Introduction
Hepatocellular carcinoma (HCC) is one of the most
common human malignancies and the leading cause of cancer-related
mortality worldwide (1,2). The development and progression of HCC
involve the deregulation of genes that are crucial to cell cycle
control, cell growth, apoptosis and cell migration. In the past few
decades, studies have focused on investigating the protein-coding
genes underlying the development and progression of HCC. Recently,
reports suggest that misexpression of microRNAs (miRNAs) also
contributes to tumorigenesis (3,4).
miRNAs are small, endogenous, non-coding RNAs known
to negatively regulate the expression of protein-coding genes
(5,6). miRNAs are involved in the regulation
of various biological processes, such as development, cell
proliferation, apoptosis and differentiation (7–9).
Deregulation of miRNAs has been observed in various types of human
cancer (10–13). Accumulating evidence indicates that
miRNAs may function as oncogenes or tumor suppressor genes (TSGs)
during tumor development and progression (4). Using an miRNA microarray, Murakami
et al(14) determined that
microRNA-200a (miR-200a) was significantly downregulated in HCC
tissues when compared with the level in adjacent non-tumor tissues.
However, the biological function of miR-200a in HCC and the
detailed mechanisms in tumorigenesis are still largely unknown.
In the present study, we confirmed that
downregulation of miR-200a occurred frequently in HCC tissues and
cell lines. Additionally, miR-200a may be an independent prognostic
factor. Moreover, ectopic expression of miR-200a dramatically
inhibited cell proliferation and clonogenicity in vitro in
HCC cells. Furthermore, gain-of-function studies revealed that
miR-200a induced G1 phase arrest of HCC cells. Further study
identified CDK6 as a novel functional target of miR-200a.
Collectively, these findings indicate that miR-200a functions as a
potential tumor suppressor in HCC, providing a potential target for
HCC therapy.
Materials and methods
Cell lines and tissue specimens
Immortalized normal liver cells (L02), human HCC
cell lines (SK-HEP-1, HepG2, MHCC97H, MHCC97L, PLC, HuH7, SMMC-7721
and Bel-7402) and human embryonic kidney cells (HEK293T) were
maintained in the recommended culture condition and incubated at
37ºC in a humidified environment containing 5% CO2.
Paired HCC and adjacent non-tumor liver tissues were collected from
patients undergoing liver transplantation or partial hepatectomy at
The First Affiliated Hospital, School of Medicine, Zhejiang
University (Hangzhou, Zhejiang, China). Written informed consent
was obtained from each patient. A total of 120 patients had a clear
histologic diagnosis of HCC with complete clinicopathological data,
and all patients were followed up for survival analysis. None of
the patients received radiotherapy or chemotherapy before surgery.
All sample data were obtained from clinical and pathologic records
and are summarized in Table I.
 | Table IAssociation of miR-200a expression
with clinicopathological parameters of the HCC cases. |
Table I
Association of miR-200a expression
with clinicopathological parameters of the HCC cases.
| Factors | Low expression group,
n (%)
(n=64) | High expression
group, n (%)
(n=56) | P-valuea |
|---|
| Age (years) | | | 0.824 |
| ≤50 | 34 (53.1) | 30 (53.6) | |
| >50 | 30 (46.9) | 26 (46.4) | |
| Gender | | | 0.578 |
| Male | 49 (76.6) | 40 (71.4) | |
| Female | 15 (23.4) | 16 (28.6) | |
| HBV | | | 0.653 |
| Positive | 52 (81.3) | 45 (80.4) | |
| Negative | 12 (18.7) | 11 (19.6) | |
| Cirrhosis | | | 0.579 |
| Positive | 51 (79.7) | 42 (75.0) | |
| Negative | 13 (20.3) | 14 (25.0) | |
| Tumor size
(cm) | | | 0.861 |
| ≤5 | 42 (65.6) | 37 (66.1) | |
| >5 | 22 (34.4) | 19 (33.9) | |
| Tumor number | | | 0.924 |
| Single | 41 (64.1) | 36 (64.3) | |
| Multiple | 23 (35.9) | 21 (35.7) | |
| Histologic
grade | | | 0.015 |
| Well and
moderate | 17 (26.6) | 26 (50.0) | |
| Poor and
others | 47 (73.4) | 26 (50.0) | |
| TNM stage | | | 0.012 |
| I | 18 (28.1) | 34 (60.7) | |
| II and III | 46 (71.9) | 22 (39.3) | |
| AFP (ng/ml) | | | 0.025 |
| ≤400 | 26 (40.6) | 33 (58.9) | |
| >400 | 38 (59.4) | 23 (41.1) | |
| PVTT | | | 0.023 |
| Positive | 24 (37.5) | 13 (23.2) | |
| Negative | 40 (62.5) | 43 (76.8) | |
Oligoribonucleotides
miR-200a mimic and the control RNA duplex (referred
to as NC) were used for the transient gain-of-function study. The
small interfering RNA (siRNA) targeting the human CDK6 transcript
was designated as siCDK6. The NC for both the miRNA mimic and siRNA
was non-homologous to any human genome sequence. All the RNA
oligoribonucleotides (Table II)
were purchased from Genepharma (Shanghai, China). All
oligoribonucleotides used in the present study are shown in
Table II.
| Table IIList of the oligonucleotides used in
the present study. |
Table II
List of the oligonucleotides used in
the present study.
| Name | Sequence
(5′→3′)a |
|---|
| miRNA and siRNA
duplexes |
| miR-200a mimic
(sense) |
UAACACUGUCUGGUAACGAUGU |
| NC (sense) |
ACUACUGAGUGACAGUAGA[dT][dT] |
| siCDK6
(sense) |
CGGACAGTAUUAGAUACAUTT |
| Primers for
RT-PCR |
| miR-200a, F |
CTCTAACACTGTCTGGTAACGATGT |
| CDK6, F |
CAGCAGCGGACAAATAAA |
| CDK6, R |
CTGGGAGTCCAATCACGT |
| GAPDH, F |
AAGGTGAAGGTCGGAGTCA |
| GAPDH, R |
GGAAGATGGTGATGGGATTT |
| Primers for 3′UTR
cloning |
| CDK6-utr, F |
CGGAGCTCTTTCTAACCTTGAATGCTGC |
| CDK6-utr, R |
CGCGTCGACAGAAAAGAAATGCTGAGGAC |
| CDK6-mut, F |
GTGTGTGTGTGTGTGTATGTGAGAGATTCTGTGATCTTTTAA
TCACAAACTTTTTGTAAACGACAAGAATAATTCAATTTTAAA |
| CDK6-mut, R |
TTTAAAATTGAATTATTCTTGTCGTTTACAAAAAGTTTGTGATT
AAAAGATCACAGAATCTCTCACATACACACACACACACAC |
RNA extraction and quantitative reverse
transcriptase-polymerase chain reaction (qRT-PCR)
Total RNA from cell lines and clinical samples was
isolated using the mirVana miRNA Isolation kit (Ambion). qRT-PCR
was performed to detect the expression of miR-200a in various cell
lines and clinical samples and the expression of CDK6 at the mRNA
level. RNA was reverse transcribed using One Step PrimeScript miRNA
cDNA Synthesis kit (Takara, Japan). The cDNA was then quantified by
real-time RT-PCR using SYBR Premix Ex Taq (Takara). All PCR
reactions were performed using the ABI7500 system (Applied
Biosystems, Foster City, CA, USA). U6 or glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used as an internal control. All primers
used are listed in Table II.
Western blotting
Western blotting was performed to detect the gene
expression at the protein level. Protein was extracted from
transfected SK-HEP-1 cells using modified RIPA buffer in the
presence of proteinase inhibitor cocktail. Equivalent quantities
(30–50 μg) of protein were separated on 10% SDS-polyacrylamide gels
and transferred to polyvinylidene difluoride membranes. Membranes
were blocked with 5% non-fat milk and then incubated overnight at
4ºC with the appropriate primary antibody at the dilutions
specified by the manufacturer. The membranes were then washed three
times in 10 ml TBST and incubated with the corresponding
horseradish peroxidase (HRP)-conjugated secondary antibody at a
dilution of 1:2,000 for 1 h. The bound secondary antibody was
detected using an Enhanced chemiluminescence (ECL) system (Pierce
Biotechnology, Inc., Rockford, IL, USA). Primary antibodies were as
follows: anti-CDK6, anti-cyclin E2, anti-Rb,
anti-phospho-Ser780-Rb, anti-cdc2, (Cell Signaling Technology),
anti-phospho-Ser795-Rb and anti-β-actin (Epitomics).
Cell transfection
All transfections were performed using
Lipofectamine® 2000 (Invitrogen) according to the
manufacturer’s instructions. Briefly, SK-HEP-1, Huh7 or HEK293T
cells were transfected with DNA, miRNA mimic, siRNA or NC. All RNA
transfections were performed at a final concentration of 50 nM.
Cells were collected for assay 48 h after transfection.
Cell proliferation assay
SK-HEP-1 and Huh7 cells seeded at a density of
4,000/well into 96-well plates were transfected with miR-200a mimic
or NC. After incubating the cells for the specified time (1, 2, 3
or 4 days), a cell proliferation assay was performed using Cell
Counting Kit-8 (CCK-8) (Dojindo) according to the manufacturer’s
instructions. The solution absorbance was measured
spectrophotometrically at 450 nm with the MRX II absorbance reader
(Dynex Technologies, Chantilly, VA, USA). The experiments were
performed in triplicate.
Colony formation assay
Aliquots of viable transfected SK-HEP-1 and Huh7
cells (1,000/well) were placed in 6-well plates 24 h after
transfection and maintained in complete medium for 2 weeks. Colony
formation was evaluated by staining the cells with 0.1% crystal
violet. The rate of colony formation was calculated using the
following equation: Colony formation rate = (number of
colonies/number of seeded cells) × 100%. The experiments were
performed in triplicate.
Cell cycle analysis
Forty-eight hours after transfection, SK-HEP-1 and
Huh7 cells were harvested, washed with PBS and fixed in 70% ethanol
at 4ºC overnight. Staining for DNA content was performed using a
DNA Prep Stain (Beckman Coulter, Fullerton, CA, USA). Cell
populations in the G0/G1, S and G2/M phases were determined using
the BD™ LSRII flow cytometry system with FACSDiva software (both
from BD Bioscience, Franklin Lakes, USA). Data were analyzed using
ModFit LT software. The experiments were performed in
triplicate.
Target prediction
To predict the target genes and the 3′ UTR binding
sites bound by the seed region of miR-200a, the TargetScan
(15) (http://www.targetscan.org/) and PicTar (16) (http://pictar.mdc-berlin.de/) databases were used.
CDK6 was chosen as the target candidate according to both
TargetScan and PicTar databases.
Vector construction and luciferase
reporter assay
The 3′ UTR of CDK6 was amplified by PCR. The
amplified sequence was subcloned and ligated into the pmirGLO
Dual-Luciferase miRNA Target Expression Vector (Promega, Madison,
WI, USA). The recombinant reporter vector was identified by
sequencing and termed the wild-type (Wt). To create the miR-200a
binding site mutants, the binding region of the seed sequence
(5′-AGTGTT-3′) was mutated to the sequence 5′-TCACAA-3′, using the
QuikChange Lightning Site-Directed Mutagenesis kit (Stratagene)
according to the manufacturer’s protocol. The recombinant vector
was confirmed by sequencing and termed the mutant type (Mut). 293T
cells plated in a 24-well plate were co-transfected with 50 nM of
either the miR-200a mimic or NC and 100 ng of pmirGLO vector
comprising Wt or Mut 3′ UTR of CDK6 using Lipofectamine®
2000. Forty-eight hours after transfection, the relative luciferase
activity was measured by the Dual-Luciferase Reporter Assay System
(Promega). All transfection experiments were performed in
triplicate.
Statistical analysis
Data are presented as the means ± SD. Comparisons
were made by using the Student’s t-test, the χ2 test and
the log-rank test. Overall survival rates were calculated
actuarially according to the Kaplan-Meier method. Variables with a
value of P<0.05 in univariate analysis were used in a subsequent
multivariate analysis based on the Cox proportional hazards model.
A value of P<0.05 was considered to indicate a statistically
significant result.
Results
miR-200a is frequently downregulated in
HCC cell lines and tissues
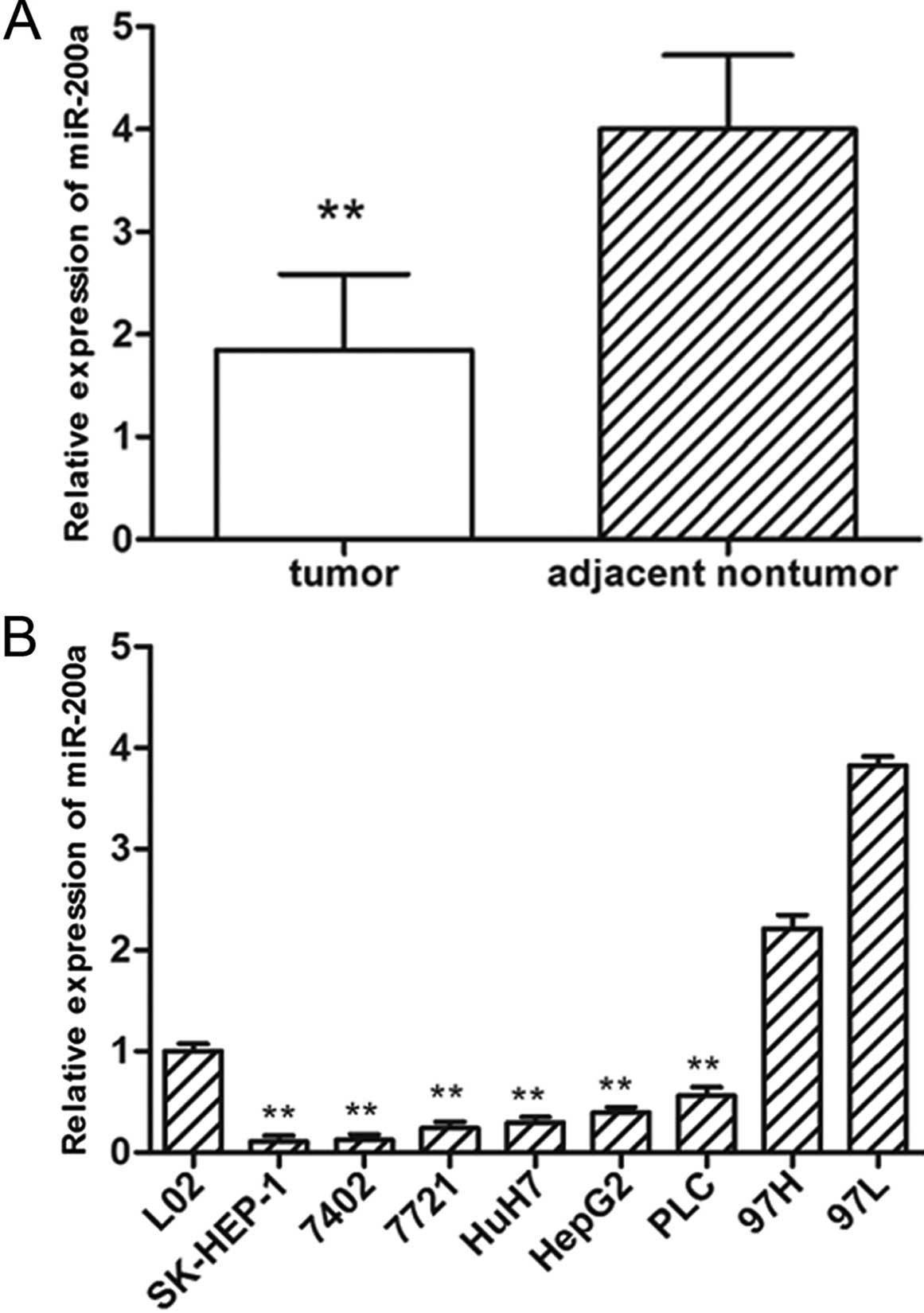
To further confirm the expression pattern of
miR-200a in HCC, we detected the expression of miR-200a in 120
paired HCC and adjacent non-cancerous liver tissues by real-time
qRT-PCR. Compared with the adjacent non-tumor tissues, significant
downregulation of miR-200a was observed in the HCC tissues
(Fig. 1A). Furthermore, the
expression of miR-200a was noticeably reduced in 6 of 8 (75.0%) HCC
cell lines examined when compared with the expression in the normal
liver cell line (L02) (Fig. 1B).
These results suggest that reduced miR-200a expression is a
frequent event in human HCC and may be involved in
tumorigenesis.
Downregulation of miR-200a is associated
with worse prognosis
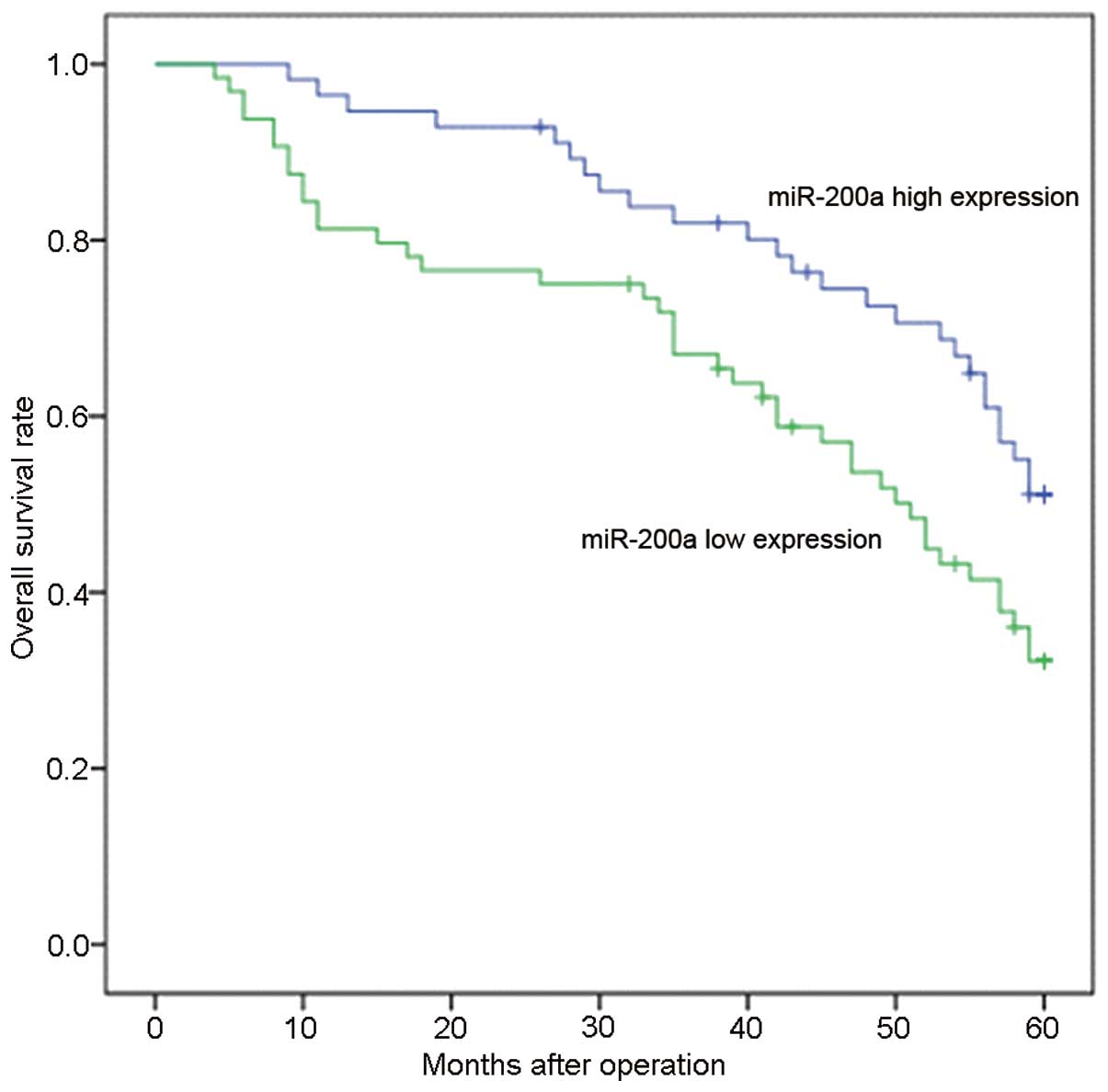
In the present study, patients with expression
values less than the average level of miR-200a in HCC tissues
(1.87, normalized to U6) were assigned to the low expression group
(n=64) whereas patients with values above the average were assigned
to the high expression group (n=56). Our data showed a significant
association between miR-200a expression and the AFP level,
histologic grade, TNM stage and PVTT (Table I). Furthermore, the Kaplan-Meier
method revealed that a lower miR-200a level was associated with
shorter overall survival (P<0.01) (Fig. 2). Multivariate analysis further
confirmed that a reduced miR-200a level was an independent and
significant prognostic factor for survival (HR, 2.621; CI,
1.116–4.642; P=0.004; Table III).
Collectively, these data suggest that deregulation of miR-200a may
contribute to the development and progression of HCC.
| Table IIIUnivariate and multivariate analysis
for overall survival of the HCC patients. |
Table III
Univariate and multivariate analysis
for overall survival of the HCC patients.
| Clinical
variables | HR (95% CI) | P-value |
|---|
| Univariate
analysis |
| Age | 1.021
(0.726–1.346) | 0.786 |
| Gender | 0.953
(0.568–1.775) | 0.653 |
| HBV | 1.157
(0.563–2.195) | 0.874 |
| Cirrhosis | 1.652
(0.838–3.688) | 0.257 |
| Tumor size | 1.568
(1.056–2.446) | 0.077 |
| Tumor no. | 2.451
(1.547–4.639) | 0.276 |
| TNM stage | 2.774
(1.674–4.797) | 0.012 |
| Histologic
grade | 1.826
(1.113–2.893) | 0.024 |
| PVTT | 3.639
(2.538–7.531) | 0.016 |
| AFP | 2.635
(1.368–5.062) | 0.017 |
| miR-200a
expression | 2.845
(1.876–4.874) | 0.002 |
| Multivariate
analysis |
| TNM stage | 2.053
(1.586–4.753) | 0.012 |
| Histologic
grade | 1.431
(0.690–2.968) | 0.336 |
| PVTT | 0.649
(0.642–1.276) | 0.172 |
| AFP | 1.153
(0.645–2.297) | 0.664 |
| miR-200a
expression | 2.621
(1.116–4.642) | 0.004 |
Ectopic expression of miR-200a
significantly inhibits cell proliferation
The frequent downregulation of miR-200a in HCC cell
lines and tissues suggests that miR-200a may serve as a potential
TSG in HCC. To test the potential tumor-suppressor role of miR-200a
in HCC, the effect of ectopic miR-200a on cell growth was evaluated
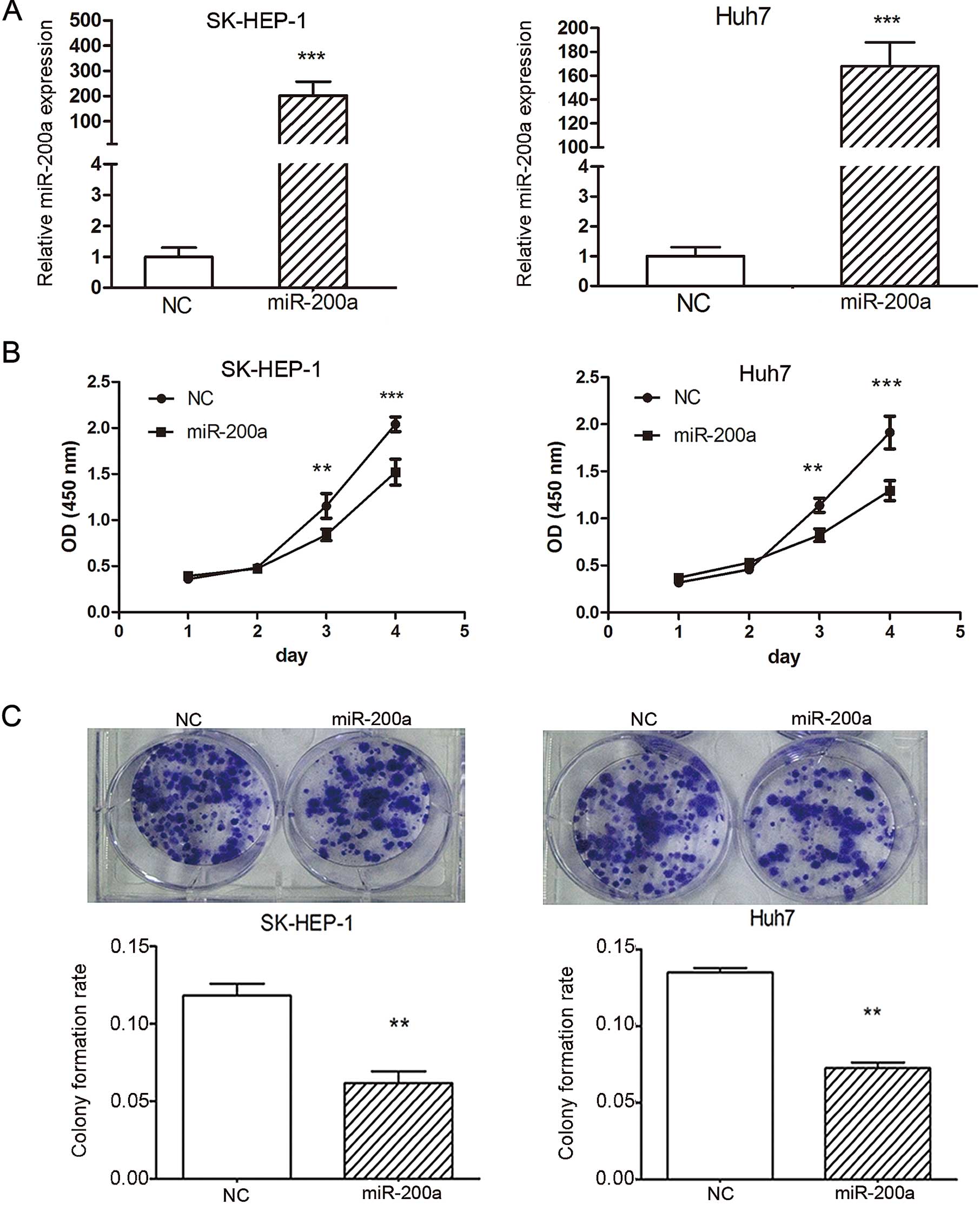
using the CCK-8 cell viability assay. The expression of miR-200a
was markedly increased in the SK-HEP-1 and Huh7 cells transfected
with 50 nM miR-200a mimic when compared with NC, normalized to U6
(Fig. 3A). Our data revealed that
miR-200a overexpression significantly inhibited cell proliferation
in SK-HEP-1 and Huh7 cells compared to NC. At day 1, the OD values
of the miR-200a mimic and NC control cells were not significantly
different. However, from day 3 onwards, the OD values of the
miR-200a-treated SK-HEP-1 and Huh7 cells were significantly lower
than the value of the control cells (Fig. 3B). These data indicated that
miR-200a inhibited cell proliferation in vitro.
miR-200a suppresses clonogenicity in
vitro
To further investigate the potential role of
miR-200a in tumorigenesis, a colony formation assay was performed
in SK-HEP-1 and Huh7 cells. Cells were transfected with the
miR-200a mimic or NC duplex, and then allowed to grow to a very low
density. Notably, SK-HEP-1 and Huh7 cells transfected with 50 nM
miR-200a mimic displayed much fewer and smaller colonies compared
with the NC duplex-transfected cells (Fig. 3C), suggesting a growth-inhibitory
role of miR-200a.
Ectopic expression of miR-200a induces
cell cycle arrest
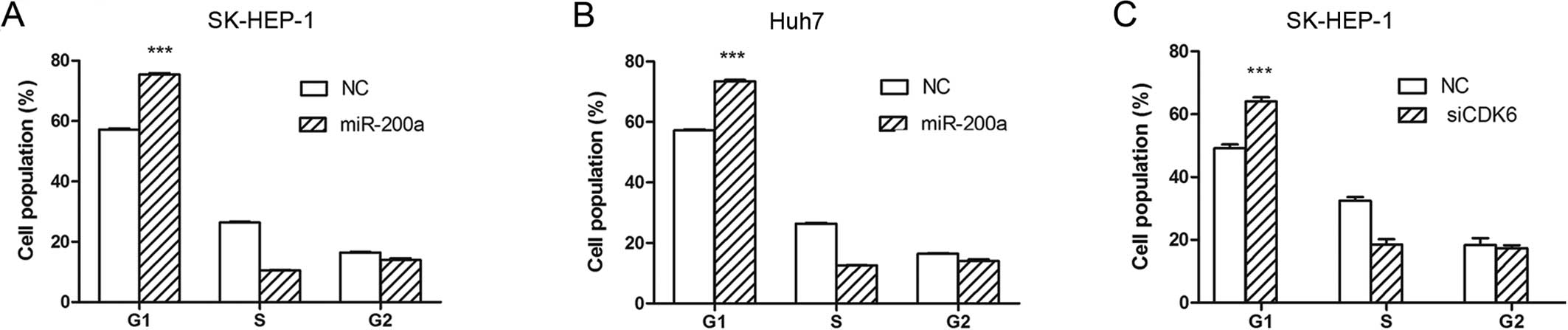
To explore the mechanisms underlying the suppression
of tumor growth by miR-200a, the effect of miR-200a on cell cycle
progression was investigated by flow cytometry. The assay showed
that overexpression of miR-200a resulted in an accumulation of a G1
population in the SK-HEP-1 (Fig.
4A) and Huh7 cells (Fig. 4B).
These results indicated that miR-200a inhibited HCC cell
proliferation by inducing cell cycle arrest at the G1 phase.
CDK6 is a direct functional target of
miR-200a
It is generally believed that miRNAs exert their
function through negatively regulating the expression of their
downstream target genes. To further elucidate the molecular
mechanism by which miR-200a inhibits G1/S transition, CDK6 was
predicted as a potential target of miR-200a by TargetScan and
PicTar. A single putative miR-200a-binding site was mapped in the
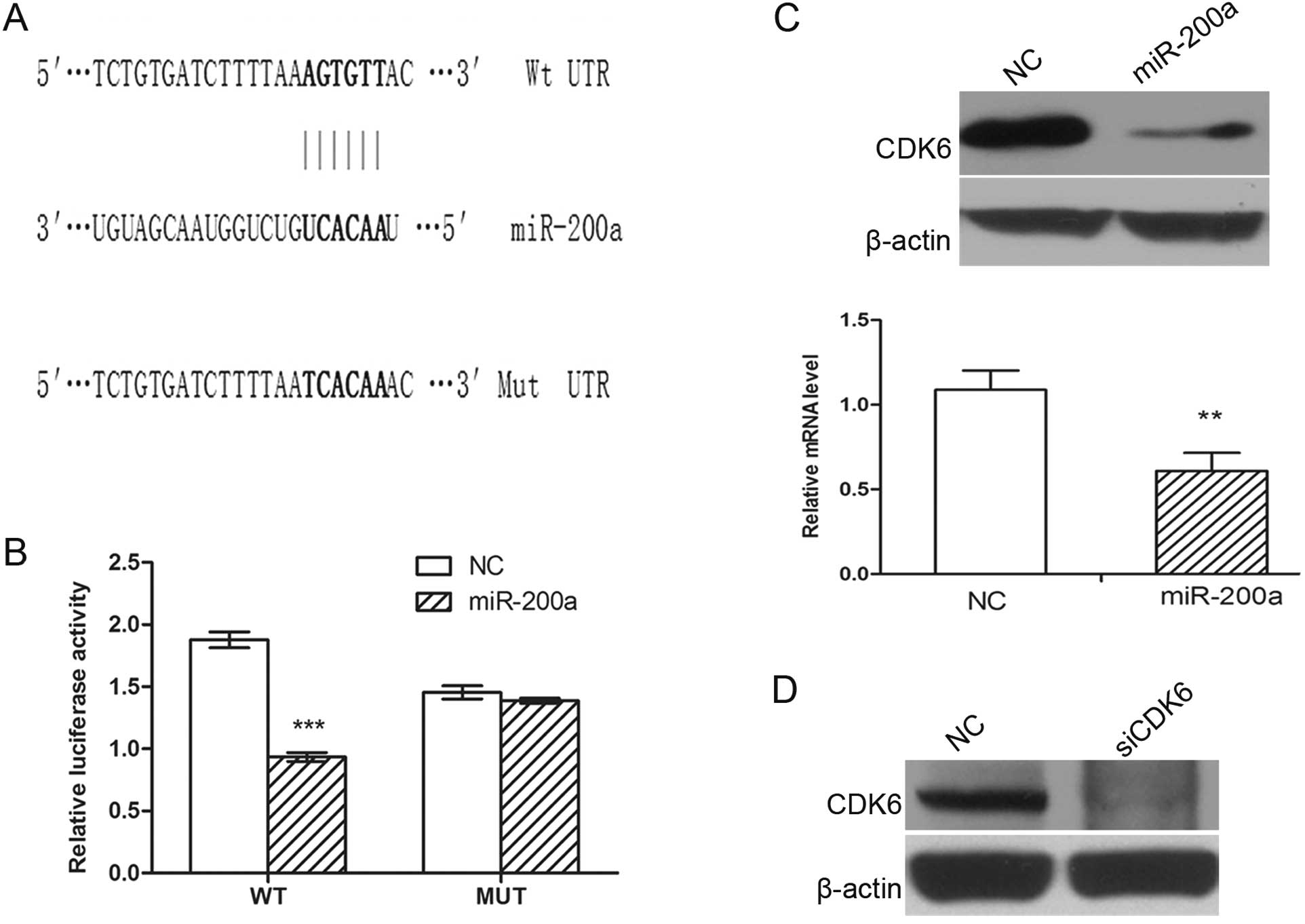
3′ UTR of CDK6 (Fig. 5A). To
validate whether CDK6 is a direct target of miR-200a, a human CDK6
3′ UTR fragment containing Wt or the mutant (Mut) miR-200a binding
sequence (Fig. 5A) was cloned
downstream of the firefly luciferase reporter gene in pmirGLO. In
HEK293 cells cotransfected with the reporter plasmids and the
miR-200a mimic or NC duplex, the luciferase activity of the
reporter that contained Wt 3′ UTR was significantly suppressed by
miR-200a mimic, but the luciferase activity of the mutant reporter
was unaffected (Fig. 5B),
indicating that miR-200a may suppress gene expression through
miR-200a binding sequence at the 3′ UTR of CDK6. Additionally,
transfection of the miR-200a mimic decreased CDK6 expression in
SK-HEP-1 cells at both the protein and mRNA levels (Fig. 5C), suggesting that CDK6 expression
is inhibited by miR-200a at the post-transcriptional level.
Collectively, the results showed that miR-200a regulated the
expression of endogenous CDK6 by directly targeting the 3′ UTR of
CDK6 mRNA and that CDK6 is a novel target of miR-200a.
miR-200a is involved in G1/S transition
through the downregulation of CDK6
To explore the role of CDK6 in the
miR-200a-regulated G1/S transition, we investigated whether
knockdown of CDK6 may phenocopy the effect of miR-200a
overexpression. SK-HEP-1 cells were transfected with siRNA duplex
targeting CDK6, which resulted in a significant reduction in
the protein levels (Fig. 5D). The
silencing of CDK6 led to G1-phase arrest (Fig. 4C), phenocopying the outcome of
miR-200a overexpression.
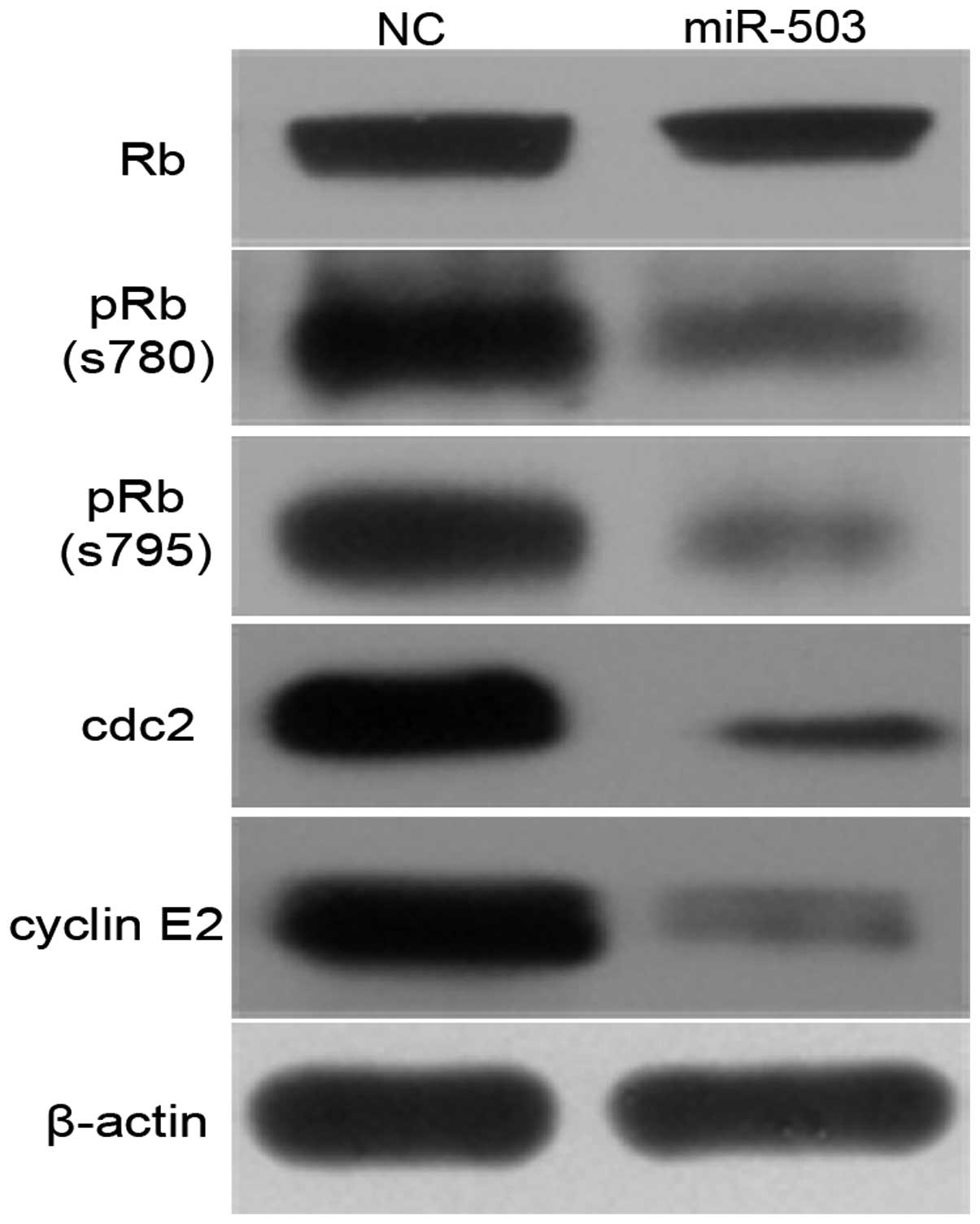
CDK6 is one of the crucial molecules that initiate
the phosphorylation of retinoblastoma (Rb), which results in the
release of E2F and subsequently the transactivation of genes
required for S-phase entry. Therefore, we investigated whether
miR-200a could attenuate these events. Ectopic expression of
miR-200a caused a reduction in phosphorylated Rb protein, however
little effect on the total Rb protein was noted (Fig. 6). Moreover, the effect of miR-200a
on endogenous genes such as cdc2 and cyclin E2, downstream genes of
E2F3, was further investigated. Overexpression of miR-200a induced
significant downregulation of cdc2 and cyclin E2 in SK-HEP-1 cells
(Fig. 6).
These data indicate that miR-200a may induce cell
cycle arrest at G1 phase by directly inhibiting CDK6 through the
Rb-E2F signaling pathway.
Discussion
Although deregulation of miRNAs has been observed in
various types of cancers (11,17),
the molecular mechanisms by which miRNAs regulate the process of
carcinogenesis and the biological behavior of cancer cells are
still largely unknown. Growing evidence has revealed that a defect
in cell cycle control is an essential step during carcinogenesis
(18,19). miR-138 was found to induce cell
cycle arrest by targeting cyclin D3 in HCC (20). miR-195 suppressed G1/S transition of
human HCC cells by targeting cyclin D1, CDK6 and E2F3 (21). Therefore, misexpression of cell
cycle-related miRNAs may facilitate tumorigenesis. A previous miRNA
microarray revealed that miR-200a was significantly downregulated
in HCC (14). Recently altered
expression of miR-200a has been observed in different types of
diseases, including cancers. Downregulation of the miR-200 family
expression was recently shown to be associated with tumor
progression (22).
Epithelial-to-mesenchymal transition (EMT), a crucial process in
the transformation of tumor cells into aggressive metastatic cancer
cells, also seems to be tightly regulated by this family (23,24).
Saydam et al(25) reported
that downregulation of miR-200a in brain tumors promoted tumor
growth by upregulating cyclin D1 and β-catenin in vitro and
in vivo. Similarly, it was reported that the overexpression
of miR-200a inhibited nasopharyngeal carcinoma cell growth,
migration and invasion by downregulation of β-catenin and Smad
interacting protein 1 (26).
Little is known concerning the role of the miR-200
family in cell growth and proliferation in HCC. Hung et
al(27) found that members of
the miR-200 family (miR-200a and miR-200b) were involved in HCC
migration by regulating E-cadherin expression. However, the role of
miR-200 family members and the relative molecular mechanism in HCC
remain largely unclear. In the present study, we attempted to
elucidate the role of miR-200a in HCC. Our findings revealed that
miR-200a was frequently downregulated in human HCC tissues. Lee
et al(28) previously
reported that the miR-200 family, including miR-200a, was
upregulated in endometrial endometrioid carcinomas when compared
with expression in normal endometrial tissues. This discrepancy may
be due to the use of different cell lines. In the present study,
multivariate analysis revealed that miR-200a is an independent
prognostic factor for survival. In addition, gain-of-function
studies indicated that overexpression of miR-200a suppressed cell
proliferation by induction of cell cycle arrest in HCC cell lines.
Moreover, we identified CDK6 as a direct target of miR-200a in HCC,
which may provide new insights into the mechanisms underlying
tumorigenesis. These results imply the important role of miR-200a
in the tumorigenesis of HCC.
The family of CDKs and their activating partners
(cyclins) are crucial molecules involved in cell cycle control. The
G1/S transition is regulated primarily by D-type cyclins in complex
with CDK4/CDK6 and E-type cyclins in complex with CDK2. These
complexes cooperate in phosphorylating and preventing Rb binding to
E2F, thus activating E2F-mediated transcription and driving cells
from G1 into S phase (29).
However, the detailed mechanisms of cell cycle regulation in HCC
require further investigation. Our results showed that miR-200a
induced cell cycle arrest of HCC cells by downregulation of CDK6 at
both the mRNA and protein levels. Further investigation revealed
that ectopic miR-200a expression significantly inhibited Rb
phosphorylation and the expression level of cdc2 and cyclin E2 at
the protein level (Fig. 6).
Collectively our results indicate that miR-200a may be involved in
the induction of cell cycle arrest in HCC through the CDK/Rb/E2F
pathway.
The underlying mechanism responsible for the
decreased expression of miR-200a in HCC remains unknown. miR-200a
is located on chromosome 1p36, a region which is often deleted in
several types of cancer (30,31),
providing a plausible mechanism for the reduced miR-200a expression
in HCC. In addition, promoter hypermethylation or transcriptional
regulation may account at least in part for the reduced miR-200a
expression in HCC. Further investigation is required to verify this
hypothesis.
In summary, we confirmed the frequent downregulation
of miR-200a in HCC and revealed the potential role of miR-200a in
tumorigenesis. Our data indicate that miR-200a may function as a
potential tumor suppressor and could be an independent prognostic
marker in HCC patients. Therefore, miR-200a may serve as a useful
therapeutic target for HCC therapy.
Acknowledgements
We thank Jilei Fu for the collection of clinical
data and Rong Su for her excellent technical assistance. The
present study was supported by the following grants and
foundations: National S&T Major Project (2012zx10002-017); NSFC
for Innovative Research Group (81121002); the National Natural
Science Foundation of China (81201944); Zhejiang Provincial Natural
Science Foundation of China for Distinguished Young Scholars (no.
R2110125).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
2
|
Aravalli RN, Steer CJ and Cressman EN:
Molecular mechanisms of hepatocellular carcinoma. Hepatology.
48:2047–2063. 2008. View Article : Google Scholar
|
|
3
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar
|
|
5
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schratt GM, Tuebing F, Nigh EA, et al: A
brain-specific microRNA regulates dendritic spine development.
Nature. 439:283–289. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brennecke J, Hipfner DR, Stark A, Russell
RB and Cohen SM: bantam encodes a developmentally regulated
microRNA that controls cell proliferation and regulates the
proapoptotic gene hid in Drosophila. Cell. 113:25–36. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Su H, Yang JR, Xu T, et al: MicroRNA-101,
down-regulated in hepatocellular carcinoma, promotes apoptosis and
suppresses tumorigenicity. Cancer Res. 69:1135–1142. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yanaihara N, Caplen N, Bowman E, et al:
Unique microRNA molecular profiles in lung cancer diagnosis and
prognosis. Cancer Cell. 9:189–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iorio MV, Visone R, Di Leva G, et al:
MicroRNA signatures in human ovarian cancer. Cancer Res.
67:8699–8707. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Murakami Y, Yasuda T, Saigo K, et al:
Comprehensive analysis of microRNA expression patterns in
hepatocellular carcinoma and non-tumorous tissues. Oncogene.
25:2537–2545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krek A, Grün D, Poy MN, et al:
Combinatorial microRNA target predictions. Nat Genet. 37:495–500.
2005. View
Article : Google Scholar
|
|
17
|
Volinia S, Calin GA, Liu CG, et al: A
microRNA expression signature of human solid tumors defines cancer
gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004.
|
|
19
|
Deshpande A, Sicinski P and Hinds PW:
Cyclins and cdks in development and cancer: a perspective.
Oncogene. 24:2909–2915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang W, Zhao LJ, Tan YX, Ren H and Qi ZT:
MiR-138 induces cell cycle arrest by targeting cyclin D3 in
hepatocellular carcinoma. Carcinogenesis. 33:1113–1120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP and
Zhuang SM: MicroRNA-195 suppresses tumorigenicity and regulates
G1/S transition of human hepatocellular carcinoma cells.
Hepatology. 50:113–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baffa R, Fassan M, Volinia S, et al:
MicroRNA expression profiling of human metastatic cancers
identifies cancer gene targets. J Pathol. 219:214–221. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ahn SM, Cha JY, Kim J, et al: Smad3
regulates E-cadherin via miRNA-200 pathway. Oncogene. 31:3051–3059.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saydam O, Shen Y, Würdinger T, et al:
Downregulated microRNA-200a in meningiomas promotes tumor growth by
reducing E-cadherin and activating the Wnt/beta-catenin signaling
pathway. Mol Cell Biol. 29:5923–5940. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xia H, Ng SS, Jiang S, et al:
miR-200a-mediated downregulation of ZEB2 and CTNNB1 differentially
inhibits nasopharyngeal carcinoma cell growth, migration and
invasion. Biochem Biophys Res Commun. 391:535–541. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hung CS, Liu HH, Liu JJ, et al:
MicroRNA-200a and -200b mediated hepatocellular carcinoma cell
migration through the epithelial to mesenchymal transition markers.
Ann Surg Oncol. Aug 7–2012.(Epub ahead of print).
|
|
28
|
Lee JW, Park YA, Choi JJ, et al: The
expression of the miRNA-200 family in endometrial endometrioid
carcinoma. Gynecol Oncol. 120:56–62. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Grillo M, Bott MJ, Khandke N, et al:
Validation of cyclin D1/CDK4 as an anticancer drug target in MCF-7
breast cancer cells: effect of regulated overexpression of cyclin
D1 and siRNA-mediated inhibition of endogenous cyclin D1 and CDK4
expression. Breast Cancer Res Treat. 95:185–194. 2006. View Article : Google Scholar
|
|
30
|
Bagchi A and Mills AA: The quest for the
1p36 tumor suppressor. Cancer Res. 68:2551–2556. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sato Y, Kobayashi H, Suto Y, et al:
Chromosomal instability in chromosome band 12p13: multiple breaks
leading to complex rearrangements including cytogenetically
undetectable sub-clones. Leukemia. 15:1193–1202. 2001. View Article : Google Scholar
|