1. Introduction
Chromosomal translocations are very common in human
cancer, particularly in hematopoietic and lymphoid tumors (1). They are involved in the initiation of
some types of cancer although the exact mechanism is not fully
understood. These translocations may provide a selective growth
advantage or chance of subsequent mutations in some stem or
progenitor cells, which may subsequently initiate the development
of some malignant tumors. For oncogenic chromosomal translocations,
gene rearrangements may change the original locations of
proto-oncogenes to generate the obvious effects on phenotype via
the two major ways (2,3). One is to generate oncogenic fusion
proteins. The best example is translocation between chromosomes 9
and 22 [t( 9;22)], i.e. Philadelphia (Ph) chromosome, in chronic
myeloid leukemia (CML), resulting in the translocation of
proto-oncogene ABL at 9q34 to BCR on chromosome 22.
The formation of BCR-ABL oncoprotein has an abnormal activity of
tyrosine kinase (TK) which is associated with the tumorigenesis of
CML and acute lymphoblastic leukemia (ALL) (4). Another way is that proto-oncogenes are
brought into proximity with the new cis-regulatory elements. The
classic example is the overexpression of proto-oncogene
c-MYC in Burkitt lymphoma due to t(8;14) to result
c-MYC juxtaposed to immunoglobulin heavy chain (IGH)
regulatory elements.
Chromosomal translocations in vivo are a
complex biological process and there are two essential steps for
the formation of chromosomal translocations. First, DNA
double-strand breaks (DSBs) occur simultaneously at the two loci.
Second, the ends of DSBs need to approach each other and are
illegitimately joined together. Aside from these essential steps,
increasing evidence shows that there are still several factors that
influence the formation of chromosomal translocations, such as
nuclear architecture, activation induced deaminase (AID)-mediated
V(D)J recombination, gene expression, and other unknown mechanisms
(5–7). In the present study, I focus on the
effects of chromosome or gene positioning on chromosomal
translocations, on the functional impacts owing to oncogenic
chromosomal translocations in human cancer.
2. Chromosomal translocations are related to
chromosome or gene positioning
Chromosomal translocations in cancer are generally
considered to be no-random. The factors that could influence
chromosomal translocation are complex and several factors, such as
the spatial positions of broken loci, recombination, DNA repair
elements, are involved. The two spatial proximal broken loci have
more probability to illegitimately join than two distant broken
loci (8). For example,
investigations have shown that chromosomes 9 and 22 neighbor in
lymphoid cells (9,10). This may partly explain why t(9;22)
easily occurs in lymphocytes. Similar to t(9;22), t(15;17),
resulting in the formation of promyelocytic leukemia-retinoic acid
receptor α (PML-RARα) fusion oncoprotein, can be detected in most
cells in acute promyelocytic leukemia (APL) (11). The study also showed that
chromosomes 15 and 17 were close to each other in lymphoid cells
(10); this may also partly explain
why t(15;17) easily occurs in hematopoietic cells. Furthermore,
intergenic distance between the PML and RARα or
BCR and ABL is shorter in hematopoietic precursors
than in B-lymphoid cells (10),
consistent with the theory that cancer originates from stem
cells.
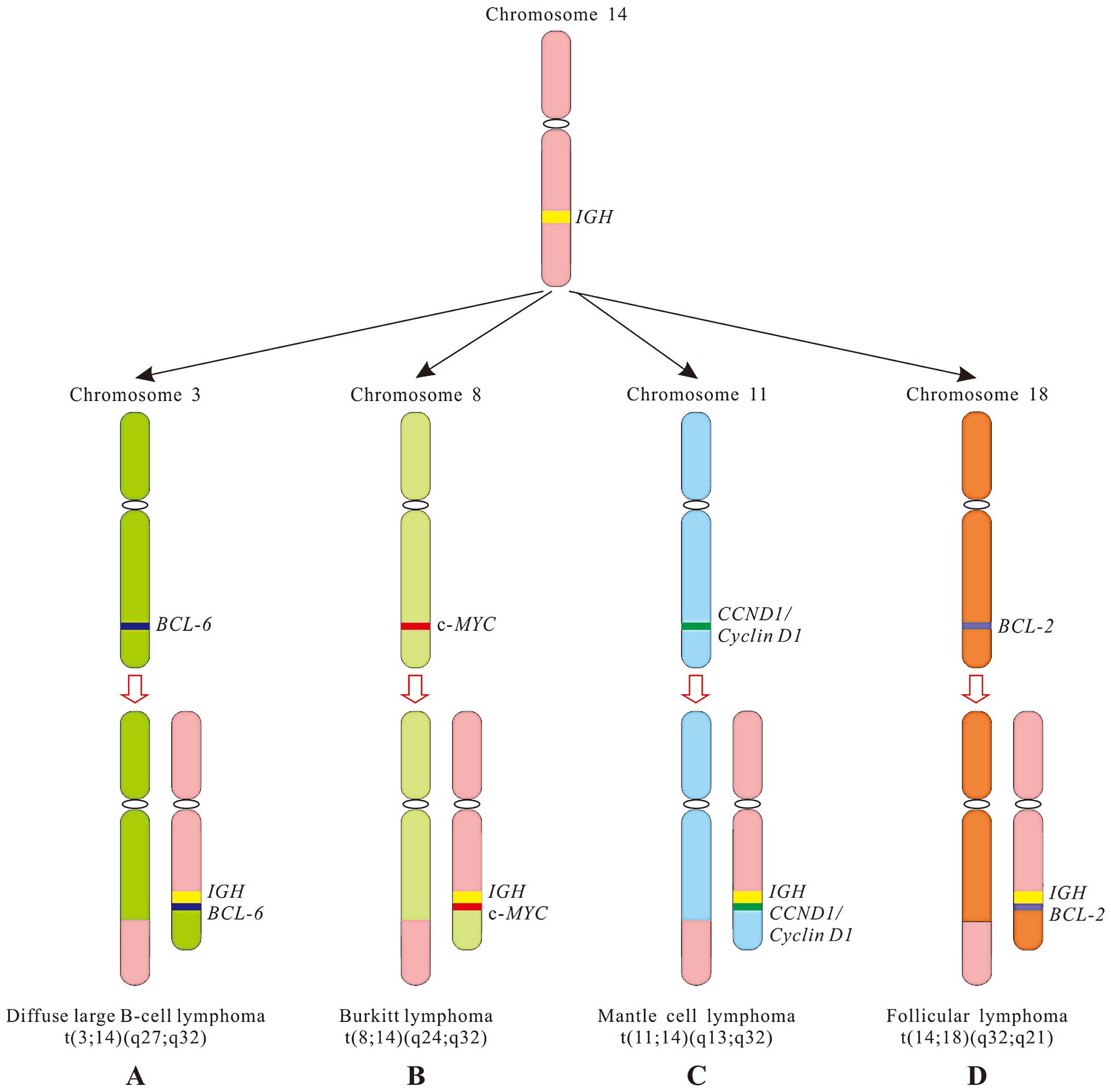
The reason why 70% of Burkitt lymphomas, a B-cell
tumor, often contains t(8;14), i.e. the c-MYC gene (8q24)
juxtaposes to IGH gene (14q32) (Fig. 1B), is because chromosome 8 closes
spatially chromosome 14 in B lymphocytes (12,13).
Research has shown that when B lymphocytes are stimulated, the
MYC gene is preferentially recruited to the same
transcription factory as the highly transcribed IGH gene.
While the c-MYC and IGH are close to each other, it
increases the incidence of specific chromosomal translocations
(14). With the exception of
t(8;14), c-MYC less often rearranges with the immunoglobulin
light chain κ (IGK) or λ (IGL) genes of chromosome 2
or 22 in Burkitt lymphoma, t(2;8)(p11.2;q24.1) or
t(8;22)(q24.1;q11.2) places c-MYC under the control of
IGK or IGL locus, respectively, resulting in the
overexpression of c-MYC. In fact, the mechanism of t(2;8) or
t(8;22) translocation is similar to that of t(8;14) in Burkitt
lymphoma, relating to spatial organization of the B cell genome
(12).
Except for t(8;14) in Burkitt lymphoma, a reciprocal
translocation between chromosomes 14 and 18 is also extremely
common in follicular lymphoma (70–95%), a B cell lymphoma with
follicular architecture. This translocation leads to the
juxtaposition of the BCL-2 gene at 18q21 and the IGH
locus, resulting in anti-apoptotic protein BCL-2 overexpression
(Fig. 1D). Measuring BCL-2
expression can be used to distinguish follicular lymphoma from
benign follicular hyperplasia, in which BCL-2 expression is low
(15). In mantle cell lymphoma, an
aggressive subtype of B cell lymphoma, most tumor cells have a
t(11;14), i.e. the cyclin D1 (CCND1) gene at 11q13 moves to
IHG locus, resulting in the overexpression of cyclin D1
(Fig. 1C) (16). Cyclin D1, a cell cycle regulator, is
not expressed in normal B cells. In diffuse large B-cell lymphoma
(DLBCL), approximately one third of patients have a t(3;14), i.e.
the oncogene BCL-6 on chromosome 3 moves to IHG
locus, resulting in the overexpression of BCL-6 (Fig. 1A), a specific transcriptional
repressor that inhibits the differentiation of B cells. The
mechanism of chromosomal translocations in follicular lymphoma,
mantle cell lymphoma and DLBCL are similar to that in Burkitt
lymphoma, relating to spatial proximity of translocation-prone gene
loci in the interphase nucleus (12).
Approximately 60% of patients with anaplastic
large-cell lymphoma (ALCL) have t(2;5), that leads to the formation
of a characteristic fusion gene between anaplastic lymphoma kinase
(ALK) at 2p23 and nucleophosmin (NPM) at 5q35. ALK, a
receptor tyrosine kinase (RTK) belonging to the insulin receptor
superfamily, has been reported to be active due to chromosomal
translocations in several types of human cancer, such as ALCL,
non-small cell lung carcinoma (NSCLC) and DLBCL (17,18).
ALK expression is generally restricted to neural tissue (19), t(2;5) leading to the expression of
truncated ALK driven by NPM promoter in lymphocytes.
Accumulating evidence suggests that DSBs and the formation of
translocation are preceded by the two gene loci being in close
proximity. For example, Mathas et al(20) found that the formation of
ALK-NPM fusion gene was related to spatial proximity of two
gene loci which was prior to the generation of translocation. This
spatial proximity of two gene loci leads to upregulation of ALK
which facilitates to induce DSBs.
Aside from interchromosomal translocations,
intrachromosomal translocations are also associated with spatial
distance of two gene loci. For example, 60–70% of papillary thyroid
carcinomas have a characteristic inv(10)(q11.2q21), i.e. breakpoint RET
(10q11.2) is relegated to opposite breakpoint the H4
(D10S170) or NCOA4 (ELE1) gene (10q21) in the same
chromosome (21). RET, an RTK, is
often found in translocation in papillary thyroid carcinoma (PTC),
particularly in patients who had radiation exposure. The H4 protein
is widely expressed in the nucleus and cytoplasm and its function
is unknown (22). According to the
different rearrangement loci, to date, PTC has 11 rearranged forms,
referred to as PTC1-11 (23).
PTC1(H4, CCDC6)-RET and PTC3(NCOA4)-RET are the most common
intrachromosomal rearrangements in PTC. By contrast, PTC2-RET and
other less common types of PTC-RET are interchromosomal
translocations (24). These
rearrangements can lead to constitutively ligand-independent RET
activity, involved in thyroid carcinogenesis. Although the
distances between RET and H4 loci are 18 Mb,
chromosome folding can offer two loci close to each other in
thyroid cells, thus increasing the probability of recombination
between them in the interphase nuclei. This chromosomal folding is
specific for thyroid cells, and this may explain why inv(10)(q11.2q21) is frequently seen in PTC
(25). The translocation of
H4 and RET occurs less in other types of cells. If it
happens in non-thyroid cells, this type of translocation may not
cause tumor.
Hormones also influence chromosomal translocations
via their receptors. Previous studies showed that ~50% of prostate
cancer cases have del(21)(q22) and
t(7;21)(1,26–28),
resulting in the translocation of an ETS (E26
transformation-specific)-regulated gene (ERG) (21q22.3) or
ETS variant 1 (ETV1) gene (7p21.2) to the transmembrane
protease serine 2 (TMPRSS2) gene (21q22.2) promoter region,
which contains androgen receptor (AR) binding sites (29). ETS is a transcription factor family
in which every family member contains ETS domain, a winged
helix-loop-helix DNA binding domain. To date, 28 members of ETS
have been identified, such as FLI (11q24), ERG,
ETV1, ETV4 (17q21), ETV5(3q) and ETV6
(12p13) (30). The translocations
of ETS are often found in human cancer, such as Ewing sarcoma
(31,32), leukemia (33,34),
prostate cancer (1,27–28)
and breast cancer (35).
TMPRSS2 is a specific expression gene in the prostate and
its expression is increased in prostate cancer (28,36).
Although it is 2.7 Mb genomic distances between ERG and
TMPRSS2 on the same chromosome and TMPRSS2 and
ETV1 are on the different chromosomes, ERG and
ETV1 regulatory regions also have AR binding sites and
androgen can induce TMPRSS2 and ERG or ETV1
spatial proximity via AR (27,28,37,38).
These studies explain why the TMPRSS2-ERG and
TMPRSS2-ETV1 translocations are easily seen in prostate
cancer as the prostate is an androgen-sensitive organ. That
hormones induce interactions between gene loci on different
chromosomes is also found in estrogen. Hu et al(39) reported that estrogen induced rapid
chromosome interactions to coordinate specific gene expression via
estrogen receptor α (ERα).
In general, when DSBs occur, the ends of DSBs are
relatively stable and mobile <250 nm (40), supporting the observation that
chromosomal translocations occur in close genes. We can image if
the broken ends are relatively stable, they may be rejoined by
themselves, thereby preventing chromosomal translocation. If the
broken ends roam, it increases the chances of illegitimate
recombination. Thus, the relative stability of the broken ends
decreases the probability of gene rearrangement and favor genomic
integrity (40,41).
3. Effects of oncogenic chromosomal
translocations
Effects of oncogenic chromosomal translocations on
cellular phenotypes are complex and diverse. Following
translocations, oncogenes may influence cellular phenotypes via the
formation of oncogenic fusion proteins or under the control of the
new regulatory elements (1–3).
Oncogenic fusion proteins
Although the products of oncogenic fusion genes are
diverse, they can primarily be classified into two groups,
transcription factors and TKs. Several oncogenic fusion proteins
are transcription factors and TKs. In fact, the products of fusion
genes are diverse; some may be neutral, some may play less
important roles in cellular phenotypes and some may cause cell
death in which we can not see this type of the translocation. The
translocations found in cancer, however, clearly have critical
functions in tumorigenesis. Generally, transcription factors and
TKs play more important roles in cellular phenotypes, and this may
partly explain why many fusion proteins detected in human cancer
are transcription factors and TKs. It should be noted, that these
so-called oncogenic fusion proteins as transcription factors and
TKs are already different from their functions of parental proteins
in several aspects and they often acquire some new functions.
It is clear that the sites of DSBs are related to
the functional consequences of fusion genes. DSBs are not random
(42) and occur preferentially in
large and evolutionarily conserved genes (43,44),
fragile sites (45), transcription
start sites (14,46,47)
and euchromatin (48,49). The breakpoints do not usually occur
in their functional domains if these genes are encoded for
transcription factors or TKs, thus fusion proteins can still retain
the activities of transcription factors or TKs (42). Several studies have shown that DSBs
preferentially occur in euchromatin, consistent with a greater
chance for translocation to occur in the sites with transcription
activity (14,46,47).
Following exposure to ionizing radiation, DSBs occur more often in
euchromatin than heterochromatin, suggesting the highly compacted
chromatin can prevent from radiation damage. From another point,
euchromatin is relatively loose and has a lack of protective
mechanism, so it is easily attacked by radiation (48,49).
In addition, the mechanisms of DSB repair in euchromatin are also
different from heterochromatin. Since the time for DSB repair in
heterochromatin is longer than euchromatin (50,51),
by extrapolation, the higher frequency of chromosomal
translocations in euchromatin than in heterochromatin is
reasonable.
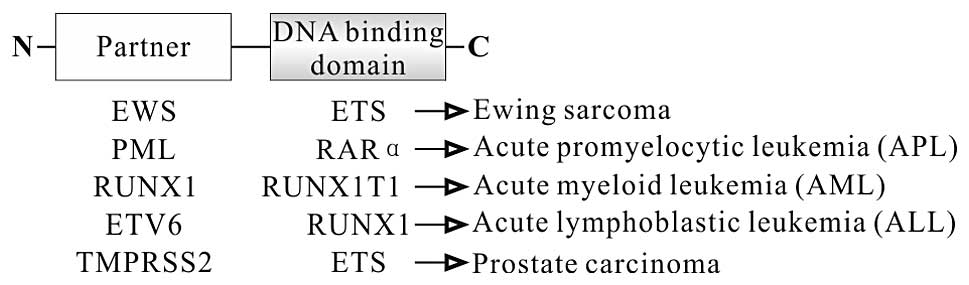
Oncogenic fusion protein as transcription
factor
The products of several oncogenic fusion genes
function as transcription factors. In this group, each fusion
protein consists of N-terminal partner fused to the DNA binding
domain at the C-terminus (Fig. 2).
For example, EWS-FLI fusion protein, a characterized protein in
Ewing sarcoma, consists of N-terminal part of EWS, a member of the
TET family at the N-terminus, and C-terminal part of FLI, a member
of the ETS family, at the C-terminus (31). As a chimeric transcription factor,
EWS-FLI fusion protein has different transcription functions
compared to its parental transcription factor FLI (32), despite identical DNA-binding domain.
This mistargeting is associated with 85% of Ewing sarcoma
development (52).
The functions of the fusion proteins as oncogenic
transcription factors are various. Some stimulate gene expression,
such as TMPRSS2-ERG and TMPRSS2-ETV1. Whether the TMPRSS2-ETS are
really fusion proteins is under debate. Some people consider that
the TMPRSS2-ETS translocations are the expression of
ETS under the influence of the TMPRSS2 promoter as
the expression of MYC under the IGH regulatory
elements in Burkitt lymphoma (2).
In fact, the TMPRSS2-ETS translocations are very
heterogeneous, both TMPRSS2 at the 5′-end and ETS at
the 3′-end have different fusion forms which generate different
fusion transcripts, including splice variants (26,29,53,54).
In most cases, the TMPRSS2 promoter and first exon or first
2 exons are juxtaposed to the ETS exons, with deletion of
the ETS promoter and first exon or first 2 exons (55). Therefore, the fusion genes are under
the control of the androgen-regulated TMPRSS2 promoter,
resulting in the high level expression of oncogenic ETS
fusion genes. For example, TMPRSS2-ERG gene fusion is the
most common among these translocations and some are composed of the
TMPRSS2 promoter and the first exon at the 5′-end and the
transcription factor domain of ERG at the 3′-end, resulting
in a truncated ERG protein lacking TMPRSS2 as the TMPRSS2
exon 1 is noncoding and does not contain an ATG (53), some are composed of the
TMPRSS2 promoter and the first 2 exons (exon 2 containing an
ATG at 142) at the 5′-end and the transcription factor
domain of ERG at the 3′-end (designed type VI), resulting in
a true fusion protein containing the first 5 amino acids of the
TMPRSS2 at the N-terminus and a slightly truncated ERG protein at
the C-terminus (Fig. 2) (53). Androgen can stimulate the
transcription of the TMPRSS2-ETS fusion since all
TMPRSS2-ETS fusions retain the TMPRSS2 promoter which
contains AR binding sites. In most cases, ETS retains DNA-binding
domain, which can stimulate the transcription of target genes for
cell growth, invasion and metastasis and promote prostate cancer
progression (26,28).
Some inhibit gene expression, such as
t(12;21)/ETV6(TEL1)-RUNX1(runt-related transcription factor 1,
previously known as AML1), t(8;21)/RUNX1-RUNXIT1(ETO),
t(15;17)/PML-RARα and inv(16)/CBFB-MYH11, which inhibit the
transcriptional activity of genes required for normal
differentiation of hematopoietic cells. Although these fusion
proteins may not be sufficient to induce leukemia alone (56), they increase the developmental risk
of acute leukemia in patients with these fusion proteins (4,34,57).
These fusion proteins repress the functions of transcription via
the different molecular mechanisms. For example, RUNX1-RUNXIT1
protein is found in ~13% of acute myeloid leukemia (AML) (58). In RUNX1-RUNXIT1 protein, the
translocation deletes the transactivation domain but retains the
runt homology domain (RHD) responsible for binding to DNA at
N-terminus of RUNX1 (Fig. 2)
(59). RUNX1-RUNXIT1 protein
interferes with wild-type RUNX1-dependent transcription via RUNXIT1
recruiting the nuclear corepressor (N-CoR)-histone deacetylase
(HDAC) complex (60). ETV6-RUNX1
protein is the most common abnormality in childhood ALL, occurring
in ~25% (4). In ETV6-RUNX1 protein,
the translocation deletes the ETS domain of ETV6, a member of the
ETS family, but retains the runt domain of RUNX1 (Fig. 2). Similar to the RUNX1-RUNXIT1
protein as a dominant negative inhibitor of RUNX1, the ETV6-RUNX1
protein represses RUNX1-dependent transcription via ETV6 recruiting
N-CoR-HDAC complex (61). RUNX1
targeting genes are required for normal hematopoietic cell
development. PML-RARα protein is linked to the development of APL,
a genetic distinct subtype of AML. This fusion protein is composed
of most of the functional domains of RARα (including the RAR
binding domain and the DNA binding domain) and the majority of PML,
including dimerization domain (Fig.
2). As a transcription factor, wild-type RARα releases
SMRT/N-CoR corepressor after binding retinoic acid (RA) and induces
the transcription of target genes that promote cell
differentiation. However, this fusion protein alters the
sensitivity to physiological levels of RA and impairs the release
of SMRT/N-CoR corepressor from RARα, therefore blocking the
differentiation of promyelocytes (62). One of the mechanisms of
pharmacologic levels of all-trans retinoic acid (ATRA) treatment
APL promotes the release of the corepressor from RARα and recovers
RA response (11). Arsenic trioxide
(As2O3) is also used to treat APL by
promoting degradation of PML-RARα protein (63).
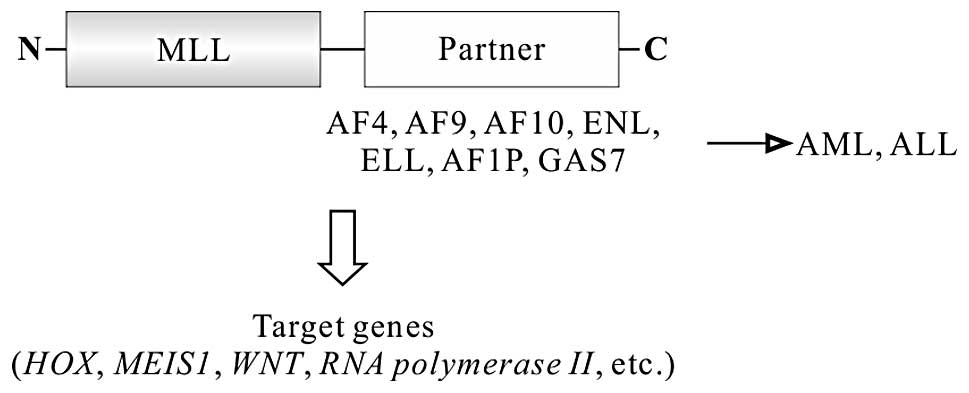
There are some mistarget gene expressions, such as
mixed lineage leukemia (MLL) fusions (64). MLL gene on 11q23 is often
rearranged with other partner genes in ALL and AML, accounting for
8% of pediatric and 10% of adult ALL (4), 15–20% of pediatric AML and <3% of
adult AML (65), or biphenotypic
(mixed lineage) leukemias. The MLL gene encodes a complex
DNA binding protein with histone H3 lysine 4 (H3K4)-specific
methyltransferase activity, which positively regulates gene
expression including HOX genes. MLL protein consists of multiple
functional domains, including the AT-hooks, DNA methyltransferase
homology domain that contains a CXXC zinc finger motif and
trithorax PHD domains at the N-terminus, the transactivation domain
(TAD) and SET domain that possesses H3K4 methyltransferase activity
at the C-terminus. Post-translationally, taspase I cleaves MLL to
generate two fragments (MLLN p300 and MLLC p180) that form a stable
complex by direct interaction of the FYRN and FYRC domains
(66). Unlike classical
sequence-specific DNA-binding transcription factor, MLL regulates
the expression of target genes via epigenetic mechanisms, such as
DNA and histone methylation modification (66).
Chromosomal translocations lead to the fusion of
5′-end portion of MLL to one of >60 different partner genes,
resulting in the formation of different fusion genes, such as
MLL-AF4 (4q21), MLL-AF9 (9p22), MLL-ENL
(19p13.3), MLL-AF10 (10p12), MLL-AF6 (6q27),
MLL-ELL (19p13.1) (Fig. 3)
(66,67). All MLL fusion proteins retain
N-terminal AT-hooks, DNA methyltransferase homology domain, thus
preserving DNA binding activity whereas the trithorax PHD domains,
TAD and SET domains are always replaced by the partners. In these
fusions, the original MLL H3K4 methyltransferase activity is
replaced by the partners which play a critical role in MLL
oncoproteins (68). Although MLL
fusion proteins lose the activity of H3K4 methylation, these fusion
proteins gain the activity of H3K79 methylation via recruiting the
H3K79 methyltransferase hDOT1L which can cause dysregulation of
whole genomic expression and is associated with MLL leukemogenesis
(67,69). Since hDOT1L plays a key role in the
development of MLL leukemia, hDOT1L is an ideal target for MLL
leukemia. Several hDOT1L inhibitors are underdeveloped. In
particular, EPZ004777, a specific hDOT1L inhibitor, seems to be a
promising drug for leukemia with MLL gene translocation (70).
Since >60 MLL fusion proteins have been found
(71), the functions of MLL fusion
proteins are very different, and the functions of some MLL fusion
proteins remain unclear or not fully understood. To date, we know
that MLL oncoproteins induce leukemia through several pathways.
First, MLL oncoproteins act as transcriptional regulators that can
bind DNA and induce aberrant expression of leukemic stem cell
target genes, such as HOX, MEIS1, WNT and
RNA polymerase II. Among MLL target genes, transcription
factor HOX genes are particular and essential for MLL
leukemogenesis (72). MLL-ENL,
MLL-ELL, MLL-AF4, MLL-AF9 and MLL-AF10 have been demonstrated to
induce acute leukemia using this pathway (Fig. 3) (67,73–75).
Second, MLL fusion partners provide a dimerization motif, such as
AF1p/Eps15 and GAS7. The MLL dimerization/oligomerization proteins
can recruit co-activators or basal transcriptional machinery to
result in the aberrant expression of target genes for inducing
acute leukemia (76,77). Third, MLL fusion partners increase
the stabilization of MLL oncoproteins. For example, all MLL-AF4,
MLL-AF9, MLL-ENL and MLL-ELL exhibit resistance to degradation
mediated by the cell cycle ubiquitin-proteasome system (71).
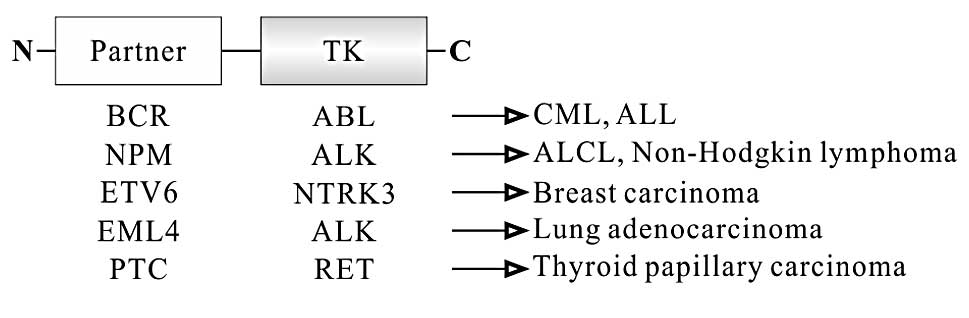
Oncogenic fusion protein as tyrosine
kinase
Another group of oncogenic fusion proteins harbor
activities of TKs. In this group, each translocation generates a
different fusion protein consisting of N-terminal partner fused to
the TK domain at the C-terminus (Fig.
4). TK domain in these fusion proteins is intact although they
are the truncated proteins (2,78,79).
For example, in ALCL, ALK breakpoints are located in the intron
flanked by exons 16 and 17, and exons 17–26 encoding the
intracytoplasmic kinase domain of ALK are intact (80). It is similar to that of RET in PTC
(81). As the regulatory parts of
kinase are often lost and replaced by unrelated sequences, the
kinase activity of these fusion proteins is determined by the
N-terminal partners. In most cases, the N-terminal partners supply
domains that promote dimerization/oligomerization, allowing fusion
kinase to be activated in the absence of physiological stimulating
signals (79,81–85).
BCR-ABL fusion protein is linked to the development
of CML and ALL (4). ABL protein has
two isoforms, 1a and b. ABL1b contains a C14 myristoyl saturated
fatty acid moiety covalently linked to the Cap region at the
N-terminus and is expressed at higher levels than ABL1a, which is
not myristoylated. The Cap region of ABL contains endogenous
autoregulatory domain which can inhibit kinase activity via
stabilizing SH3 and SH2 domains of ABL (86,87).
This fusion protein is composed of the majority of BCR at the
N-terminus and most of the functional domains of ABL except Cap
domain at the C-terminus (Fig. 4),
resulting in the disregulatory activation of BCR-ABL TK (88). In addition, oligomerization domain
and GRB2-binding site at tyrosine 177 (Y177) in BCR partner are
also essential for BCR-ABL-mediated CML (82,89).
Imatinib/Gleevec®, a specific BCR-ABL inhibitor, was the
first molecular target drug approved by the US Food and Drug
Administration (FDA) to treat patients with CML in 1996. Dasatinib
and nilotinib, second generation inhibitors of ABL, have also been
approved to treat patients with imatinib-resistant CML (90).
H4-RET fusion protein is the most common chromosomal
translocation in PTC, and accounts for 60–70% of PTC. This protein
consists of the N-terminal promoter and leucine zipper domain of H4
at the N-terminus and the TK domains of RET at the C-terminus
(Fig. 4). RET lacks the signal
peptide and transmembrane domain in this chimeric oncoprotein, thus
the aberrant TK activity of RET fusion is controlled by H4 partner
which provides an active promoter and dimerization domain for
ligand-independent activation of the fusion protein (91).
Approximately 5% of NSCLCs have inv(2)(p21;p23),
resulting in the formation of echinoderm microtubule-associated
protein-like 4 (EML4)-ALK fusion gene (85). EML4-ALK protein consists of various
length EML4 containing the coiled-coil domain at the N-terminus and
the intracellular catalytic domain of ALK at the C-terminus
(Fig. 4). As ALK lacks the
extracellular and transmembrane domain in this oncoprotein, so EML4
partner constitutively activates the TK of ALK via the dimerization
of EML4-ALK, involved in the carcinogenesis of NSCLC (85). EML4-ALK is most commonly detected in
non-smokers with NSCLC. NSCLC with EML4-ALK has unique pathological
and clinical features, such as Asian patients, younger,
adenocarcinoma and lack of EGFR and KRAS mutations (92). Crizotinib, an ALK inhibitor, was
recently approved by the FDA to treat patients with ALK-positive
NSCLC (93).
Oncogenes under the control of a stronger
promoter
Proto-oncogenes are brought into proximity with the
new cis-regulatory elements, leading to their overexpression
which is seen in several types of lymphoma and leukemia,
particularly in B and T cell malignancies. This is because V(D)J
recombination exists during B and T cell development, which
generates antibody and T cell receptor (TCR) diversity. However,
V(D)J recombination may also increase the risk of chromosomal
translocation in the same regions, which may partly explain why
chromosomal translocation frequently occurs in several types of
lymphoma and leukemia. For example, the overexpression of oncogenes
c-MYC, BCL-2, CCND1 and BCL-6 in B cell
lymphomas may be associated with errors in V(D)J recombination
(Fig. 1) (16,94–96),
suggesting the mechanism of chromosomal translocations in these B
cell lymphomas is similar.
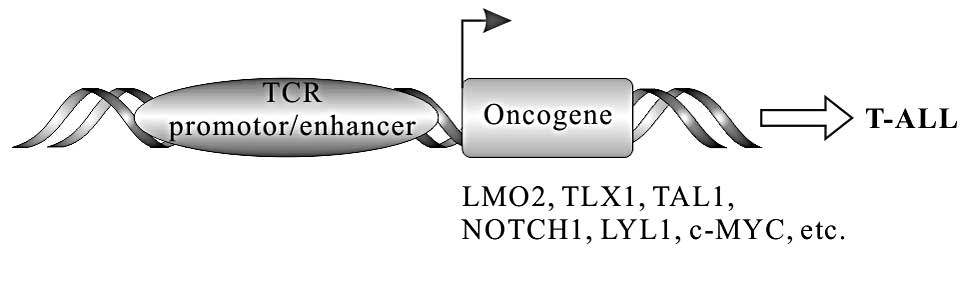
In a subset of T cell ALL (T-ALL), chromosomal
translocation can make proto-oncogenes under the control of TCR
regulatory elements, resulting in the deregulated transcription of
these proto-oncogenes, such as TLX1 (HOX11), TLX3
(HOX11L2), LMO1, LMO2, c-MYC, LYL1,
T-cell acute lymphocytic leukemia-1/stem cell leukemia
(TAL1/SCL), TAL2 and NOTCH1 (Fig. 5). TLX1 and TLX3 belong to homeobox
transcription factors. LIM domain only (LMO) 1 and LMO2 belong to
LIM transcription factors containing LIM zinc-finger motifs. c-MYC,
LYL1, TAL1/SCL and TAL2 belong to basic helix-loop-helix (bHLH)
transcription factors. NOTCH1, one of NOTCH family, is a
transmembrane protein.
The TCR is composed of two different protein
chains. In 95% of T cells, TCR consists of αβ chains, whereas in 5%
of T cells, TCR consists of γδ chains. TCRα (TCRA) and δ
(TCRD) chain genes are localized on 14q11.2, TCRβ
(TCRB) and TCRγ (TCRG) loci are localized on 7q34 and
7p15, respectively. The breakpoints often occur in TCRA/D or
TCRB. The t(11;14)(p13;q11) and t(7;11)(q34;p13) have been
found in 3% T-ALL (97). Both
translocations lead to LMO2 (11p13) under the control of the
TCRD or TCRB locus, resulting in LMO2 overexpression
which may be involved in the T-ALL development (98). Proto-oncogene TLX1 (T-cell
leukemia homeobox 1, previously known as HOX11 or
TCL3) on 10q24 is normally not expressed in T cells and its
expression is often deregulated in T-ALL (99). This deregulated expression of
TLX1 is related to t(7;10)(q34;q24) and t(10;14)(q24;q11)
which account for 7% of childhood and 31% of adult T-ALL (97). These translocations make TLX1
under the control of the TCRB or TCRA locus,
resulting in the overexpression of TLX1 which may contribute
to T-ALL via blocking apoptosis of developing T cell in the thymus
(100). TLX1 overexpression has
also been demonstrated in the absence of a 10q24 rearrangement,
suggesting that other mechanisms, such as epigenetic alterations,
can lead to this aberrant expression of TLX1(101,102). The situation is similar to
TAL1 (1p32). Approximately 7% of childhood T-ALL and 12% of
adult T-ALL have t(1;14)(p32;q11), leading to deregulated
expression of TAL1 under control of the TCRA/D loci
(4). However, the overexpression of
TAL1 in T-ALL also occurs in the absence of TAL1
rearrangement, suggesting that other mechanisms may influence the
overexpression of TAL1(103).
NOTCH1 plays crucial roles in cell development,
hematopoietic stem cell maintenance and T cell fate specification
in the mature organism (104).
NOTCH1 is regarded as an oncoprotein. In a low number of human
T-ALL patients, they had t(7;9)(q34;q34.3) which results to fuse
the 3′ end of NOTCH1 (9q34.3) to TCRB locus, leading
to overexpression of a truncated NOTCH1 protein that lack the
negative regulatory region (NRR) (105). NRR is NOTCH1 extracellular domain
and responsible for preventing ligand-independent receptor
activation.
4. Conclusion
Chromosomal translocations in human cancer are not
random and tend to occur in some specific sites with spatial
proximity in genome organization. The oncogenic chromosomal
translocations may provide a selective growth advantage or chance
of secondary mutations in some stem or progenitor cells via
different pathways, such as the formation of oncogenic fusion
proteins and under the control of the new regulatory elements.
Understanding the mechanisms of chromosomal translocations in
cancer may help us to develop new approaches in early the diagnosis
and target therapy of cancer.
Acknowledgements
Dr Peng Gao is acknowledged for the images and
comments on this review. This study was in part supported by a
grant from the Ministry of Education, China (no.
20110092110043).
References
|
1
|
Mitelman F, Johansson B and Mertens F: The
impact of translocations and gene fusions on cancer causation. Nat
Rev Cancer. 7:233–245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nambiar M, Kari V and Raghavan SC:
Chromosomal translocations in cancer. Biochim Biophys Acta.
1786:139–152. 2008.PubMed/NCBI
|
|
3
|
Fröhling S and Döhner H: Chromosomal
abnormalities in cancer. N Engl J Med. 359:722–734. 2008.
|
|
4
|
Pui CH, Relling MV and Downing JR: Acute
lymphoblastic leukemia. N Engl J Med. 350:1535–1548. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aplan PD: Causes of oncogenic chromosomal
translocation. Trends Genet. 22:46–55. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Raghavan SC and Lieber MR: DNA structures
at chromosomal translocation sites. Bioessays. 28:480–494. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hakim O, Resch W, Yamane A, et al: DNA
damage defines sites of recurrent chromosomal translocations in B
lymphocytes. Nature. 484:69–74. 2012.PubMed/NCBI
|
|
8
|
Meaburn KJ, Misteli T and Soutoglou E:
Spatial genome organization in the formation of chromosomal
translocations. Semin Cancer Biol. 17:80–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kozubek S, Lukásová E, Marecková A, et al:
The topological organization of chromosomes 9 and 22 in cell nuclei
has a determinative role in the induction of t(9,22) translocations
and in the pathogenesis of t(9,22) leukemias. Chromosoma.
108:426–435. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Neves H, Ramos C, da Silva MG, Parreira A
and Parreira L: The nuclear topography of ABL, BCR, PML, and RARα
genes: evidence for gene proximity in specific phases of the cell
cycle and stages of hematopoietic differentiation. Blood.
93:1197–1207. 1999.PubMed/NCBI
|
|
11
|
Collins SJ: Retinoic acid receptors,
hematopoiesis and leukemogenesis. Curr Opin Hematol. 15:346–351.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roix JJ, McQueen PG, Munson PJ, Parada LA
and Misteli T: Spatial proximity of translocation-prone gene loci
in human lymphomas. Nat Genet. 34:287–291. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Misteli T: The inner life of the genome.
Sci Am. 304:66–73. 2011. View Article : Google Scholar
|
|
14
|
Osborne CS, Chakalova L, Mitchell JA, et
al: Myc dynamically and preferentially relocates to a
transcription factory occupied by Igh. PLoS Biol.
5:e1922007. View Article : Google Scholar
|
|
15
|
Cornfield DB, Mitchell DM, Almasri NM,
Anderson JB, Ahrens KP, Dooley EO and Braylan RC: Follicular
lymphoma can be distinguished from benign follicular hyperplasia by
flow cytometry using simultaneous staining of cytoplasmic bcl-2 and
cell surface CD20. Am J Clin Pathol. 114:258–263. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Welzel N, Le T, Marculescu R, et al:
Templated nucleotide addition and immunoglobulin
JH-gene utilization in t(11;14) junctions:
implications for the mechanism of translocation and the origin of
mantle cell lymphoma. Cancer Res. 61:1629–1636. 2001.PubMed/NCBI
|
|
17
|
Palmer RH, Vernersson E, Grabbe C and
Hallberg B: Anaplastic lymphoma kinase: signalling in development
and disease. Biochem J. 420:345–361. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barreca A, Lasorsa E, Riera L, et al:
Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol.
47:R11–R23. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iwahara T, Fujimoto J, Wen D, et al:
Molecular characterization of ALK, a receptor tyrosine kinase
expressed specifically in the nervous system. Oncogene. 14:439–449.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mathas S, Kreher S, Meaburn KJ, et al:
Gene deregulation and spatial genome reorganization near
breakpoints prior to formation of translocations in anaplastic
large cell lymphoma. Proc Natl Acad Sci USA. 106:5831–5836. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gandhi M, Evdokimova V and Nikiforov YE:
Mechanisms of chromosomal rearrangements in solid tumors: the model
of papillary thyroid carcinoma. Mol Cell Endocrinol. 321:36–43.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Merolla F, Pentimalli F, Pacelli R,
Vecchio G, Fusco A, Grieco M and Celetti A: Involvement of
H4(D10S170) protein in ATM-dependent response to DNA damage.
Oncogene. 26:6167–6175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nikiforov YE: Thyroid carcinoma: molecular
pathways and therapeutic targets. Mod Pathol. 21(Suppl 2): S37–S43.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ciampi R, Giordano TJ, Wikenheiser-Brokamp
K, Koenig RJ and Nikiforov YE: HOOK3-RET: a novel type of RET/PTC
rearrangement in papillary thyroid carcinoma. Endocr Relat Cancer.
14:445–452. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gandhi M, Medvedovic M, Stringer JR and
Nikiforov YE: Interphase chromosome folding determines spatial
proximity of genes participating in carcinogenic RET/PTC
rearrangements. Oncogene. 25:2360–2366. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clark J, Merson S, Jhavar S, et al:
Diversity of TMPRSS2-ERG fusion transcripts in the human prostate.
Oncogene. 26:2667–2673. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin C, Yang L, Tanasa B, et al: Nuclear
receptor-induced chromosomal proximity and DNA breaks underlie
specific translocations in cancer. Cell. 139:1069–1083. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Squire JA, Park PC, Yoshimoto M, Alami J,
Williams JL, Evans A and Joshua AM: Prostate cancer as a model
system for genetic diversity in tumors. Adv Cancer Res.
112:183–216. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kumar-Sinha C, Tomlins SA and Chinnaiyan
AM: Recurrent gene fusions in prostate cancer. Nat Rev Cancer.
8:497–511. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seth A and Watson DK: ETS transcription
factors and their emerging roles in human cancer. Eur J Cancer.
41:2462–2478. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sankar S and Lessnick SL: Promiscuous
partnerships in Ewing’s sarcoma. Cancer Genet. 204:351–365.
2011.PubMed/NCBI
|
|
32
|
Patel M, Simon JM, Iglesia MD, Wu SB,
McFadden AW, Lieb JD and Davis IJ: Tumor-specific retargeting of an
oncogenic transcription factor chimera results in dysregulation of
chromatin and transcription. Genome Res. 22:259–270. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zelent A, Greaves M and Enver T: Role of
the TEL-AML1 fusion gene in the molecular pathogenesis of
childhood acute lymphoblastic leukaemia. Oncogene. 23:4275–4283.
2004.
|
|
34
|
Bohlander SK: ETV6: a versatile player in
leukemogenesis. Semin Cancer Biol. 15:162–174. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li Z, Tognon CE, Godinho FJ, et al:
ETV6-NTRK3 fusion oncogene initiates breast cancer from
committed mammary progenitors via activation of AP1 complex. Cancer
Cell. 12:542–558. 2007. View Article : Google Scholar
|
|
36
|
Vaarala MH, Porvari K, Kyllönen A,
Lukkarinen O and Vihko P: The TMPRSS2 gene encoding
transmembrane serine protease is overexpressed in a majority of
prostate cancer patients: detection of mutated TMPRSS2 form
in a case of aggressive disease. Int J Cancer. 94:705–710.
2001.
|
|
37
|
Mani RS, Tomlins SA, Callahan K, et al:
Induced chromosomal proximity and gene fusions in prostate cancer.
Science. 326:12302009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bastus NC, Boyd LK, Mao X, et al:
Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate
epithelial cells. Cancer Res. 70:9544–9548. 2010.PubMed/NCBI
|
|
39
|
Hu Q, Kwon YS, Nunez E, et al: Enhancing
nuclear receptor-induced transcription requires nuclear motor and
LSD1-dependent gene networking in interchromatin granules. Proc
Natl Acad Sci USA. 105:19199–19204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Soutoglou E, Dorn JF, Sengupta K, et al:
Positional stability of single double-strand breaks in mammalian
cells. Nat Cell Biol. 9:675–682. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Parada LA, McQueen PG and Misteli T:
Tissue-specific spatial organization of genomes. Genome Biol.
5:R442004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ortiz de Mendíbil I, Vizmanos JL and Novo
FJ: Signatures of selection in fusion transcripts resulting from
chromosomal translocations in human cancer. PLoS One.
4:e48052009.PubMed/NCBI
|
|
43
|
Bickmore WA and Teague P: Influences of
chromosome size, gene density and nuclear position on the frequency
of constitutional translocations in the human population.
Chromosome Res. 10:707–715. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Narsing S, Jelsovsky Z, Mbah A and Blanck
G: Genes that contribute to cancer fusion genes are large and
evolutionarily conserved. Cancer Genet Cytogenet. 191:78–84. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Burrow AA, Williams LE, Pierce LC and Wang
YH: Over half of breakpoints in gene pairs involved in
cancer-specific recurrent translocations are mapped to human
chromosomal fragile sites. BMC Genomics. 10:592009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Branco MR and Pombo A: Intermingling of
chromosome territories in interphase suggests role in
translocations and transcription-dependent associations. PLoS Biol.
4:e1382006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chiarle R, Zhang Y, Frock RL, et al:
Genome-wide translocation sequencing reveals mechanisms of
chromosome breaks and rearrangements in B cells. Cell. 147:107–119.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Obe G, Pfeiffer P, Savage JR, et al:
Chromosomal aberrations: formation, identification and
distribution. Mutat Res. 504:17–36. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cowell IG, Sunter NJ, Singh PB, Austin CA,
Durkacz BW and Tilby MJ: γH2AX foci form preferentially in
euchromatin after ionising-radiation. PLoS One. 2:e10572007.
|
|
50
|
Lorat Y, Schanz S, Schuler N, Wennemuth G,
Rübe C and Rübe CE: Beyond repair foci: DNA double-strand break
repair in euchromatic and heterochromatic compartments analyzed by
transmission electron microscopy. PLoS One. 7:e381652012.
View Article : Google Scholar
|
|
51
|
Murray JM, Stiff T and Jeggo PA: DNA
double-strand break repair within heterochromatic regions. Biochem
Soc Trans. 40:173–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Turc-Carel C, Aurias A, Mugneret F, et al:
Chromosomes in Ewing’s sarcoma. I An evaluation of 85 cases of
remarkable consistency of t(11;22)(q24;q12). Cancer Genet
Cytogenet. 32:229–238. 1988.
|
|
53
|
Wang J, Cai Y, Ren C and Ittmann M:
Expression of variant TMPRSS2/ERG fusion messenger RNAs is
associated with aggressive prostate cancer. Cancer Res.
66:8347–8351. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang J, Cai Y, Shao LJ, et al: Activation
of NF-κB by TMPRSS2/ERG fusion isoforms through toll-like
receptor-4. Cancer Res. 71:1325–1333. 2011.
|
|
55
|
Tomlins SA, Bjartell A, Chinnaiyan AM,
Jenster G, Nam RK, Rubin MA and Schalken JA: ETS gene fusions in
prostate cancer: from discovery to daily clinical practice. Eur
Urol. 56:275–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yuan Y, Zhou L, Miyamoto T, et al:
AML1-ETO expression is directly involved in the development of
acute myeloid leukemia in the presence of additional mutations.
Proc Natl Acad Sci USA. 98:10398–10403. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Licht JD and Sternberg DW: The molecular
pathology of acute myeloid leukemia. Hematology Am Soc Hematol Educ
Program. 2005:137–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Rubnitz JE, Raimondi SC, Halbert AR, et
al: Characteristics and outcome of t(8;21)-positive childhood acute
myeloid leukemia: a single institution’s experience. Leukemia.
16:2072–2077. 2002.PubMed/NCBI
|
|
59
|
Okumura AJ, Peterson LF, Okumura F,
Boyapati A and Zhang DE: t(8;21)(q22;q22) Fusion proteins
preferentially bind to duplicated AML1/RUNX1 DNA-binding sequences
to differentially regulate gene expression. Blood. 112:1392–1401.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang J, Wang M and Liu JM: Domains
involved in ETO and human N-CoR interaction and ETO transcription
repression. Leuk Res. 28:409–414. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang L and Hiebert SW: TEL contacts
multiple co-repressors and specifically associates with histone
deacetylase-3. Oncogene. 20:3716–3725. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mengeling BJ, Phan TQ, Goodson ML and
Privalsky ML: Aberrant corepressor interactions implicated in
PML-RARα and PLZF-RARα leukemogenesis reflect an altered
recruitment and release of specific NCoR and SMRT splice variants.
J Biol Chem. 286:4236–4247. 2011.PubMed/NCBI
|
|
63
|
Zhang XW, Yan XJ, Zhou ZR, et al: Arsenic
trioxide controls the fate of the PML-RARα oncoprotein by directly
binding PML. Science. 328:240–243. 2010.PubMed/NCBI
|
|
64
|
Mueller D, García-Cuéllar MP, Bach C, Buhl
S, Maethner E and Slany RK: Misguided transcriptional elongation
causes mixed lineage leukemia. PLoS Biol. 7:e10002492009.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Balgobind BV, Zwaan CM, Pieters R and Van
den Heuvel-Eibrink MM: The heterogeneity of pediatric
MLL-rearranged acute myeloid leukemia. Leukemia. 25:1239–1248.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu H, Cheng EH and Hsieh JJ: MLL fusions:
pathways to leukemia. Cancer Biol Ther. 8:1204–1211. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Krivtsov AV and Armstrong SA: MLL
translocations, histone modifications and leukaemia stem-cell
development. Nat Rev Cancer. 7:823–833. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Dobson CL, Warren AJ, Pannell R, Forster A
and Rabbitts TH: Tumorigenesis in mice with a fusion of the
leukaemia oncogene Mll and the bacterial lacZ gene.
EMBO J. 19:843–851. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bernt KM, Zhu N, Sinha AU, et al:
MLL-rearranged leukemia is dependent on aberrant H3K79
methylation by DOT1L. Cancer Cell. 20:66–78. 2011. View Article : Google Scholar
|
|
70
|
Daigle SR, Olhava EJ, Therkelsen CA, et
al: Selective killing of mixed lineage leukemia cells by a potent
small-molecule DOT1L inhibitor. Cancer Cell. 20:53–65. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu H, Cheng EH and Hsieh JJ: Bimodal
degradation of MLL by SCFSkp2 and APCCdc20
assures cell cycle execution: a critical regulatory circuit lost in
leukemogenic MLL fusions. Genes Dev. 21:2385–2398. 2007.PubMed/NCBI
|
|
72
|
Ayton PM and Cleary ML: Transformation of
myeloid progenitors by MLL oncoproteins is dependent on
Hoxa7 and Hoxa9. Genes Dev. 17:2298–2307. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zeisig BB, Schreiner S, García-Cuéllar MP
and Slany RK: Transcriptional activation is a key function encoded
by MLL fusion partners. Leukemia. 17:359–365. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Okada Y, Feng Q, Lin Y, et al: hDOT1L
links histone methylation to leukemogenesis. Cell. 121:167–178.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wong P, Iwasaki M, Somervaille TC, So CW
and Cleary ML: Meis1 is an essential and rate-limiting
regulator of MLL leukemia stem cell potential. Genes Dev.
21:2762–2774. 2007. View Article : Google Scholar
|
|
76
|
So CW, Lin M, Ayton PM, Chen EH and Cleary
ML: Dimerization contributes to oncogenic activation of MLL
chimeras in acute leukemias. Cancer Cell. 4:99–110. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
So CW and Cleary ML: Dimerization: a
versatile switch for oncogenesis. Blood. 104:919–922. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bischof D, Pulford K, Mason DY and Morris
SW: Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s
lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein
in oncogenesis. Mol Cell Biol. 17:2312–2325. 1997.
|
|
79
|
Goldman JM and Melo JV: Chronic myeloid
leukemia - advances in biology and new approaches to treatment. N
Engl J Med. 349:1451–1464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ladanyi M and Cavalchire G: Molecular
variant of the NPM-ALK rearrangement of Ki-1 lymphoma involving a
cryptic ALK splice site. Genes Chromosomes Cancer. 15:173–177.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jhiang SM: The RET proto-oncogene in human
cancers. Oncogene. 19:5590–5597. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhao X, Ghaffari S, Lodish H, Malashkevich
VN and Kim PS: Structure of the Bcr-Abl oncoprotein oligomerization
domain. Nat Struct Biol. 9:117–120. 2002.PubMed/NCBI
|
|
83
|
Alberti L, Carniti C, Miranda C, Roccato E
and Pierotti MA: RET and NTRK1 proto-oncogenes in human diseases. J
Cell Physiol. 195:168–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mizuki M, Ueda S, Matsumura I, Ishiko J,
Schwäble J, Serve H and Kanakura Y: Oncogenic receptor tyrosine
kinase in leukemia. Cell Mol Biol. 49:907–922. 2003.PubMed/NCBI
|
|
85
|
Mano H: Non-solid oncogenes in solid
tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci.
99:2349–2355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Nagar B, Hantschel O, Seeliger M, Davies
JM, Weis WI, Superti-Furga G and Kuriyan J: Organization of the
SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine
kinase. Mol Cell. 21:787–798. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Chen S, Dumitrescu TP, Smithgall TE and
Engen JR: Abl N-terminal cap stabilization of SH3 domain dynamics.
Biochemistry. 47:5795–5803. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Mian AA, Oancea C, Zhao Z, Ottmann OG and
Ruthardt M: Oligomerization inhibition, combined with allosteric
inhibition, abrogates the transformation potential of
T315I-positive BCR/ABL. Leukemia. 23:2242–2247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
He Y, Wertheim JA, Xu L, Miller JP,
Karnell FG and Choi JK: The coiled-coil domain and Tyr177 of bcr
are required to induce a murine chronic myelogenous leukemia-like
disease by bcr/abl. Blood. 99:2957–2968. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Reddy EP and Aggarwal AK: The ins and outs
of bcr-abl inhibition. Genes Cancer. 3:447–454. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tong Q, Xing S and Jhiang SM: Leucine
zipper-mediated dimerization is essential for the PTC1
oncogenic activity. J Biol Chem. 272:9043–9047. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Pillai RN and Ramalingam SS: The biology
and clinical features of non-small cell lung cancers with EML4-ALK
translocation. Curr Oncol Rep. 14:105–110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Shaw AT, Yeap BY, Solomon BJ, et al:
Effect of crizotinib on overall survival in patients with advanced
non-small-cell lung cancer harbouring ALK gene rearrangement: a
retrospective analysis. Lancet Oncol. 12:1004–1012. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Willis TG and Dyer MJ: The role of
immunoglobulin translocations in the pathogenesis of B-cell
malignancies. Blood. 96:808–822. 2000.PubMed/NCBI
|
|
95
|
Dadi S, Le Noir S, Asnafi V, Beldjord K
and Macintyre EA: Normal and pathological V(D)J recombination:
contribution to the understanding of human lymphoid malignancies.
Adv Exp Med Biol. 650:180–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Martinez-Climent JA, Fontan L, Gascoyne
RD, Siebert R and Prosper F: Lymphoma stem cells: enough evidence
to support their existence? Haematologica. 95:293–302. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Graux C, Cools J, Michaux L, Vandenberghe
P and Hagemeijer A: Cytogenetics and molecular genetics of T-cell
acute lymphoblastic leukemia: from thymocyte to lymphoblast.
Leukemia. 20:1496–1510. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Van Vlierberghe P, van Grotel M, Beverloo
HB, et al: The cryptic chromosomal deletion del(11)(p12p13) as a
new activation mechanism of LMO2 in pediatric T-cell acute
lymphoblastic leukemia. Blood. 108:3520–3529. 2006.PubMed/NCBI
|
|
99
|
Brake RL, Kees UR and Watt PM: Multiple
negative elements contribute to repression of the HOX11
proto-oncogene. Oncogene. 17:1787–1795. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Riz I, Hawley TS, Johnston H and Hawley
RG: Role of TLX1 in T-cell acute lymphoblastic leukaemia
pathogenesis. Br J Haematol. 145:140–143. 2009.
|
|
101
|
Kees UR, Heerema NA, Kumar R, et al:
Expression of HOX11 in childhood T-lineage acute
lymphoblastic leukaemia can occur in the absence of cytogenetic
aberration at 10q24: a study from the Children’s Cancer Group
(CCG). Leukemia. 17:887–893. 2003.
|
|
102
|
Dadi S, Le Noir S, Payet-Bornet D, et al:
TLX homeodomain oncogenes mediate T cell maturation arrest in T-ALL
via interaction with ETS1 and suppression of TCRα gene expression.
Cancer Cell. 21:563–576. 2012.PubMed/NCBI
|
|
103
|
De Keersmaecker K, Marynen P and Cools J:
Genetic insights in the pathogenesis of T-cell acute lymphoblastic
leukemia. Haematologica. 90:1116–1127. 2005.
|
|
104
|
Grabher C, von Boehmer H and Look AT:
Notch 1 activation in the molecular pathogenesis of T-cell acute
lymphoblastic leukaemia. Nat Rev Cancer. 6:347–359. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
South AP, Cho RJ and Aster JC: The
double-edged sword of Notch signaling in cancer. Semin Cell Dev
Biol. 23:458–464. 2012. View Article : Google Scholar : PubMed/NCBI
|