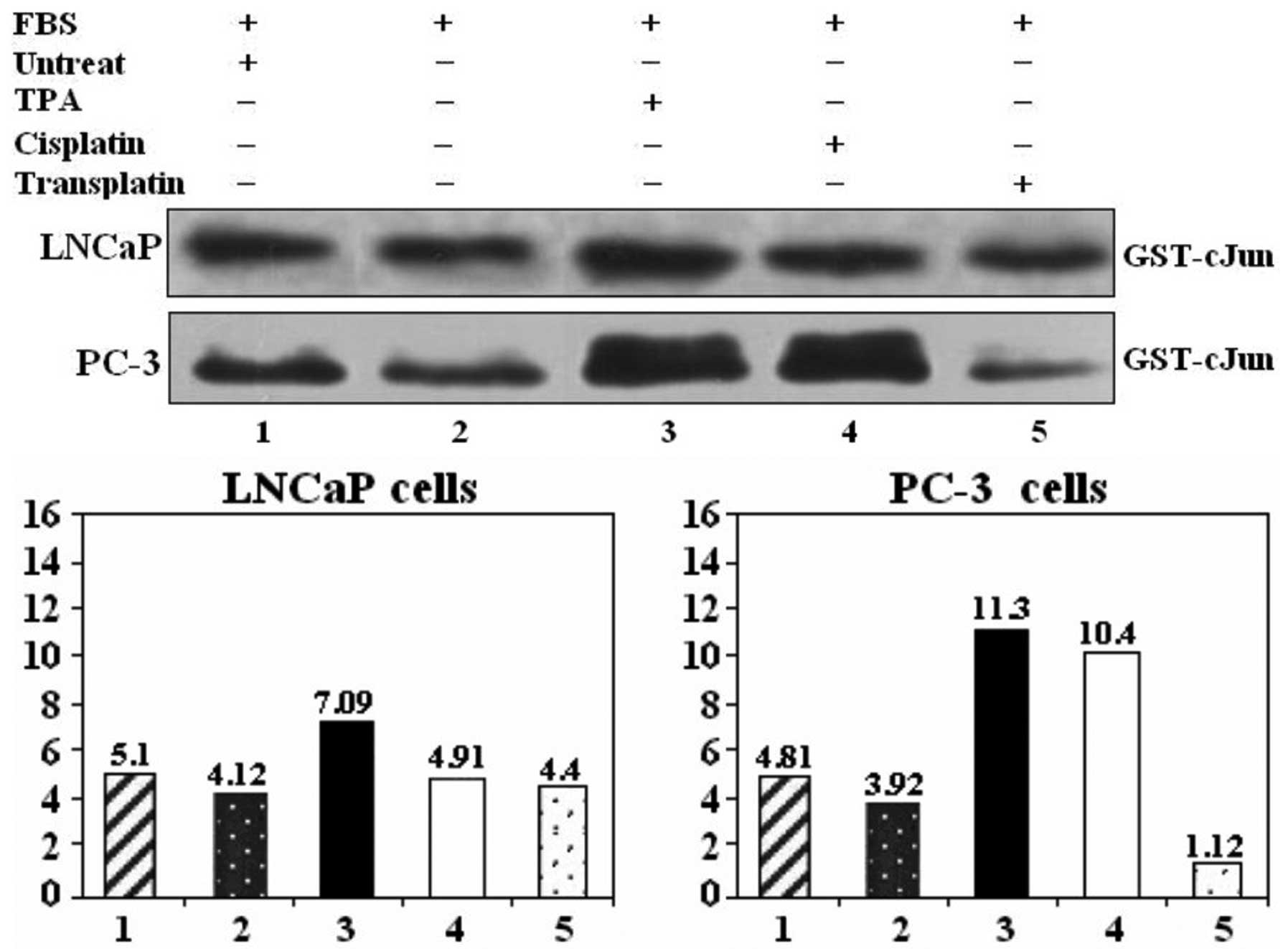
se. To determine whether TPA and cisplatin activate the JNK
pathway in PC-3 and LNCaP cells, cells were cultured in 10% PBS and
treated with different stimuli (Fig.
1) such as TPA (30 nM), cisplatin (300 μg/ml) and transplatin
as the control. JNK-1 activity was measured 1 h after the
treatments, as described in Materials and methods. Results showed
that addition of TPA and cisplatin activated JNK-1 and JNK-2
activities, while transplatin did not have any effect in inducing
JNK activity (Fig. 1).
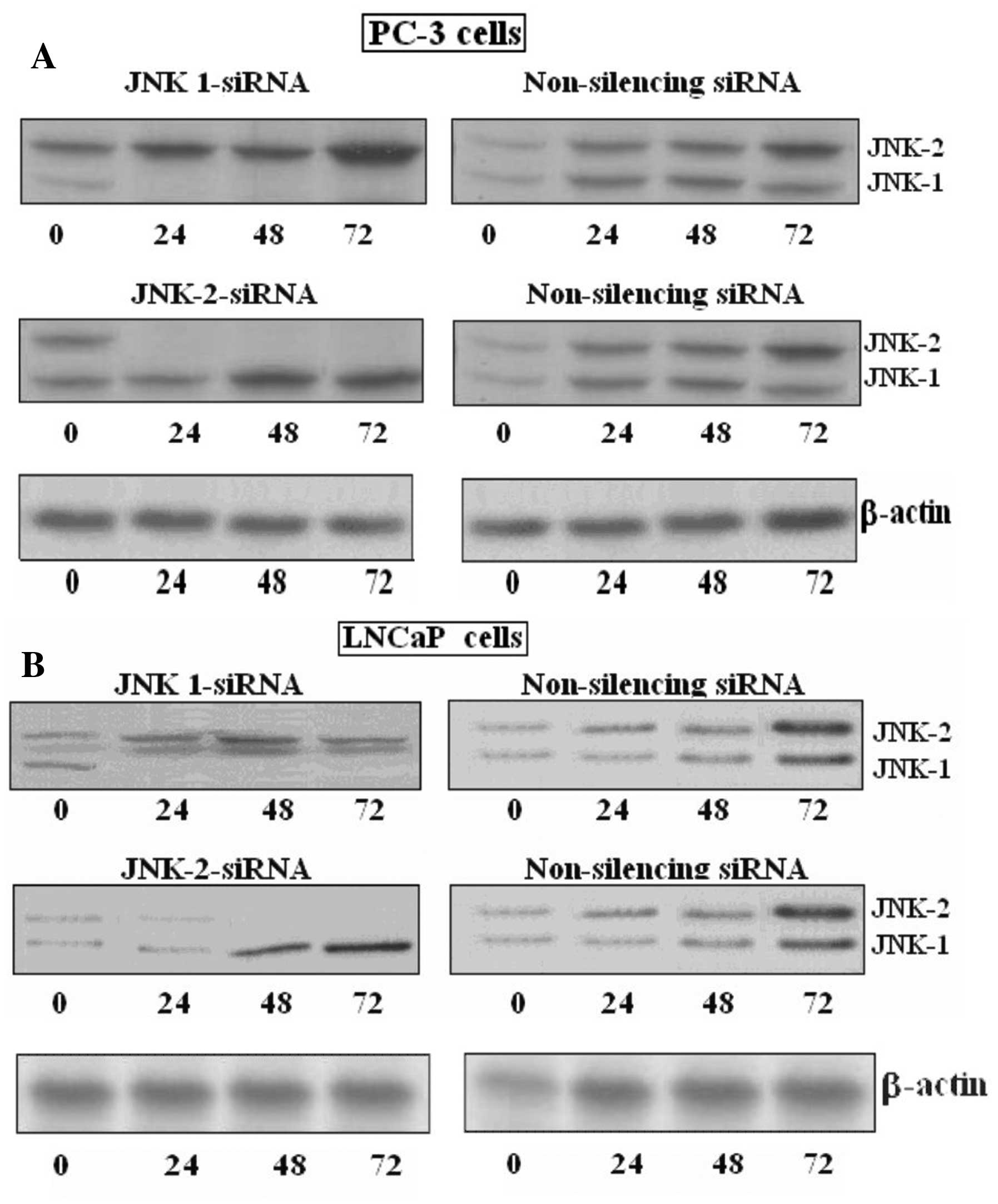
Jnk-1-siRNA and Jnk-2-siRNA specifically
inhibit the expression of JNK-1 and JNK-2 protein,
respectively
In order to determine whether a causal link exists
between JNK activity and protein expression, we used specific
siRNAs against JNK-1 and JNK-2 complementary to mRNA sequences
common to either JNK-1 or JNK-2. To examine the specific effect of
the abovementioned siRNAs on prostate cancer cells, the proteins
and mRNA levels were determined by western blotting and RT-PCR
analysis, respectively. Results are displayed in Fig. 2A and B. JNK-1 and JNK-2 protein and
mRNA were strongly expressed in the LNCaP prostate carcinoma cell
lines as reflected by western blotting. JNK-1 and JNK-2 protein
expression was strongly inhibited by siRNA against the named
kinases. The inhibition was completely compared with the control or
non-silencing siRNA.
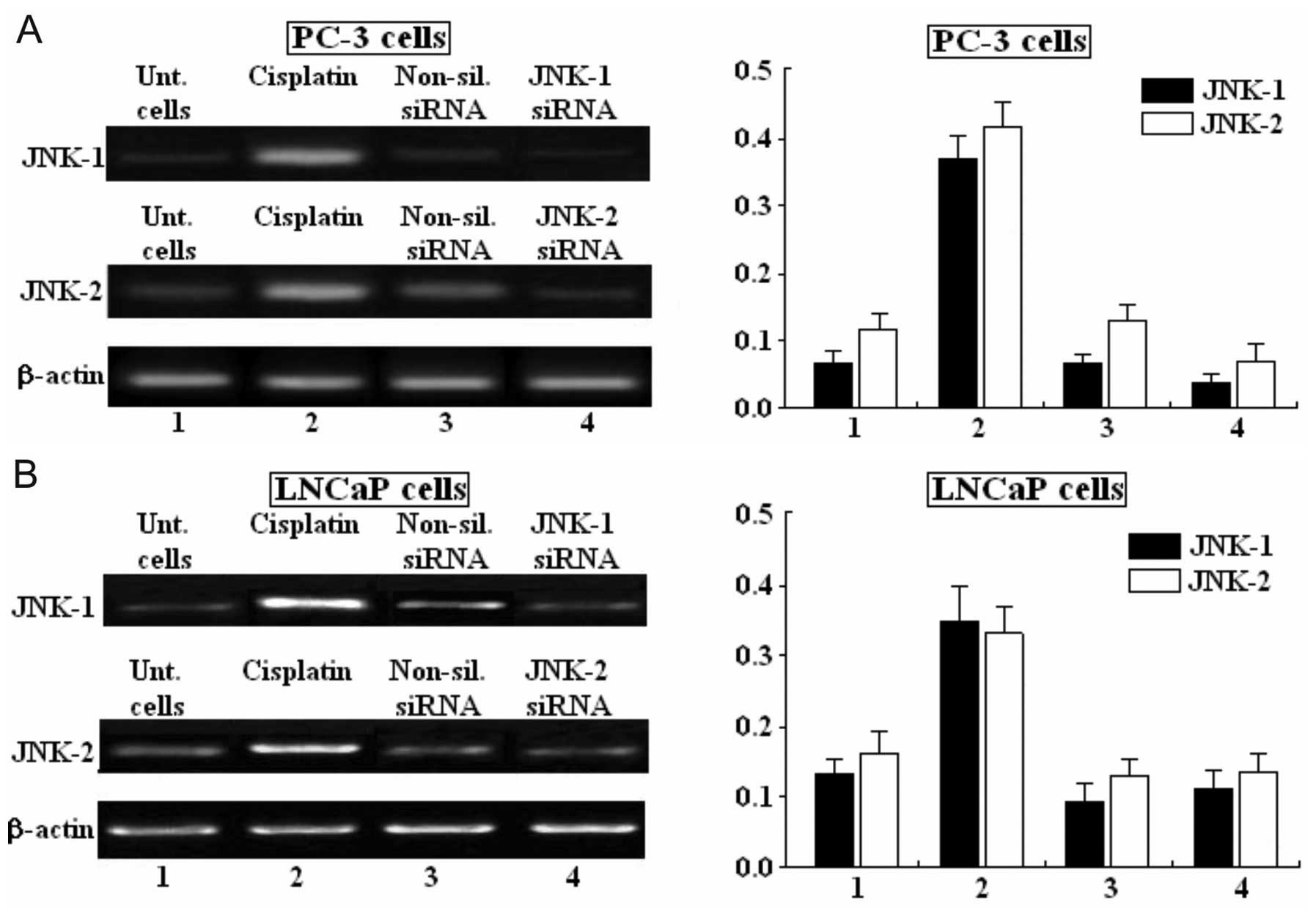
Semi-quantitative analysis of JNK-1 and
JNK-2 mRNA expression by RT-PCR in human PC-3 and LNCaP prostate
carcinoma cell lines
JNK-1, JNK-2 and β-actin in the indicated cell lines
was analyzed by RT-PCR, and ethidium bromide stained agarose gels
are shown. PC-3 cells were either untreated (lane 1), treated with
cisplatin (lane 2), treated with non-silencing siRNA (lane 3) or
treated with siRNA-JNK-1 or siRNA-JNK-2 (lane 4) (Fig. 3A). LNCaP cells were also untreated
(lane 1), treated with cisplatin (lane 2), treated with
non-silencing siRNA (lane 3) and treated with siRNA-JNK-1 or
siRNA-JNK-2 (lane 4) (Fig. 3B).
Cisplatin was able to markedly increase the mRNA expression of
JNK-1 and JNK-2 in both PC-3 and LNCaP cell lines. Non-silencing
siRNA did not have any effect on the expression of mRNA. Moreover,
siRNAs against JNK-1 and JNK-2 were able to decrease the expression
of mRNA in both PC-3 and LNCaP prostate carcinoma cell lines. The
integrity of each RNA sample was confirmed by using primers to the
human β-actin gene (Fig. 3A and B
bottom lanes).
Transfection with siRNA-JNK-1 and
siRNA-JNK-2 blocks the effect of TPA (30 nM) and cisplatin on the
transcriptional activity of the reporter gene
To show that the blocking of JNK-1/JNK-2 activity
inhibits the activation of promoters bearing a tandem repeat of
AP-1 response elements, transient transfection studies were carried
out using a reporter construct bearing the consensus or classic
AP-1 site repeated in tandem (4x) driving expression of a
luciferase reporter gene. Expression of this reporter in untreated
or PC-3 and LNCaP cells transfected with non-silencing siRNA
(Fig. 4) led to little activation,
whereas TPA (30 nM) and cisplatin treatment strongly enhanced
expression of this reporter. The TPA or cisplatin-induced
expression of the reporter gene was inhibited following
transfection with siRNA-JNK-1, suggesting that siRNA against JNK-1
enhances the sensitivity of PC-3 and LNCaP for cisplatin. However,
TPA in the presence of JNK-1-siRNA (bar 8, Fig. 4) was unable to reverse the
inhibitory effects of JNK-1-siRNA (bar 5, Fig. 4) compared with the effect of TPA
alone (bar 3, Fig. 4), and observed
in the transcriptional activity expressed as luciferase activity
(Fig. 4). The results are
consistent with the fact that activation of the JNK pathway leads
to phosphorylation of at least 3 principal transcription factor
including Elk-1, ATF-1 and c-Jun. Thus, phosphorylation of c-Jun at
serines 63 and 73 leads to increased formation of AP-1 complexes
consisting of c-Jun and a member of the Fos family of transcription
factors.
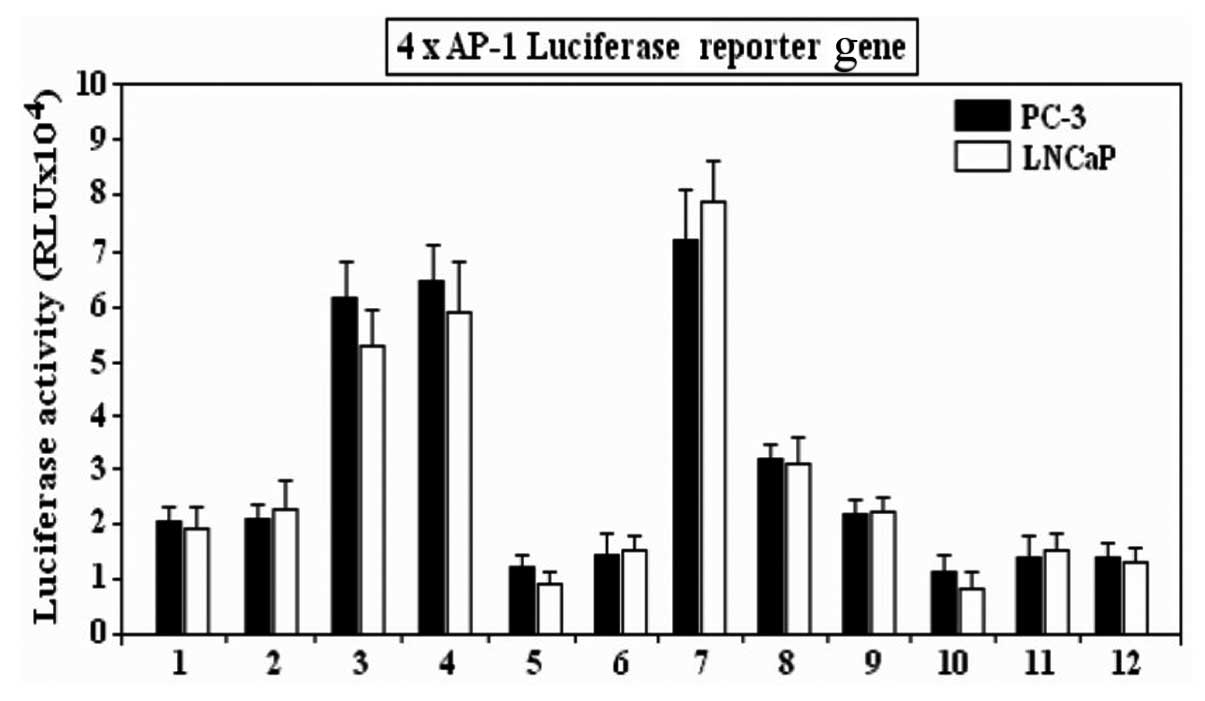 | Figure 4Inhibition of AP-1-dependent
transcription induced by TPA and cisplatin stimulation, by siRNA
against JNK-1 (JNK-1-siRNA). The reporter, which contains 4× AP-1
response elements in tandem directing expression of the luciferase
gene, was transfected into PC-3 or LNCaP cells. The graph shows
luciferase activity compared to the controls with a reporter
lacking the AP-1 sites. The AP-1 reporter requires an activated
c-Jun and was, therefore, activated by increased expression of
JNK-1. The reporters were inhibited by Jnk-1-siRNA. Bar 1,
untreated cells; 2, non-silencing siRNA; 3, TPA (30 nM); 4,
cisplatin; 5, JNK-1-siRNA; 6, JNK-2-siRNA; 7, TPA + cisplatin; 8,
TPA + JNK-1-siRNA; 9, TPA + JNK-2-siRNA; 10, cisplatin +
JNK-1-siRNA; 11, cisplatin + JNK-2-siRNA; 12,
JNK-1-siRNA/JNK-2-siRNA. |
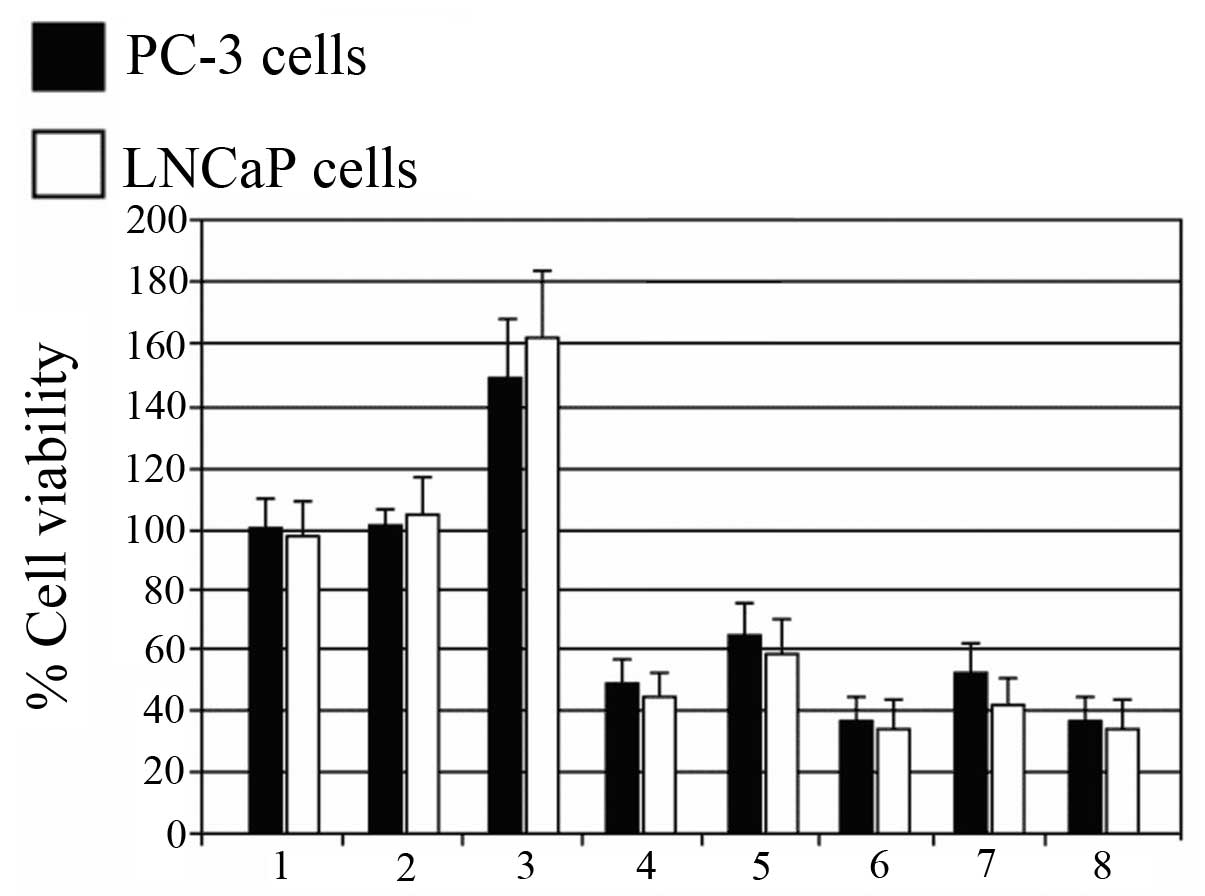
siRNA-JNK-1/2 increases the sensitivity
of PC-3 and LNCaP cells to the antiproliferative effect of
cisplatin
In light of the important role of JNK in the
antitumor action of cisplatin, we hypothesized that concurrent
inhibition of JNK-1/JNK-2 signaling by siRNAs could sensitize PC-3
and LNCaP cisplatin-resistant cells to the antiproliferative
activity of this chemotherapeutic drug. To test this hypothesis,
PC-3 and LNCaP cells were exposed to an indicated concentration of
cisplatin (300 μg/ml) alone or in combination with siRNA-JNK-1 and
siRNA-JNK-2 for 48 h. Fig. 5
demonstrates the ability of JNK-1 and JNK-2 siRNAs to improve the
sensitivity of PC-3 and LNCaP cells to cisplatin-induced cell
death. Cisplatin alone caused an increase in viability of ~40 and
50% (lane 3), respectively; a reduction in cell viability was
observed after siRNA-JNK-1 or siRNA-JNK-2 treatment (lanes 4 and
5). Further reduction in cell viability was observed in cells
treated with both cisplatin and siRNA-JNK-1 or siRNA-JNK-2 (lanes 6
and 7) and the combination of siRNA-JNK-1 and siRNA-JNK-2 achieved
~63% cell death (lane 8), similar to either, siRNA-JNK-1 or
siRNA-JNK-2. Notably, siRNA against Jnk-1 or Jnk-2 was able to
overcome the resistance of PC-3 and LNCaP cells to cisplatin.
Discussion
We analyzed the role of the JNK pathway to mediate
cisplatin-driven transformation in prostate carcinoma cell lines.
To test the transforming role of JNK in prostate carcinoma, we
examined two prostate cell lines, PC-3 and LNCaP. We employed
siRNAs that abrogated the expression of either JNK-1 or JNK-2.
siRNA methods were applied in vitro and confirmed that
inhibition of the JNK pathway blocked expression of the transformed
phenotype as manifested by activity, protein expression, mRNA,
transcription and cell viability. It has been demonstrated that the
kinases JNK-1 and JNK-2 are expressed in a high proportion of most
types of human cancers, including breast, prostate,
gastrointestinal cancer, lymphoma, melanoma and myeloid leukemia
(38–40). Expression of the nonphosphorylatable
mutant, c-Jun 63–73, reduces growth by 30–50% in human tumor cells
(42,43) and the altered expression of JNK-1 or
JNK-2 seems to define a common event associated with pathogenesis
(44). Although some studies
previously revealed that the effects of inactivation of JNK-1 and
JNK-2 in some cell lines were modest (45), other groups using different
approaches to reduce the protein levels, found that a decrease in
JNK-1 and JNK-2 expression inhibited the growth of several types of
tumor cells, including breast tumor cells. In fact, the oncogenic
role of JNK has been recognized in human breast cancer cells
(23,46), such as the mediation of the
transformation by Bcr-Abl (46). In
this study, we observed that treatment of serum-stimulated prostate
carcinoma cells with siRNA-JNK-1 was more effective than
siRNA-JNK-2. In contrast, treatment with siRNA-JNK-2 had little
effect on both PC-3 and LNCaP cells similar to treatment with the
non-silencing siRNA or the untreated cells. These results are in
agreement with other studies which demonstrated the role of the JNK
pathway and its effect on several types of human cancers (27–29)
including the observations suggesting that the JNK/SAPK pathway may
play a major role in the transformation process (43,44).
Our results demonstrated that siRNAs can effectively downregulate
the endogenous JNK-1 or JNK-2 expression with great specificity.
This downregulation may involved the participation of several
genes, demonstrating autoregulation of the pathway since c-Jun is
known to be activated by genotoxic stress, including cisplatin
(45). As JNK inhibition by siRNA
results in decreased activation and expression of JNK-1 and JNK-2,
we next determined the effect of JNK inhibition on AP-1 activation
by transfection of the AP-1-luciferase plasmid, which contains four
tandem copies of the AP-1 consensus sequence. Since activation of
the JNK pathway leads to phosphorylation of at least 3 principal
transcription factors including Elk-1, ATF-1 and c-Jun, we aimed to
ascertain whether siRNAs against JNK-1 and JNK-2 affect the
transcriptional activity of c-Jun. As we know, phosphorylation of
c-Jun at serines 63 and 73 leads to increased formation of AP-1
complexes consisting of c-Jun and a member of the Fos family of
transcription factors (3,4,31,46).
Thus, transient transfection of 4× AP-1-luciferase in PC-3 and
LNCaP cells treated with either TPA or cisplatin significantly
increased AP-1 reporter activity, but this increase in
transcription activity was strongly reversed by JNK-1-siRNA, but
only partially by JNK-2-siRNA. Similarly, a viability assay showed
that cell transfection with either JNK-1-siRNA or JNK-2-siRNA was
able to induce growth arrest in cells treated with cisplatin. Given
the fact that it has been previously demonstrated that inhibition
of c-Jun by a dominant-negative c-Jun sensitizes cells to
cisplatinum toxicity (3,4,31) and
that JNK-1 and JNK-2 is upstream on c-Jun in the JNK pathway, it is
reasonable to believe that these small interfering RNAs would be
effective in sensitizing resistant cells to chemotherapeutic
treatment. In summary, the present results suggest that blocking
the JNK pathway may increase cisplatin sensitivity in PC-3 and
LNCaP prostate carcinoma cell lines. Moreover, an optimum result
was observed for siRNA-JNK-1 treatment in both PC-3 and LNCaP
cells.
Acknowledgements
We thank Dr Dan Mercola (The Sidney Kimmel Cancer
Center, San Diego, CA, USA) for providing the prostate cell lines
PC-3 and LNCaP, and for providing us with the vector for expressing
Jnk-1. This study was supported by the Laboratory of Experimental
Biomedicine, University of Tarapaca, UTA, Chile.
References
|
1
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kralova J, Sheely JI, Liss AS and Bose HR
Jr: ERK and JNK activation is essential for oncogenic
transformation by v-Rel. Oncogene. 29:6267–6279. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Binétruy B, Smeal T and Karin M: Ha-Ras
augments c-Jun activity and stimulates phosphorylation of its
activation domain. Nature. 351:122–127. 1991.PubMed/NCBI
|
|
4
|
Smeal T, Binétruy B, Mercola DA, Birrer M
and Karin M: Oncogenic and transcriptional cooperation with Ha-Ras
requires phosphorylation of c-Jun on serines 63 and 73. Nature.
354:494–496. 1991. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mechta F, Lallemand D, Pfarr CM and Yaniv
M: Transformation by ras modifies AP1 composition and activity.
Oncogene. 14:837–847. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Minden A and Karin M: Regulation and
function of the JNK subgroup of MAP kinases. Biochim Biophys Acta.
1333:F85–F104. 1997.PubMed/NCBI
|
|
7
|
Verheij M, Ruiter GA, Zerp SF, van
Blitterswijk WJ, Fuks Z, Haimovitz-Friedman A and Bartelink H: The
role of the stress-activated protein kinase (SAPK/JNK) signaling
pathway in radiation-induced apoptosis. Radiother Oncol.
47:225–232. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paul A, Wilson S, Belham CM, Robinson CJ,
Scott PH, Gould GW and Plevin R: Stress-activated protein kinases:
activation, regulation and function. Cell Signal. 9:403–410. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gupta S, Barrett T, Whitmarsh AJ, Cavanagh
J, Sluss HK, Dérijard B and Davis RJ: Selective interaction of JNK
protein kinase isoforms with transcription factors. EMBO J.
15:2760–2770. 1996.PubMed/NCBI
|
|
10
|
Adler V, Fuchs SY, Kim J, Kraft A, King
MP, Pelling J and Ronai Z: jun-NH2-terminal kinase
activation mediated by UV-induced DNA lesions in melanoma and
fibroblast cells. Cell Growth Differ. 6:1437–1446. 1995.
|
|
11
|
Amdjadi K and Sefton BM: Ultraviolet
light-induced stimulation of the JNK mitogen-activated protein
kinase in the absence of src family tyrosine kinase activation. J
Biol Chem. 275:22520–22525. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Potapova O, Haghighi A, Bost F, Liu C,
Birrer MJ, Gjerset R and Mercola D: The Jun kinase/stress-activated
protein kinase pathway functions to regulate DNA repair and
inhibition of the pathway sensitizes tumor cells to cisplatin. J
Biol Chem. 272:14041–14044. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saleem A, Datta R, Yuan ZM, Kharbanda S
and Kufe D: Involvement of stress-activated protein kinase in the
cellular response to 1-β-D-arabinofuranosylcytosine and other
DNA-damaging agents. Cell Growth Differ. 6:1651–1658. 1995.
|
|
14
|
Osborn MT and Chambers TC: Role of the
stress-activated/c-Jun NH2-terminal protein kinase
pathway in the cellular response to adriamycin and other
chemotherapeutic drugs. J Biol Chem. 271:30950–30955.
1996.PubMed/NCBI
|
|
15
|
Hashimoto A, Kurosaki M, Gotoh N, Shibuya
M and Kurosaki T: Shc regulates epidermal growth factor-induced
activation of the JNK signaling pathway. J Biol Chem.
274:20139–20143. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bost F, McKay R, Dean N and Mercola D: The
JUN kinase/stress-activated protein kinase pathway is required for
epidermal growth factor stimulation of growth of human A549 lung
carcinoma cells. J Biol Chem. 272:33422–33429. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang SA, Lee ES, Yoon HY, Randazzo PA and
Lee ST: PTK6 inhibits down-regulation of EGF receptor through
phosphorylation of ARAP1. J Biol Chem. 285:26013–26021. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Dam H, Wilhelm D, Herr I, Steffen A,
Herrlich P and Angel P: ATF-2 is preferentially activated by
stress-activated protein kinases to mediate c-jun induction in
response to genotoxic agents. EMBO J. 14:1798–1811. 1995.PubMed/NCBI
|
|
19
|
Pulverer BJ, Kyriakis JM, Avruch J,
Nikolakaki E and Woodgett JR: Phosphorylation of c-jun mediated by
MAP kinases. Nature. 353:670–674. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanabe M, Izumi H, Ise T, Higuchi S,
Yamori T, Yasumoto K and Kohno K: Activating transcription factor 4
increases the cisplatin resistance of human cancer cell lines.
Cancer Res. 63:8592–8595. 2003.PubMed/NCBI
|
|
21
|
Crul M, Schellens JH, Beijnen JH and
Maliepaard M: Cisplatin resistance and DNA repair. Cancer Treat
Rev. 23:341–366. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parra E and Ferreira J: Knockdown of the
c-Jun-N-terminal kinase expression by siRNA inhibits MCF-7 breast
carcinoma cell line growth. Oncol Rep. 24:1339–1345. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsuiki H, Tnani M, Okamoto I, Kenyon LC,
Emlet DR, Holgado-Madruga M, Lanham IS, et al: Constitutively
active forms of c-Jun NH2-terminal kinase are expressed
in primary glial tumors. Cancer Res. 63:250–255. 2003.PubMed/NCBI
|
|
24
|
Vivas-Mejia P, Benito JM, Fernandez A, Han
HD, Mangala L, Rodriguez-Aguayo C, et al:
c-Jun-NH2-kinase-1 inhibition leads to antitumor
activity in ovarian cancer. Clin Cancer Res. 16:184–194. 2010.
|
|
25
|
Lovejoy KS, Todd RC, Zhang S, McCormick
MS, D’Aquino JA, Reardon JT, Sancar A, Giacomini KM and Lippard SJ:
cis-Diammine (pyridine)chloroplatinum(II), a monofunctional
platinum(II) antitumor agent: uptake, structure, function, and
prospects. Proc Natl Acad Sci USA. 105:8902–8907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levresse V, Marek L, Blumberg D and
Heasley LE: Regulation of platinum-compound cytotoxicity by the
c-Jun N-terminal kinase and c-Jun signaling pathway in small-cell
lung cancer cells. Mol Pharmacol. 62:689–697. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Helbig L, Damrot J, Hülsenbeck J, Köberle
B, Brozovic A, Osmak M, Fiket Z, Kaina B and Fritz G: Late
activation of stress-activated protein kinases/c-Jun N-terminal
kinases triggered by cisplatin-induced DNA damage in
repair-defective cells. J Biol Chem. 286:12991–13001. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fritz G and Kaina B: Late activation of
stress kinases (SAPK/JNK) by genotoxins requires the DNA repair
proteins DNA-PKcs and CSB. Mol Biol Cell. 17:851–861. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brozovic A, Fritz G, Christmann M,
Zisowsky J, Jaehde U, Osmak M and Kaina B: Long-term activation of
SAPK/JNK, p38 kinase and fas-L expression by cisplatin is
attenuated in human carcinoma cells that acquired drug resistance.
Int J Cancer. 112:974–985. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hayakawa J, Depatie C, Ohmichi M and
Mercola D: The activation of c-Jun NH2-terminal kinase
(JNK) by DNA-damaging agents serves to promote drug resistance via
activating transcription factor 2 (ATF2)-dependent enhanced DNA
repair. J Biol Chem. 278:20582–20592. 2003.
|
|
31
|
Smeal T, Binetruy B, Mercola D,
Grover-Bardwick A, Heidecker G, Rapp UR and Karin M:
Oncoprotein-mediated signalling cascade stimulates c-Jun activity
by phosphorylation of serines 63 and 73. Mol Cell Biol.
12:3507–3513. 1992.PubMed/NCBI
|
|
32
|
Boyle WJ, Smeal T, Defize LH, Angel P,
Woodgett JR, Karin M and Hunter T: Activation of protein kinase C
decreases phosphorylation of c-Jun at sites that negatively
regulate its DNA-binding activity. Cell. 64:573–584. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Parra E, Ortega A and Saenz L:
Down-regulation of Egr-1 by siRNA inhibits growth of human prostate
carcinoma cell line PC-3. Oncol Rep. 22:1513–1518. 2009.PubMed/NCBI
|
|
34
|
Parra E, Ferreira F and Saenz L:
Inhibition of Egr-1 by siRNA in prostate carcinoma cell lines is
associated with decreased expression of AP-1 and NF-κB. Int J Mol
Med. 28:847–853. 2011.PubMed/NCBI
|
|
35
|
Parra E, Ferreira J and Ortega A:
Overexpression of EGR-1 modulates the activity of NF-κB and AP-1 in
prostate carcinoma PC-3 and LNCaP cell lines. Int J Oncol.
39:345–352. 2011.PubMed/NCBI
|
|
36
|
Inostroza J, Sáenz L, Calaf G, Cabello G
and Parra E: Role of the phosphatase PP4 in the activation of JNK-1
in prostate carcinoma cell lines PC-3 and LNCaP resulting in
increased AP-1 and EGR-1 activity. Biol Res. 38:163–178. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu JJ, Li Q and Reed E: Comparison of two
human ovarian carcinoma cell lines (A2780/CP70 and MCAS) that are
equally resistant to platinum, but differ at codon 118 of the ERCC1
gene. Int J Oncol. 16:555–560. 2000.PubMed/NCBI
|
|
38
|
Ma FY, Flanc RS, Tesch GH, Han Y, Atkins
RC, Bennett BL, Friedman GC, Fan JH and Nikolic-Paterson DJ: A
pathogenic role for c-Jun amino-terminal kinase signaling in renal
fibrosis and tubular cell apoptosis. J Am Soc Nephrol. 18:472–484.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vivanco I, Palaskas N, Tran C, Finn SP,
Getz G, Kennedy NJ, Jiao J, Rose J, Xie W, Loda M, et al:
Identification of the JNK signaling pathway as a functional target
of the tumor suppressor PTEN. Cancer Cell. 11:555–569. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cellurale C, Girnius N, Jiang F,
Cavanagh-Kyros J, Lu S, Garlick DS, Mercurio AM and Davis RJ: Role
of JNK in mammary gland development and breast cancer. Cancer Res.
72:472–481. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Burgess GS, Williamson EA, Cripe LD,
Litz-Jackson S, Bhatt JA, Stanley K, Stewart MJ, Kraft AS,
Nakshatri H and Boswell HS: Regulation of the c-jun gene in p210
BCR-ABL transformed cells corresponds with activity of JNK, the
c-jun N-terminal kinase. Blood. 92:2450–2460. 1998.PubMed/NCBI
|
|
42
|
Raitano AB, Halpern JR, Hambuch TM and
Sawyers CL: The Bcr-Abl leukemia oncogene activates Jun kinase and
requires Jun for transformation. Proc Natl Acad Sci USA.
92:11746–11750. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu J, Minemoto Y and Lin A: c-Jun
N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for
tumor necrosis factor alpha-induced c-Jun kinase activation and
apoptosis. Mol Cell Biol. 24:10844–10856. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hayakawa J, Mittal S, Wang Y, Korkmaz KS,
Adamson E, English C, Ohmichi M, McClelland M and Mercola D:
Identification of promoters bound by c-Jun/ATF2 during rapid
large-scale gene activation following genotoxic stress. Mol Cell.
16:521–535. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Halazonetis TD, Georgopoulos K, Greenberg
ME and Leder P: c-Jun dimerizes with itself and with c-Fos, forming
complexes of different DNA binding affinities. Cell. 55:917–924.
1988. View Article : Google Scholar : PubMed/NCBI
|