Introduction
The c-Jun NH2-terminal kinases (JNKs)
belong to a subfamily of mitogen-activated protein kinases
implicated in the transduction of a wide array of intracellular
signals associated with, for instance, cellular proliferation,
differentiation, development, the inflammatory response and
apoptosis (1,2). Reflecting such versatile functions of
JNK in signal transduction, a growing body of evidence now suggests
that JNKs may also play a significant role in the biology of human
cancers (3–5). Indeed, aberrant activation of JNK has
been increasingly reported in a variety of human cancers, and the
roles of JNK in the control of fundamental cellular properties of
cancer cells, such as proliferation, survival and migration, have
been documented (2–5). Significantly, adding to such roles of
JNK in cancer cells in general, we and others have recently
identified a specific role of JNK in the control of cancer stem
cells (CSCs) of glioblastoma (5–7), a
particular subpopulation of cancer cells now presumed to have a
pivotal role in cancer relapse and/or metastasis as well as in
therapy resistance (8,9). Most importantly, we demonstrated for
the first time in our recent study that ‘sustained’ JNK activity is
not only required for the prevention of differentiation and
maintenance of the ‘stem cell state’ of glioblastoma CSCs but also
for the maintenance of their ‘tumor-initiating capacity’.
Furthermore, we showed in the study that transient, short-term
inhibition of JNK in vivo is sufficient to exert a
long-lasting suppressive effect on the tumor-initiating capacity of
glioblastoma cells, suggesting that JNK could make an excellent
therapeutic target to achieve long-term control of glioblastoma,
one of the deadliest of all human cancers (5,6).
However, to date, it remains totally unknown whether such a role of
JNK in the control of the stemness and/or the tumor-initiating
capacity of tumor cells is unique to glioblastoma or is commonly
shared by other human cancers.
Lung cancer is currently the leading cause of
cancer-related mortality in the world (10), with non-small cell lung cancer
(NSCLC) accounting for the majority of lung cancer cases (~80%)
(10,11). Similarly to glioblastomas (12–15),
aberrant activation of JNK has been observed frequently in NSCLC
tumors (16,17), and a number of studies do provide
evidence supporting the idea that JNK has a positive role in
promoting the growth of NSCLC tumors (16–18).
Nevertheless, evidence is not yet sufficient, with the exact
role(s) of JNK in NSCLC cells, in particular in vivo,
remaining poorly understood. Here in this study, therefore, we
investigated the role of JNK in NSCLC using a human NSCLC cell line
A549 (19). Intriguingly, the
results of the xenograft analyses of the present study demonstrated
that JNK is more specifically involved in tumor initiation than in
bulk tumor growth (i.e., growth of already established tumors) of
A549 cells. Our findings thus suggest that the maintenance of the
tumor-initiating population may be one of the primary roles of JNK
in NSCLC tumors in vivo and that such a role of JNK is
shared by human cancers other than glioblastoma.
Materials and methods
Antibodies and reagents
SP600125 was purchased from Calbiochem (La Jolla,
CA, USA) and was dissolved in dimethylsulfoxide (DMSO) to prepare a
50 mM stock solution. Anti-c-Jun (#9165) and anti-phospho-c-Jun
(Ser63) (#9261) antibodies were purchased from Cell Signaling
Technology, Inc. (Beverly, MA, USA). Anti-β-actin (A1978) was from
Sigma (St. Louis, MO, USA). Anti-JNK1 (sc-474) and anti-JNK2
(sc-7345) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA).
Cell culture
The human NSCLC cell line A549 was obtained from the
Riken BioResource Center. To verify the authenticity of the A549
cells used in this study, the A549 cells in actual use were
subjected to genotyping by short tandem repeat (STR) profiling
(Bio-Synthesis, Inc., Lewisville, TX, USA) at the completion of
this study. For the serum-free culture of A549, a culture condition
reported to allow the selection of undifferentiated cancer stem
cells including those of lung cancer (20–22),
cells were cultured on collagen-I-coated dishes (IWAKI) in
serum-free culture medium [Dulbecco’s modified Eagle’s medium
(DMEM)/F12 supplemented with B27 (both from Gibco-BRL, Carlsbad,
CA, USA)], 20 ng/ml EGF, 20 ng/ml FGF2 (Peprotech, Inc., Rocky
Hill, NJ, USA), D-(+)-glucose (final concentration, 26.2 mM), 2 mM
L-glutamine (final concentration, 4.5 mM), 100 units/ml penicillin,
and 100 μg/ml streptomycin). The serum-free culture medium was
changed every 3 days, and EGF and FGF2 were added to the culture
medium every day. For the serum culture of A549 cells, cells were
maintained in DMEM/F12 with 10% fetal bovine serum and antibiotics
(100 units/ml penicillin, 100 μg/ml streptomycin). Unless otherwise
indicated, A549 cells were maintained and subjected to experiments
using the serum-free culture condition. Throughout the study, the
cell number was determined using a hemocytometer, and the cell
viability was examined by the dye exclusion method (0.2% trypan
blue). Cell viability (%) was defined as 100 × ‘the number of
viable cells’/(‘the number of viable cells’ + ‘the number of dead
cells’).
Gene silencing by siRNA
siRNAs against human JNK1 (HSS108547), JNK2
(HSS108550), and Stealth RNAi™ siRNA negative control duplexes
(Medium GC Duplexes #2) were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). Transfection of siRNAs was
performed using Lipofectamine RNAiMAX (Invitrogen Life
Technologies) according to the manufacturer’s instructions. To
achieve sustained knockdown of the target genes, siRNA transfection
was repeated 4 days after the initial transfection.
Immunoblot analysis
Cells were washed with ice-cold phosphate-buffered
saline (PBS) and lysed in the RIPA buffer [10 mM Tris-HCl (pH 7.4),
0.1% SDS, 1% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1.5 mM
Na3VO4, 10 mM NaF, 10 mM sodium
pyrophosphate, 10 mM sodium β-glycerophosphate and 1% protease
inhibitor cocktail set III (Calbiochem)]. After centrifugation for
10 min at 14,000 × g at 4°C, the supernatants were recovered as the
cell lysates, and the protein concentration of the cell lysates was
determined by the BCA protein assay kit (Pierce Biotechnology,
Inc., Rockford, IL, USA). Cell lysates containing equal amounts of
protein were separated by SDS-PAGE and transferred to a
polyvinylidene difluoride membrane. The membrane was probed with a
primary antibody and then with an appropriate HRP-conjugated
secondary antibody according to the protocol recommended by the
manufacturer of each antibody. Immunoreactive bands were visualized
using Immobilon Western Chemiluminescent HRP Substrate (Millipore,
Billerica, MA, USA).
Mouse studies
Mouse xenograft studies were carried out essentially
as previously described (6,23). For subcutaneous implantation of A549
cells, cells were suspended in 200 μl of PBS and injected into the
flank region of 5- to 7-week-old male BALB/cAJcl-nu/nu mice
(Clea Japan, Inc.). After implantation, the recipient mice were
monitored for general health status and the presence of
subcutaneous tumors. Tumor volume was determined by measuring tumor
diameters (measurement of 2 perpendicular axes of tumors) using a
caliper and calculated as 1/2 × (larger diameter) × (smaller
diameter)2. For serial transplantation, excised tumors
were washed in chilled sterile PBS and transferred into DMEM/F12,
minced with scissors, and incubated in Accutase for 30 min at 37°C.
After washing with DMEM/F12, the cells were resuspended in DMEM/F12
and filtered through a 70-μm strainer. After determination of cell
number and viability, the single-cell suspension of the tumor cells
was subjected to subcutaneous injection. For systemic
administration of SP600125, the SP600125 stock solution (50 mM in
DMSO) was diluted in PBS to prepare 200 μl solutions of SP600125
for each injection. The SP600125 solutions were injected
intraperitoneally into nude mice. Control mice received 200 μl of
DMSO diluted in PBS. Note that all the control- and
SP600125-treated mice received an equal volume of DMSO per body
weight (3.6 ml/kg/day). All animal experiments were performed under
a protocol approved by the Animal Research Committee of Yamagata
University.
Statistical analysis
Results are expressed as the means ± SD, and
differences were compared using the 2-tailed Student’s t-test.
P-values <0.05 were considered statistically significant and
indicated with asterisks (*) in the figures.
Results
Rapid and potent inhibition of A549 cell
proliferation by JNK inhibition in vitro
To examine the role of JNK in A549 cells, we first
tested the effect of JNK inhibition on A549 cell growth in
vitro using SP600125, an ATP-competitive, reversible inhibitor
of JNK (24). When A549 cells
cultured in the presence of serum were treated with SP600125 at 20
μM, the JNK inhibitor caused a pronounced proliferation block on
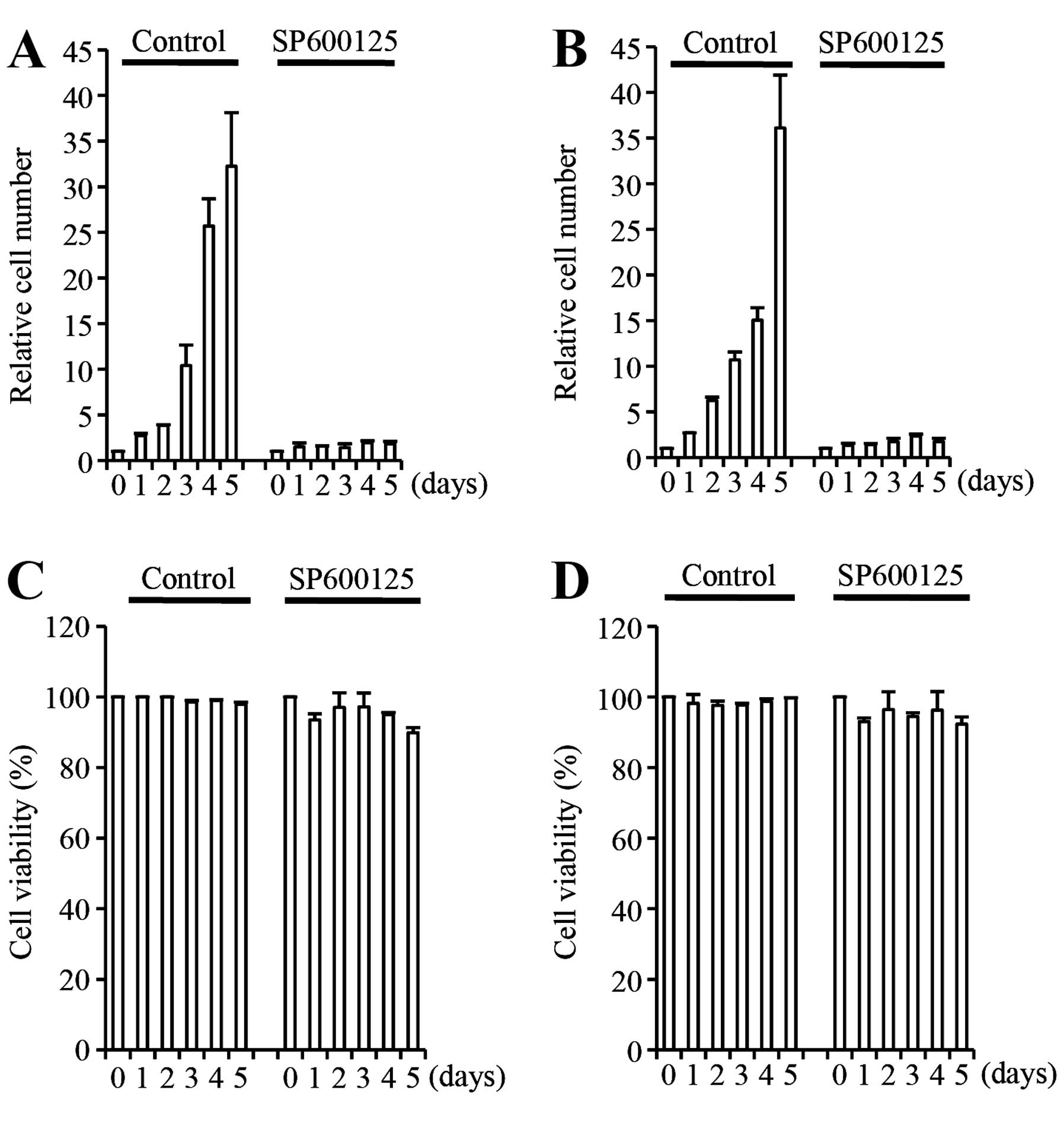
A549 cells without causing significant toxicity (Fig. 1A and C). Growth suppression by
SP600125 was rapid and potent, being apparent as early as 1 day
after the initiation of the inhibitor treatment and was already
marked by 3 days of inhibitor treatment (Fig. 1A). Identical experiments were
conducted with A549 cells maintained in serum-free culture
condition, reported to allow selective expansion of the
undifferentiated, stem cell population of lung cancer cells
(21). The results were similar to
those obtained from serum-cultured A549 cells (Fig. 1B and D), suggesting that the JNK
activity is required for the proliferation of A549 cells
irrespective of the culture condition, at least in vitro.
These results were essentially consistent with those of previous
studies that demonstrated the essential role of JNK in the in
vitro growth of NSCLC cells using NSCLC cell lines other than
A549 (16–18).
JNK inhibition fails to inhibit the
growth of established A549 tumors in vivo
Given the marked growth inhibitory effect of
SP600125 on A549 cells observed in vitro, we next examined
whether it has a similar inhibitory effect on the growth of A549
tumors in vivo. To this end, we implanted A549 cells
subcutaneously into nude mice, and when the implanted A549 cells
formed discrete subcutaneous tumors (~7 mm in diameter), we treated
the tumor-bearing mice by systemic, intraperitoneal administration
of SP600125 (40 mg/kg) for 10 consecutive days and monitored the
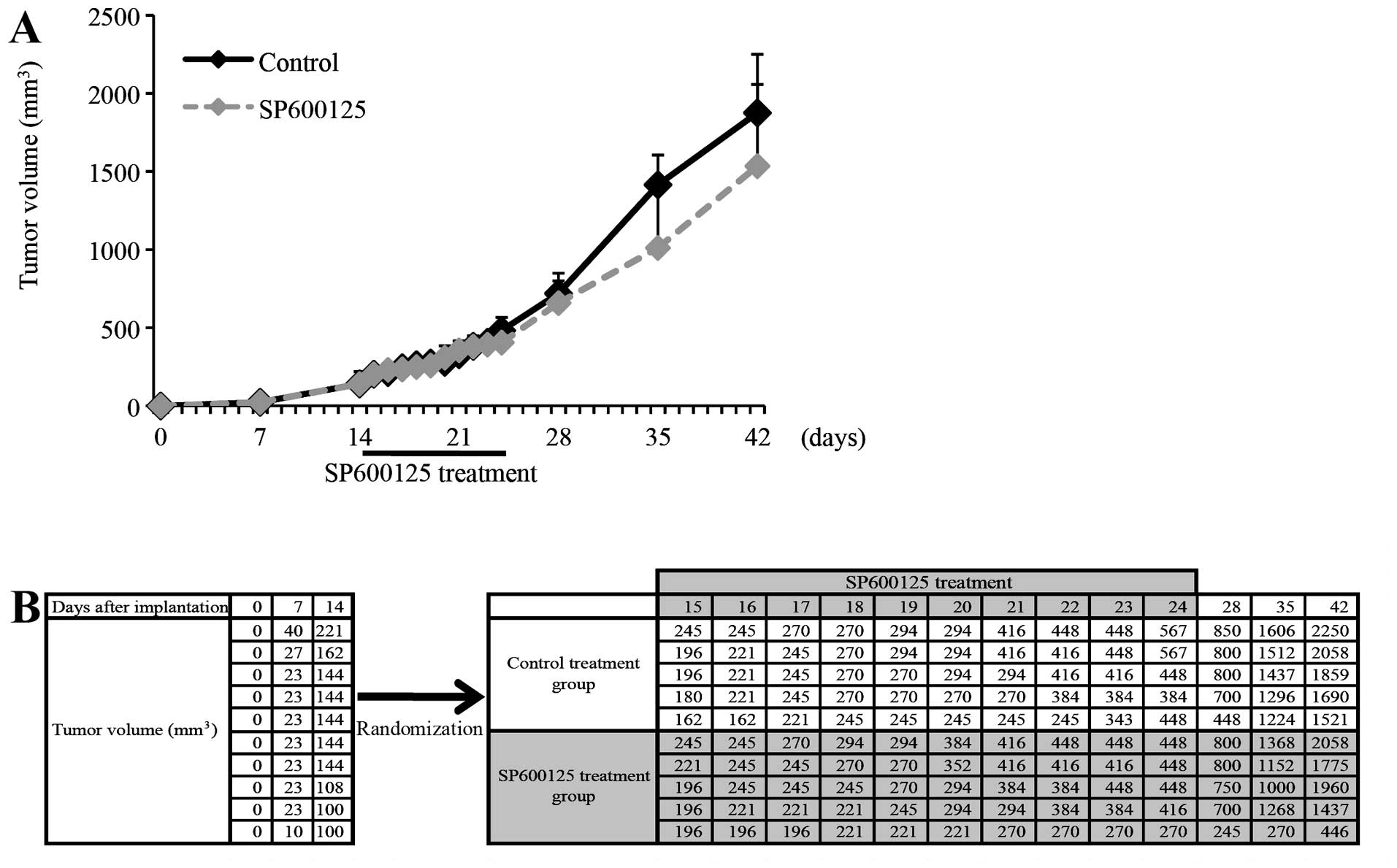
growth of tumors during and after the treatment. Strikingly, we
observed no significant inhibition of A549 tumor growth by SP600125
under this treatment condition (Fig.
2). Although the average tumor volume of the SP600125-treated
tumors was somewhat smaller than that of the control-treated tumors
in this particular set of experiment (Fig. 2A), this was due to a single unique
tumor that ‘exceptionally’ showed retarded growth in the course of
SP600125 treatment, in sharp contrast to the other 9 tumors (4
SP600125-treated and 5 control-treated tumors) that all grew at a
similar rate (Fig. 2B). Such a
retarded growth of established A549 tumors during SP600125
treatment was never observed in any of the other sets of similar
experiment, and the average tumor volume of SP600125-treated tumors
was even larger (although not statistically significant) than that
of the control-treated tumors in some experiments (Fig. 3A). These results clearly indicate
that SP600125 is not effective at inhibiting the growth of
established A549 tumors in vivo, at least under the
treatment condition used in this study.
Selective elimination of the
tumor-initiating cell population within established A549 tumors by
JNK inhibition in vivo
Although SP600125 thus failed to control the growth
of the entire bulk A549 tumors, it was nevertheless still possible
that SP600125 selectively reduced the tumor-initiating cell
population within the tumors, just as we demonstrated previously
using glioblastoma xenografts (6).
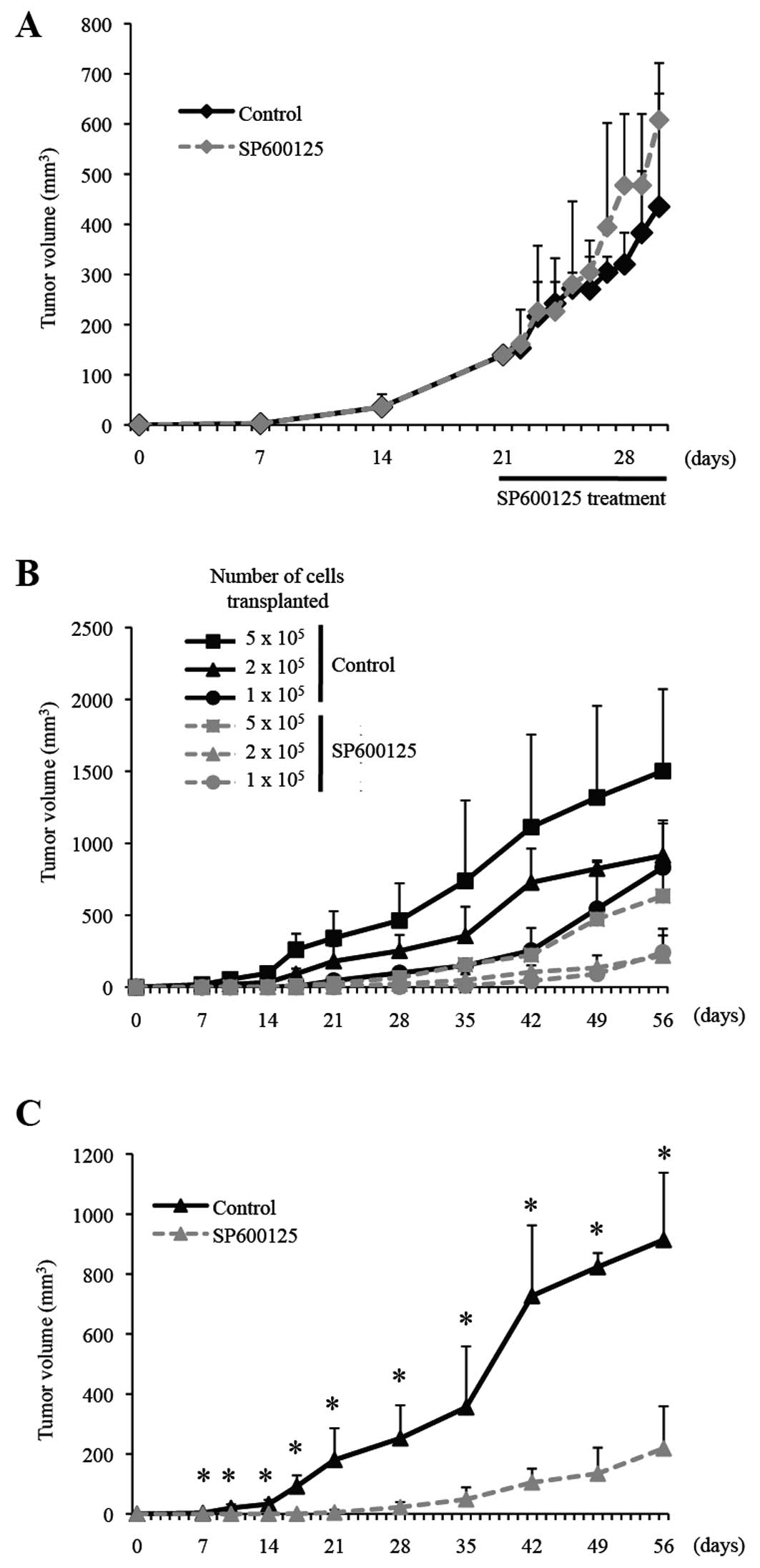
To explore this possibility, we next conducted serial
transplantation experiments. Mice implanted subcutaneously with
A549 cells were treated by an intraperitoneal injection of SP600125
(40 mg/kg) for a consecutive 10 days when the subcutaneous tumors
reached the size of 6–7 mm, similarly as in Fig. 2. The treated subcutaneous tumors
were then excised, and after dissociation into single cells, serial
dilutions (5, 2 and 1×105) of the dissociated cells were
transplanted again subcutaneously into nude mice to initiate tumor
formation. Whereas there was no significant difference in the
growth of SP600125- and control-treated A549 tumors before and
during the course of the treatment (Fig. 3A), tumor initiation was
substantially inhibited and delayed for cells transplanted from
SP600125-treated tumors as compared to those from control-treated
tumors (Fig. 3B and C). We also
found that the growth curve of tumors formed by transplantation of
5×105 cells from SP600125-treated tumors closely
overlapped with that of tumors formed by transplantation of
1×105 cells from control-treated tumors. The results
suggest that the SP600125 treatment reduced the tumor-initiating
cell population within A549 tumors to ~1/5 in vivo.
Inhibition of tumor initiation of A549
cells by JNK inhibition in vivo
Having shown that SP600125 successfully eliminates
the tumor-initiating population of A549 cells within established
tumors, we next determined whether SP600125 blocks tumor
initiation, albeit not bulk tumor growth, of A549 cells in
vivo. To this end, we treated nude mice receiving a
subcutaneous implantation of A549 cells with SP600125 using the
identical treatment condition (intraperitoneal injection of 40
mg/kg SP600125 for 10 consecutive days), except that the treatment
was initiated on the following day of A549 cell implantation
instead of initiating SP600125 treatment after tumor formation.
Remarkably, the identical SP600125 treatment condition, which
failed to inhibit the growth of established A549 tumors,
effectively and significantly inhibited the formation of A549
tumors (Fig. 4). Thus,
collectively, the results of the xenograft analyses conducted thus
far suggest that systemically administered SP600125 exerts its
antitumor activity against A549 cells through selective inhibition
of their tumor initiation.
Essential role for the cell-intrinsic JNK
activity of A549 cells in the maintenance of their tumor-initiating
capacity
Since SP600125 was delivered systemically, there
were two possible explanations, not mutually exclusive, for the
mechanism of the antitumor activity of SP600125 observed in
vivo. One was a direct mechanism, in which the cell-intrinsic
JNK activity of A549 cells was required for the maintenance of
their tumor-initiating capacity. The other was an indirect
mechanism, in which the tumor-initiating capacity of A549 cells
depended on the function(s) of other cells (i.e., cells of the host
animals) sensitive to JNK inhibition. To determine the role of the
‘cell-intrinsic JNK activity’ in the maintenance of the
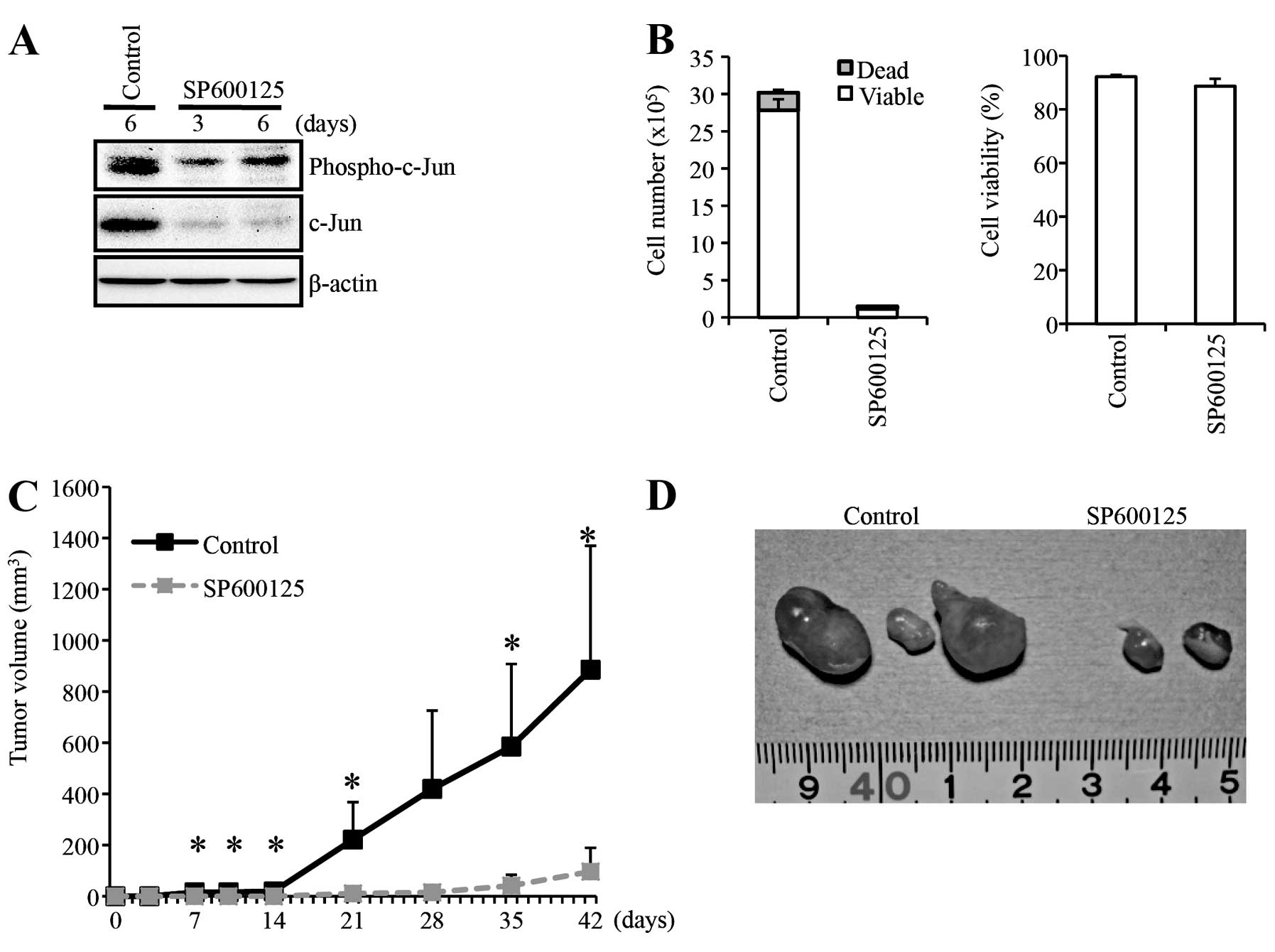
tumor-initiating capacity of A549 cells, we examined the
tumor-initiating capacity of A549 cells pre-treated in vitro
with SP600125 before implantation into nude mice. For this
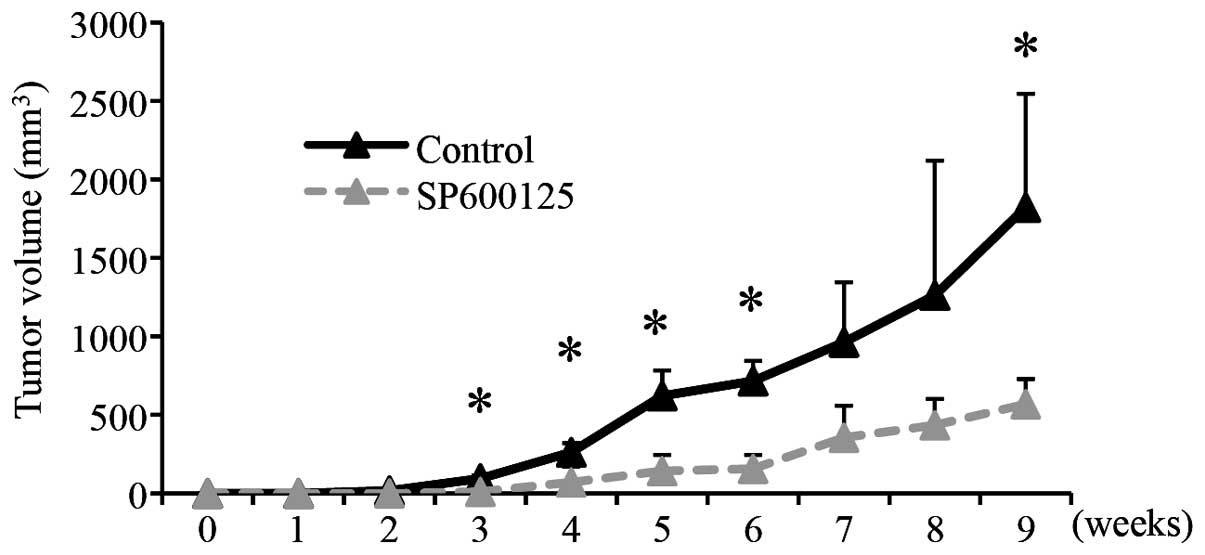
xenograft analysis, A549 cells were treated with SP600125 at 20 μM
to inhibit JNK, for 6 days before implantation (Fig. 5A). This SP600125 treatment condition
suppressed the cellular proliferation of A549 cells without causing
significant reduction of their viability (Fig. 5B) as shown earlier in this study
(Fig. 1). Then, after washout of
SP600125, the same number (5×105 viable cells) of the
SP600125-treated and control-treated A549 cells was implanted
subcutaneously into nude mice. The results of the xenograft
analysis clearly indicated that the SP600125 pre-treatment
inhibited tumor formation by A549 cells (Fig. 5C). Notably, whereas all 3 mice
receiving control-treated A549 cells developed large tumors, the
three mice receiving SP600125-treated cells developed significantly
smaller tumors, with one of them even surviving in a tumor-free
state throughout the entire observation period (Fig. 5D). These results are in support of
the idea that the cell-intrinsic activity of JNK is directly
required for the maintenance of the tumor-initiating capacity of
A549 cells, although they do not necessarily exclude the
possibility that systemic JNK inhibition deprived A549 cells of
their tumor initiation capacity through other indirect
mechanisms.
Finally, to definitively establish that the
cell-intrinsic JNK is required for the tumor-initiating capacity of
A549 cells through a non-pharmacological genetic approach, we
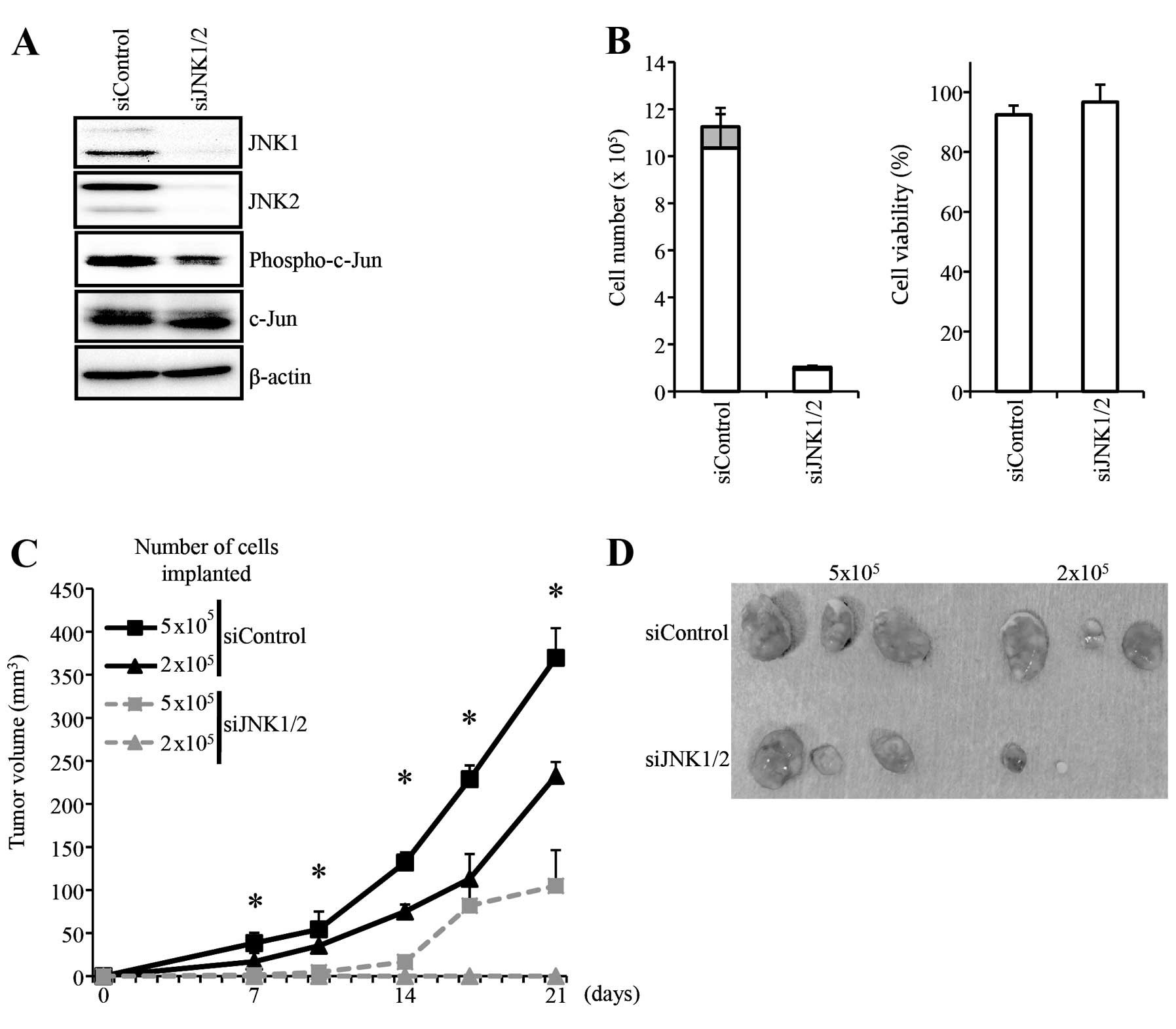
conducted knockdown experiments. To this end, we transiently
transfected A549 cells with a combination of siRNAs against JNK1
and JNK2. Under the transfection condition, the knockdown of JNK1
and JNK2 was efficient, as indicated also by the reduced level of
phosphorylated c-Jun (Fig. 6A).
Similarly to SP600125, the JNK knockdown caused marked blockage of
proliferation but not any significant reduction in the viability of
the A549 cells by the time they were subjected to xenograft
analysis (Fig. 6B). Subcutaneous
implantation of control A549 cells and cells in which JNK1 and JNK2
were transiently knocked down revealed that the JNK knockdown
significantly impaired the tumor-initiating capacity of A549 cells
(Fig. 6C and D). Collectively, the
results demonstrated that the cell-intrinsic JNK activity is
essential for the maintenance of the tumor-initiating capacity of
A549 cells.
Discussion
Since its characterization as a key molecule in the
transduction of pro-apoptotic signaling (25,26),
JNK has long drawn considerable attention as a ‘negative regulator’
of tumor growth, for example, as an important mediator of cell
death signals elicited by cancer therapies such as chemotherapy and
radiotherapy (27,28). On the other hand, accumulating
evidence has now come to support the emerging idea that JNK also
plays significant roles in the ‘promotion’ of tumor growth
(3–5). Only recently, an essential role of JNK
in the maintenance of the tumor-initiating capacity of glioblastoma
stem cells has been documented and added as one of such ‘pro-tumor’
roles of JNK (6,7). In line with such roles of JNK,
aberrant activation of JNK has been increasingly observed in human
cancers (3–5). NSCLC, the major subtype of lung
cancer, is among such human cancers in which activated JNK has been
implicated as a positive regulator of tumor growth (16–18).
However, since the cellular functions of JNK in NSCLC cells have
been studied primarily in vitro(16–18),
the exact role(s) of JNK in NSCLC cells in vivo still
remains poorly understood. To address this issue, therefore, we
conducted both in vitro and in vivo analyses of JNK
functions in A549 NSCLC cells. Strikingly, in sharp contrast to the
results of the in vitro analysis in which inhibition of JNK
caused rapid and potent blockage of A594 cell proliferation, JNK
inhibition in vivo completely failed to inhibit the growth
of already established A549 tumors (Figs. 2 and 3A). However, the identical treatment
condition quite efficiently deprived the tumor cells of their
capacity to initiate tumors upon secondary transplantation
(Fig. 3B and C), indicating that
the JNK inhibitor treatment was effective and selectively targeted
the tumor-initiating capacity of A549 cells while sparing their
proliferative activity in vivo. The idea that JNK plays a
role in the maintenance of the tumor-initiating capacity of A549
cells was further corroborated by the demonstration that the
identical JNK inhibitor treatment (SP600125 40 mg/kg/day for 10
consecutive days) that failed to inhibit the bulk tumor growth
(Figs. 2 and 3A) did inhibit tumor initiation by A549
cells in vivo (Fig. 4).
Apparently, the cell-intrinsic JNK activity was essential for the
maintenance of the tumor-initiating capacity of A549 cells since
JNK inhibition in vitro, i.e., in the absence of cells from
host animals, was sufficient to deprive A549 cells of their
tumor-initiating capacity (Figs. 5
and 6). Collectively, these
findings give rise to and support the idea that the primary role of
JNK in A549 cells in vivo, in particular in established
tumors, may be to maintain their tumor-initiating capacity rather
than to simply sustain cellular proliferation and survival. It
needs to be emphasized here, however, that our results do not
necessarily exclude the possibility that more intensified SP600125
treatment conditions would have some inhibitory effect on the
growth of established A549 tumors. Nevertheless, they do dictate
that we should be prudent enough not to remove JNK from the list of
potential therapeutic targets even if SP600125 should prove to be
ineffective at controlling bulk tumor growth in any conditions.
Another key observation we made in this study is
that short-term, ‘transient’ inhibition of JNK was sufficient to
cause ‘sustained’ loss of the tumor-initiating capacity of A549
cells. Except for the experiment whereby the systemic
administration of SP600125 was started on the following day after
A549 cell implantation (Fig. 4),
the xenograft experiments examining the effect of SP600125 on the
tumor-initiating capacity of A549 cells were conducted in the
complete absence of SP600125 (Figs. 3B,
C and 5), which is a reversible
inhibitor of JNK (24). This
implies that the state of ‘lost tumor-initiating capacity’ is a
stable one that does not require continuous inhibition of JNK,
whereas continuous JNK activity was apparently required for the
maintenance of the tumor-initiating capacity. In this regard, the
JNK-dependent tumor-initiating capacity of A549 shares the key
characteristics of what we recently described as
‘stemness-associated tumor-initiating capacity (STATIC)’ (5). By definition, STATIC is a type of
tumor-initiating capacity regulated in close association with the
stemness/differentiation of the cells most likely through an
epigenetic mechanism (5). Although
we need to admit that the A549 cells used in this study may not be
typical bona fide CSCs since they failed to grow as non-adherent
spheres in the serum-free, stem cell culture condition (data not
shown), the results of the present study strongly suggest that the
A549 cells analyzed in this study share with glioblastoma stem
cells the core mechanism of tumor-initiating capacity, which we
have just shown to be STATIC (5,6). This
idea is quite in good agreement with our prediction/proposal that
STATIC may not always be associated with the stemness of the cells,
for instance, in such a case where the regulatory mechanism
governing stemness is selectively disrupted downstream of the core
mechanism of STATIC that orchestrates stemness and tumor-initiating
capacity (5). As such, this study
may be the first instance to show that STATIC is not a property
unique to CSCs, and to the best of our knowledge, is the first
study to demonstrate that the cell-intrinsic JNK activity plays a
pivotal role in the control of tumor-initiating capacity of cancer
cells other than glioblastoma cells.
Although we clearly demonstrated using a mouse
xenograft model the essential role of JNK in the maintenance of the
tumor-initiating capacity of A549 cells, it still remains to be
shown whether the mechanism is relevant to naturally occurring
NSCLCs. In this respect, it is notable that JNK was recently shown
to be essential for the development of lung tumors in a mouse model
of lung adenocarcinoma driven by K-ras (29,30),
which is frequently activated by mutations in human lung
adenocarcinomas (31,32) and also in A549 cells derived from a
human lung adenocarcinoma, a subtype of NSCLC (33,34).
Although it remains unclear how the deletion of the JNK genes (JNK1
+ JNK2) contributed to the reduced formation of lung
hyperplastic/neoplastic lesions and whether ‘continued’ as opposed
to ‘transient’ loss of JNK (because the JNK genes were deleted
genetically and therefore irreversibly in the study) is required
for the suppression of lung tumor formation observed in the study
(29), the findings provided by the
study in conjunction with our current results may lend support to
the idea that JNK contributes to K-ras-driven NSCLC carcinogenesis
by preventing loss of tumor-initiating capacity of
K-ras-transformed NSCLC cells. If this is actually the case, then
JNK would be a highly promising molecular target to control
recurrence of NSCLC tumors, be it either local or distant (i.e.,
metastasis), since any form of recurrence may involve the process
of tumor initiation by tumor-initiating cells. Future studies to
investigate the prevalence and significance of this JNK-regulated
mechanism of tumor initiation in human NSCLCs in general,
therefore, are warranted.
In conclusion, we demonstrated in the present study
that JNK is specifically involved in the maintenance of the
tumor-initiating capacity of A549 NSCLC cells. Of therapeutic
importance, short-term, transient inhibition of JNK was sufficient
to stably control the tumor-initiating capacity of A549 cells,
making JNK targeting an attractive candidate as a therapeutic
approach to gain long-term control over NSCLCs. Our demonstration
that JNK is involved in the control of the tumor-initiating
capacity of human cancer cells other than glioblastoma cells
indicates that the mechanism is not unique to glioblastomas and
therefore justifies further investigation of the role of JNK in the
control of the tumor-initiating capacity of other human
cancers.
References
|
1
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Cell Biol. 19:142–149. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kennedy NJ and Davis RJ: Role of JNK in
tumor development. Cell Cycle. 2:199–201. 2003.PubMed/NCBI
|
|
3
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen F: JNK-induced apoptosis,
compensatory growth, and cancer stem cells. Cancer Res. 72:379–386.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kitanaka C, Sato A and Okada M: JNK
signaling in the control of the tumor-initiating capacity
associated with cancer stem cells. Genes Cancer. January
22–2013.(Epub ahead of print). View Article : Google Scholar
|
|
6
|
Matsuda K, Sato A, Okada M, et al:
Targeting JNK for therapeutic depletion of stem-like glioblastoma
cells. Sci Rep. 2:5162012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoon CH, Kim MJ, Kim RK, et al: c-Jun
N-terminal kinase has a pivotal role in the maintenance of
self-renewal and tumorigenicity in glioma stem-like cells.
Oncogene. 31:4655–4666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu Y, Ramena G and Elble RC: The role of
cancer stem cells in relapse of solid tumors. Front Biosci.
4:1528–1541. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Malik B and Nie D: Cancer stem cells and
resistance to chemo and radio therapy. Front Biosci. 4:2142–2149.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
11
|
Collins LG, Haines C, Perkel R and Enck
RE: Lung cancer: diagnosis and management. Am Fam Physician.
75:56–63. 2007.
|
|
12
|
Li JY, Wang H, May S, et al: Constitutive
activation of c-Jun N-terminal kinase correlates with histologic
grade and EGFR expression in diffuse gliomas. J Neurooncol.
88:11–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Antonyak MA, Kenyon LC, Godwin AK, et al:
Elevated JNK activation contributes to the pathogenesis of human
brain tumors. Oncogene. 21:5038–5046. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsuiki H, Tnani M, Okamoto I, et al:
Constitutively active forms of c-Jun NH2-terminal kinase
are expressed in primary glial tumors. Cancer Res. 63:250–255.
2003.PubMed/NCBI
|
|
15
|
Cui J, Han SY, Wang C, et al: c-Jun
NH2-terminal kinase 2α2 promotes the tumorigenicity of
human glioblastoma cells. Cancer Res. 66:10024–10031. 2006.
|
|
16
|
Khatlani TS, Wislez M, Sun M, et al: c-Jun
N-terminal kinase is activated in non-small-cell lung cancer and
promotes neoplastic transformation in human bronchial epithelial
cells. Oncogene. 26:2658–2666. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nitta RT, Del Vecchio CA, Chu AH, Mitra
SS, Godwin AK and Wong AJ: The role of the c-Jun N-terminal kinase
2-α-isoform in non-small cell lung carcinoma tumorigenesis.
Oncogene. 30:234–244. 2011.
|
|
18
|
Lee JJ, Lee JH, Ko YG, Hong SI and Lee JS:
Prevention of premature senescence requires JNK regulation of Bcl-2
and reactive oxygen species. Oncogene. 29:561–575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perez EA, Hack FM, Fletcher TS and Chou
TC: Modulation of intrinsic in vitro resistance to carboplatin by
edatrexate in the A549 human nonsmall cell lung cancer cell line.
Oncol Res. 6:151–156. 1994.PubMed/NCBI
|
|
20
|
Eramo A, Ricci-Vitiani L, Zeuner A, et al:
Chemotherapy resistance of glioblastoma stem cells. Cell Death
Differ. 13:1238–1241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eramo A, Lotti F, Sette G, et al:
Identification and expansion of the tumorigenic lung cancer stem
cell population. Cell Death Differ. 15:504–514. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ricci-Vitiani L, Lombardi DG, Pilozzi E,
et al: Identification and expansion of human colon
cancer-initiating cells. Nature. 445:111–115. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sato A, Sunayama J, Okada M, et al:
Glioma-initiating cell elimination by metformin activation of FOXO3
via AMPK. Stem Cells Transl Med. 1:811–824. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bennett BL, Sasaki DT, Murray BW, et al:
SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase.
Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Verheij M, Ruiter GA, Zerp SF, et al: The
role of the stress-activated protein kinase (SAPK/JNK) signaling
pathway in radiation-induced apoptosis. Radiother Oncol.
47:225–232. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fan M and Chambers TC: Role of
mitogen-activated protein kinases in the response of tumor cells to
chemotherapy. Drug Resist Updat. 4:253–267. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cellurale C, Sabio G, Kennedy NJ, et al:
Requirement of c-Jun NH2-terminal kinase for
Ras-initiated tumor formation. Mol Cell Biol. 31:1565–1576.
2011.PubMed/NCBI
|
|
30
|
Jackson EL, Willis N, Mercer K, et al:
Analysis of lung tumor initiation and progression using conditional
expression of oncogenic K-ras. Genes Dev. 15:3243–3248.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vachtenheim J: Occurrence of ras mutations
in human lung cancer. Minireview Neoplasma. 44:145–149.
1997.PubMed/NCBI
|
|
32
|
Kadara H, Kabbout M and Wistuba II:
Pulmonary adenocarcinoma: a renewed entity in 2011. Respirology.
17:50–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kurtze I, Sonnemann J and Beck JF:
KRAS-mutated non-small cell lung cancer cells are responsive to
either co-treatment with erlotinib or gefitinib and histone
deacetylase inhibitors or single treatment with lapatinib. Oncol
Rep. 25:1021–1029. 2011.PubMed/NCBI
|
|
34
|
Okudela K, Hayashi H, Ito T, et al: K-ras
gene mutation enhances motility of immortalized airway cells and
lung adenocarcinoma cells via Akt activation: possible contribution
to non-invasive expansion of lung adenocarcinoma. Am J Pathol.
164:91–100. 2004. View Article : Google Scholar
|