Introduction
Histone deacetylase (HDAC) is a class of enzymes
that removes acetyl groups from lysine amino acid on histone,
leading to the control of transcription (1). Dysregulation of HDAC activity causes
the silence of tumor suppressor genes such as p53 and contributes
to cancer initiation and development (2,3). It
was reported that HDAC activity and expression are increased in
several types of human cancer, including breast and prostate cancer
(4,5). Therefore, HDAC inhibitors can be
considered as novel strategic agents in cancer therapeutics. In
fact, vorinostat and romidepsin have been used for the treatment of
cutaneous T-cell lymphoma (6). HDAC
inhibitors have been known to induce cell cycle arrest, cell
differentiation, apoptotic and autophagic cell death in various
cancer cells (7–9). In addition, it is reported that HDAC
inhibitor and tumor necrosis factor (TNF)-family members
synergistically induce apoptosis in many cancer cells such as
breast, liver and lymphoma cells (10–12).
HDAC inhibitors also generate reactive oxygen species (ROS) in
solid tumor and leukemia cells (13). Excessive production of ROS, known as
oxidative stress, has been recognized to induce cell death.
Cervical cancer is a major cause of mortality in
women worldwide and its occurrence results from both genetic and
epigenetic events. Overexpression of HDAC2 is observed in cervical
cancer cells (14). Furthermore, it
was reported that the acetylated form of histone H3 in cytologic
smears is related to the progression of cervical cancer (15). Originally, valproic acid (VPA) was
clinically used in epilepsy and bipolar disorder. However, it was
recently reported that VPA has an anticancer effect on ovarian and
liver cancer cells in vitro and in vivo(16,17).
However, little is known about the anticancer effect of VPA on
cervical cancer cells in view of changes in ROS and GSH levels.
Therefore, in the present study, we investigated the effects of VPA
on cell growth and death in human cervical HeLa cells in relation
to ROS and GSH levels.
Materials and methods
Cell culture
Human cervix adenocarcinoma HeLa cells were obtained
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
and maintained in a humidified incubator containing 5%
CO2 at 37°C. The HeLa cells were cultured in RPMI-1640
medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10%
fetal bovine serum (FBS; Sigma-Aldrich) and 1%
penicillin-streptomycin (Gibco-BRL, Grand Island, NY, USA). The
cells were routinely grown in 100-mm plastic tissue culture dishes
(Nunc, Roskilde, Denmark) and harvested with a solution of
trypsin-EDTA while in a logarithmic phase of growth.
Reagents
The VPA was purchased from Sigma-Aldrich, and was
dissolved in water at 1 M as a stock solution. The pan-caspase
inhibitor (Z-VAD-FMK;
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone), the caspase-3
inhibitor (Z-DEVD-FMK;
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone), the
caspase-8 inhibitor (Z-IETD-FMK;
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone) and the
caspase-9 inhibitor (Z-LEHD-FMK;
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone) were obtained
from R&D Systems, Inc. (Minneapolis, MN, USA) and were
dissolved in DMSO at 10 mM to serve as stock solutions. TNF-α was
also obtained from R&D Systems and were dissolved in water at
10 μg/ml as a stock solution. NAC and BSO were also obtained from
Sigma-Aldrich, and NAC was dissolved in buffer [20 mM HEPES (pH
7.0)] at 100 mM as a stock solution. BSO was dissolved in water at
100 mM as a stock solution. Cells were pretreated with 15 μM
caspase inhibitors, 2 mM NAC or 100 μM BSO for 1 h prior to VPA
treatment.
Growth inhibition assay
The effect of VPA on cell growth was determined by
measuring 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT; Sigma-Aldrich) absorbance in living cells, as
previously described (18). In
brief, 5×103 cells were seeded in 96-well microtiter
plates (Nunc) for MTT assays. After exposure to the designated
doses of VPA for the indicated times, MTT solution [20 ml: 2 mg/ml
in phosphate-buffered saline (PBS)] was added to each well of the
96-well plates. The plates were additionally incubated for 3 h at
37°C. Medium was withdrawn from the plates by pipetting and 200 ml
DMSO was added to each well to solubilize the formazan crystals.
The optical density was measured at 570 nm using a microplate
reader (Synergy™ 2, BioTek Instruments Inc., Winooski, VT,
USA).
Nuclear/cytosol fractionation
The isolation of nuclear and cytosol extract was
performed with the nuclear/cytosol fractionation kit (BioVision,
San Francisco, CA, USA) according to the manufacturer’s
instructions. In brief, 1×106 cells in a 60-mm culture
dish (Nunc) were incubated with the indicated doses of VPA for 24
h. The cells were then washed in PBS and suspended in 200 μl
cytosol extraction buffer provided with the kit on ice for 10 min.
After 5 min centrifugation, the supernatant (cytosol fraction) was
collected and the pellets were resuspended in the nuclear
extraction buffer provided with the kit. Protein concentrations
were determined using the Bradford method.
Measurement of HDAC activity
HDAC activity was assessed using the HDAC assay kit
(Millipore, Billerica, MA, USA) according to the manufacturer’s
instructions. In brief, 1×106 cells in a 60-mm culture
dish (Nunc) were incubated with the indicated doses of VPA for 24
h. The cells were then washed in PBS and suspended in 5 volumes of
lysis buffer (R&D Systems). Protein concentrations were
determined using the Bradford method. Supernatant samples
containing 30 μg of total, cytosol and nuclear protein were used
for determination of HDAC activity. These samples were added to
each well in 96-well microtiter plates (Nunc) with HDAC substrate
provided by the assay kit at 37°C for 1 h. The optical density of
each well was measured at 405 nm using a microplate reader
(Synergy™ 2; BioTek Instruments).
Western blot analysis
The expression of proteins was evaluated using
western blot analysis, as previously described (19). In brief, 1×106 cells in a
60-mm culture dish (Nunc) were incubated with the designated doses
of VPA for 24 h. The cells were then washed in PBS and suspended in
five volumes of lysis buffer (20 mM HEPES, pH 7.9, 20% glycerol,
200 mM KCl, 0.5 mM EDTA, 0.5% NP40, 0.5 mM DTT, 1% protease
inhibitor cocktail). Supernatant protein concentrations were
determined using the Bradford method. Supernatant samples
containing 30 μg total protein were resolved by 15% SDS-PAGE gels
depending on the sizes of target proteins, transferred to
Immobilon-P PVDF membranes (Millipore) by electroblotting, and then
probed with anti-acetylated H3 (Millipore), anti-PARP, anti-c-PARP,
anti-Bcl-2 (Cell Signaling Technology Inc., Danvers, MA, USA)
anti-β-actin antibodies (Santa Cruz Biotechnology, Santa Cruz, CA,
USA). Membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies. Blots were developed
using an ECL kit (Amersham, Arlington Heights, IL, USA).
Cell cycle and sub-G1 cell analysis
Cell cycle and sub-G1 cell analysis were determined
by propidium iodide (PI, Ex/Em = 488/617 nm; Sigma-Aldrich)
staining, as previously described (20). In brief, 1×106 cells in a
60-mm culture dish (Nunc) were incubated with the designated doses
of VPA with or without 15 μM caspase inhibitors, 2 mM NAC or 100 μM
BSO for 24 h. Cells were washed again with PBS, then incubated with
PI (10 μg/ml) with simultaneous RNase treatment at 37°C for 30 min.
Cellular DNA content was measured using a FACStar flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA) and analyzed by using
Lysis II and CellFit software (Becton-Dickinson).
Annexin V-FITC/PI staining for cell death
detection
Apoptotic cell death was determined by staining
cells with Annexin V-fluorescein isothiocyanate (FITC; Invitrogen
Life Technologies, Camarillo, CA, USA; Ex/Em = 488/519 nm), as
previously described (20). In
brief, 1×106 cells in a 60-mm culture dish (Nunc) were
incubated with the designated doses of VPA with or without 15 μM
caspase inhibitors, 10 ng/ml TNF-α, 2 mM NAC or 100 μM BSO for 24
h. Cells were washed twice with cold PBS and then resuspended in
500 μl of binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5
mM CaCl2) at a concentration of 1×106
cells/ml. Annexin V-FITC (5 μl) and PI (1 μg/ml) were then added
and the cells were analyzed with a FACStar flow cytometer.
Quantification of caspase-3, -8 and -9
activity
The activity of caspase-3, -8 and -9 was assessed
using the caspase-3, -8 and -9 colorimetric assay kits (R&D
Systems), respectively (21). In
brief, 1×106 cells in a 60-mm culture dish (Nunc) were
incubated with 10 mM VPA for 24 h. The cells were then washed in
PBS and suspended in 5 volumes of lysis buffer provided with the
kit. Protein concentrations were determined using the Bradford
method. Supernatants containing 50 μg total protein were used to
determine caspase-3, -8 and -9 activities. The supernatants were
added to each well in 96-well microtiter plates (Nunc) with
DEVD-pNA, IETD-pNA or LEHD-pNA as caspase-3, -8 and -9 substrates
and the plates were incubated at 37°C for 1 h. The optical density
of each well was measured at 405 nm using a microplate reader
(Synergy™ 2; BioTek Instruments). The activity of caspase-3, -8 and
-9 was expressed in arbitrary absorbance units.
Measurement of MMP (ΔΨm)
The MMP (ΔΨm) levels were measured by a
Rhodamine 123 fluorescent dye (Sigma-Aldrich; Ex/Em = 485/535 nm)
as previously described (20,22).
In brief, 1×106 cells in a 60-mm culture dish (Nunc)
were incubated with the designated doses of VPA for 24 h. Cells
were washed twice with PBS and incubated with Rhodamine 123 (0.1
μg/ml) at 37°C for 30 min. Rhodamine 123 staining intensity was
determined by a FACStar flow cytometer. The cells that were
Rhodamine 123 negative were indicated to have lost MMP
(ΔΨm). MMP (ΔΨm) levels in cells except MMP
(ΔΨm) loss cells were expressed as mean fluorescence
intensity (MFI), which was calculated by the CellQuest
software.
Lactate dehydrogenase (LDH) activity for
the detection of necrosis
Necrosis in cells treated with VPA and/or TNF-α was
evaluated by an LDH kit (Sigma-Aldrich). In brief, 1×106
cells in a 60-mm culture dish (Nunc) were incubated with the
indicated doses of VPA and/or TNF-α for 24 h. After treatment, the
culture media were collected and centrifuged for 5 min at 1,500
rpm. Media supernatant (50 μl) was added to a fresh 96-well plate
along with LDH assay reagent and then incubated at room temperature
for 30 min. The absorbance values were measured at 490 nm using a
microplate reader. LDH release was expressed as the percentage of
extracellular LDH activity compared with the control cells.
Detection of intracellular ROS level
Intracellular ROS such as
H2O2, •OH and ONOO• was
detected by means of an oxidation-sensitive fluorescent probe dye,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA,
Invitrogen Molecular Probes, Eugene, OR, USA; Ex/Em = 495 nm/529
nm) (20). In brief,
1×106 cells in a 60-mm culture dish (Nunc) were
incubated with the designated doses of VPA for the indicated times.
Cells were then washed in PBS and incubated with 20 μM
H2DCFDA at 37°C for 30 min. DCF fluorescence was
detected using a FACStar flow cytometer. ROS level was expressed as
MFI, which was calculated by the CellQuest software
(Becton-Dickinson).
Detection of intracellular GSH level
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Ex/Em = 522/595 nm;
Invitrogen Life Technologies) as previously described (23). In brief, 1×106 cells were
incubated in a 60-mm culture dish (Nunc) with the designated doses
of VPA with or without 2 mM NAC or 100 μM BSO for 24 h. Cells were
then washed with PBS and incubated with 5 μM CMFDA at 37°C for 30
min. CMF fluorescence intensity was determined using a FACStar flow
cytometer. Negative CMF staining (GSH depletion) of cells was
expressed as the percentage of (-) CMF cells.
Statistical analysis
The results represent the mean of at least three
independent experiments (mean ± SD). Data were analyzed using
InStat software (GraphPad Prism 4; GraphPad San Diego, CA, USA).
The Student’s t-test or one-way analysis of variance (ANOVA) with
post hoc analysis using Tukey’s multiple comparison test was used
for parametric data. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of VPA on cell growth and HDAC
activity in HeLa cells
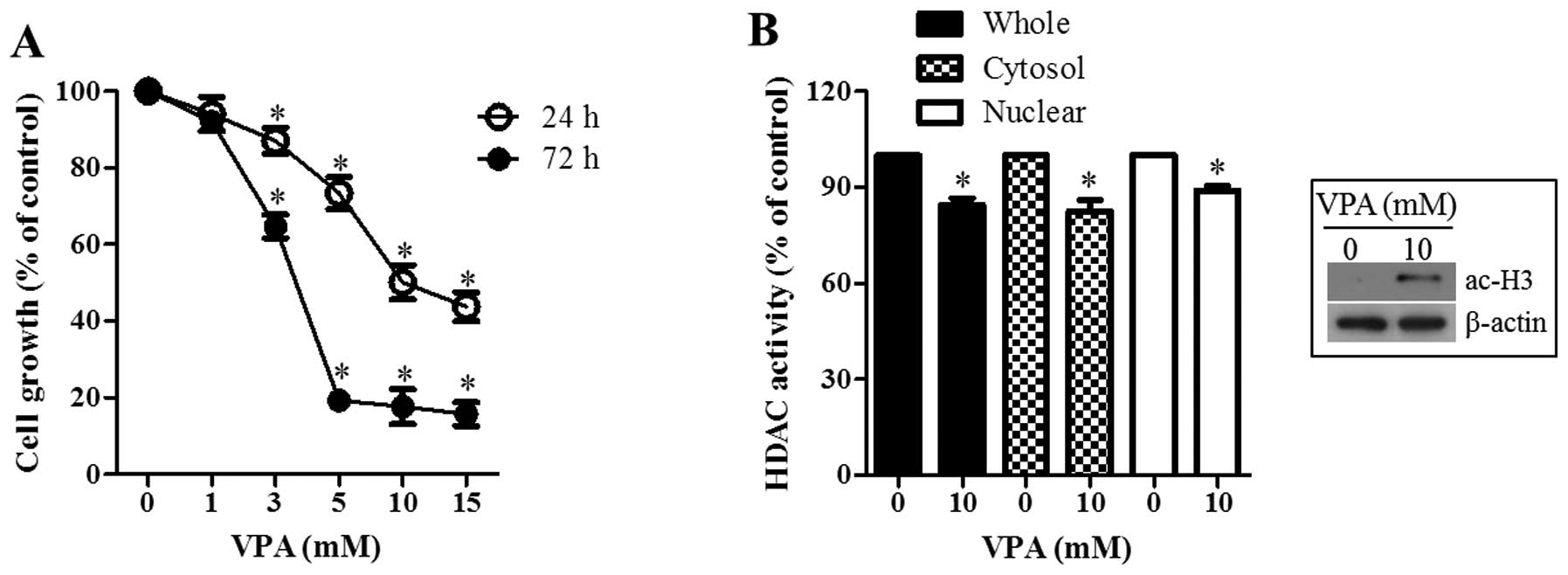
The effect of VPA on the growth inhibition of HeLa
cells was examined using MTT assays. After exposure to the various
concentrations of VPA for various times, HeLa cell growth was dose-
and time-dependently decreased with an IC50 of ~10 and 4
mM at 24 and 72 h, respectively (Fig.
1A). When testing whether VPA as a HDAC inhibitor indeed
inhibited HDAC activity, VPA significantly attenuated the
activities of total, cytosol and nuclear HDACs at 24 h (Fig. 1B). Furthermore, it was observed that
VPA increased the form of acetylated histone 3 in HeLa cells
(Fig. 1B).
Effects of VPA on cell cycle distribution
and sub-G1 cells in HeLa cells
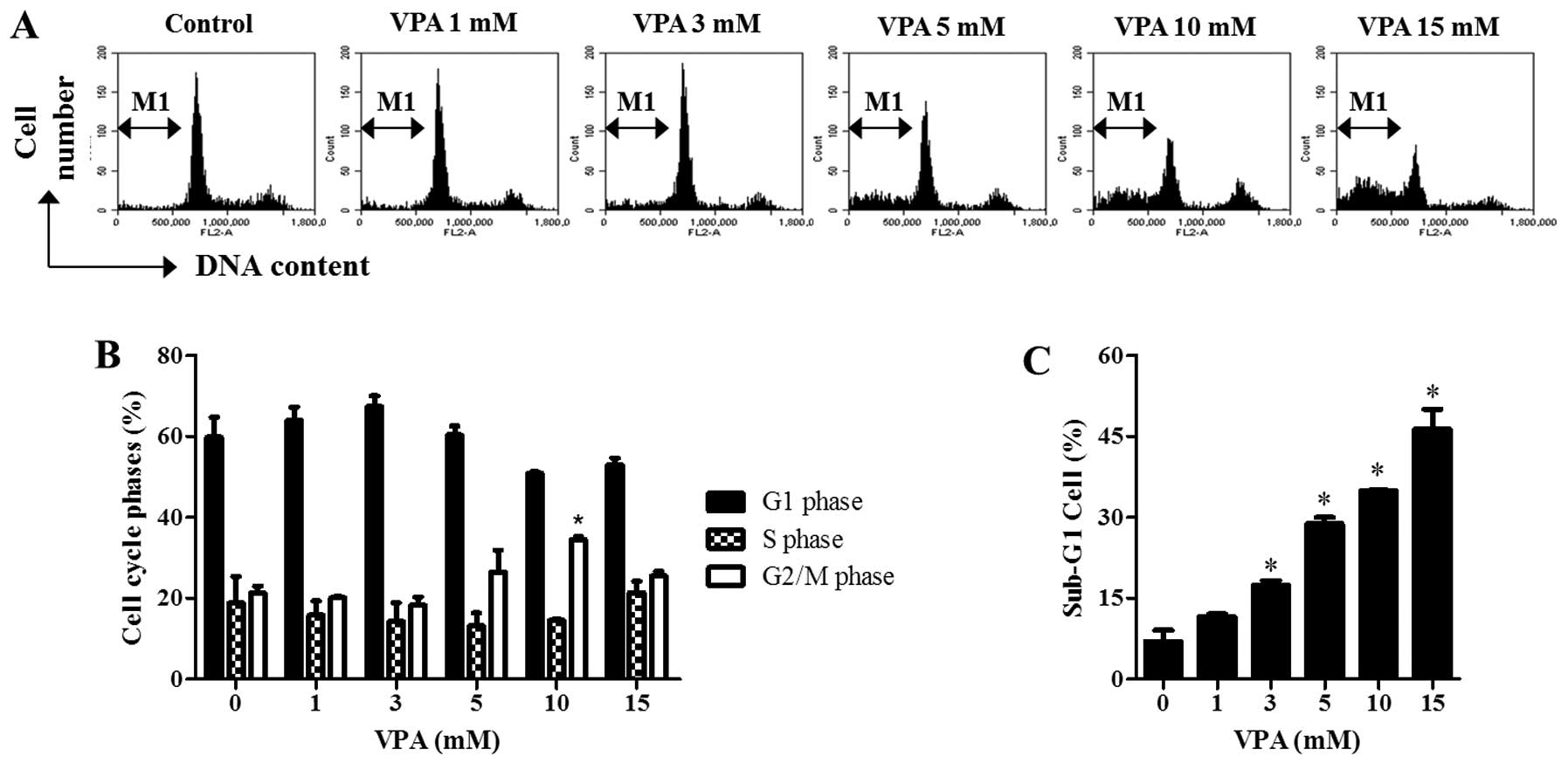
Since the growth inhibition of HeLa cells by VPA
could be explained by an arrest during the cell cycle progression,
cell cycle distributions were examined at 24 h. As shown in
Fig. 2A and B, DNA flow cytometric
analysis indicated that 1–3 mM VPA seemed to induce a G1 phase
arrest while 10 mM VPA significantly induced a G2/M phase arrest of
cell cycle in HeLa cells. In addition, VPA increased the percentage
of sub-G1 cells in HeLa cells in a dose-dependent manner at 24 h
(Fig. 2A and C).
Effects of VPA on cell death, MMP
(ΔΨm) and LDH release in HeLa cells
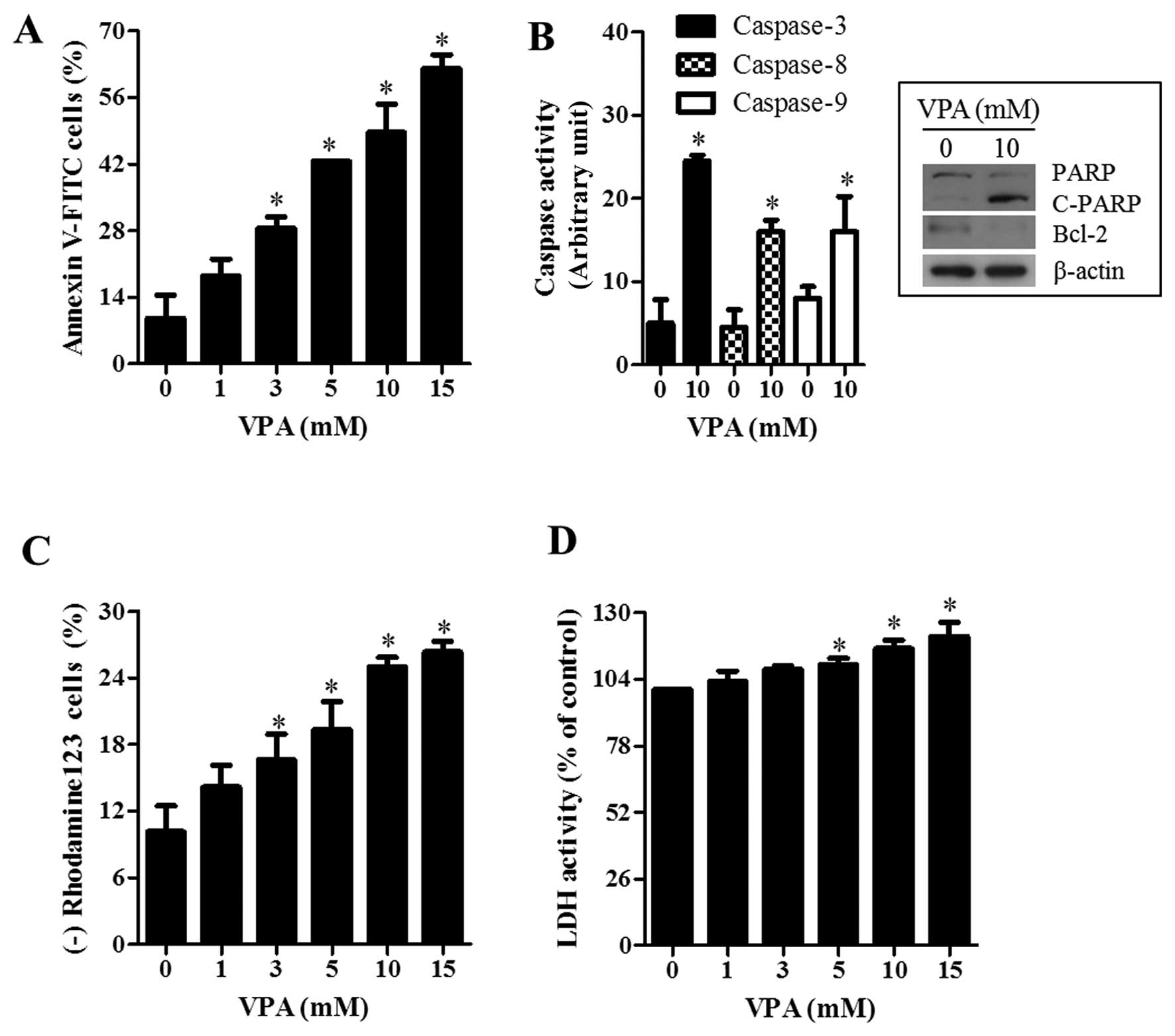
VPA also increased the number of Annexin V-FITC
positive cells in HeLa cells (Fig.
3A). In addition, the activity of caspase-3, -8 and -9 was
increased in 10 mM VPA-treated HeLa cells (Fig. 3B). The examination of the
expressions in apoptotic-related proteins showed that the intact
form of poly (ADP-ribose) polymerase (PARP) was reduced and instead
its cleavage form was induced by VPA (Fig. 3B). The level of Bcl-2 was also
downregulated by VPA in HeLa cells (Fig. 3B). Cell death is closely related to
the collapse of the MMP (ΔΨm) (24). As expected, loss of MMP
(ΔΨm) was observed in VPA-treated HeLa cells (Fig. 3C). Since VPA induced necrosis in
HeLa cells, the status of necrosis was assessed using an LDH
release. Treatment with 5–15 mM VPA significantly increased LDH
release (Fig. 3D).
Effects of caspase inhibitors and TNF-α
in VPA-treated HeLa cells
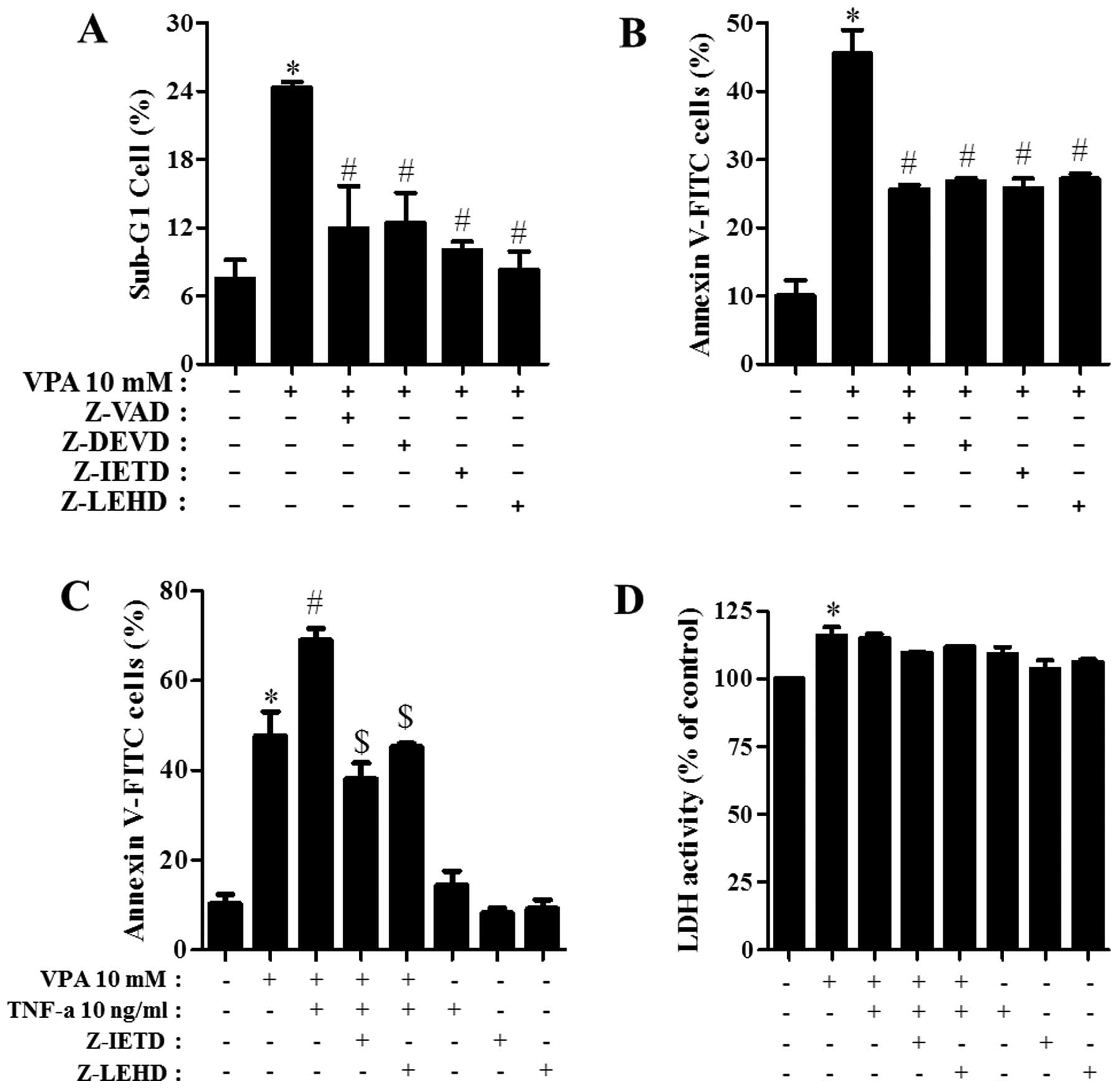
It was determined which caspases were involved in
the death of VPA-treated HeLa cells. For this experiment, we chose
10 mM VPA as a suitable dose to differentiate the level of cell
death in the presence or absence of each caspase inhibitor
[pan-caspase inhibitor (Z-VAD), caspase-3 inhibitor (Z-DEVD),
caspase-8 inhibitor (Z-IETD), or caspase-9 inhibitor (Z-LEHD)]. A
concentration of 15 μM of each caspase inhibitor was used as an
optimal dose since it did not affect cell death in the control HeLa
cells (25). All the caspase
inhibitors attenuated the percentage of sub-G1 cells in VPA-treated
HeLa cells (Fig. 4A) and they
prevented apoptotic cell death in these cells (Fig. 4B). Therefore, the activation of
various caspases seemed to be involved in apoptotic HeLa cell death
caused by VPA. Moreover, TNF-α enhanced apoptotic cell death in
VPA-treated HeLa cells (Fig. 4C).
When Z-IETD (caspase-8 inhibitor) or Z-LEHD (caspase-9 inhibitor)
was co-incubated in HeLa cells co-treated with VPA and TNF-α, these
inhibitors significantly prevented apoptosis caused by co-treatment
with VPA and TNF-α (Fig. 4C).
However, TNF-α did not augment LDH release in VPA-treated and
-untreated HeLa cells (Fig. 4D).
Neither Z-IETD nor Z-LEHD affected LDH release in VPA-treated and
-untreated HeLa cells (Fig.
4D).
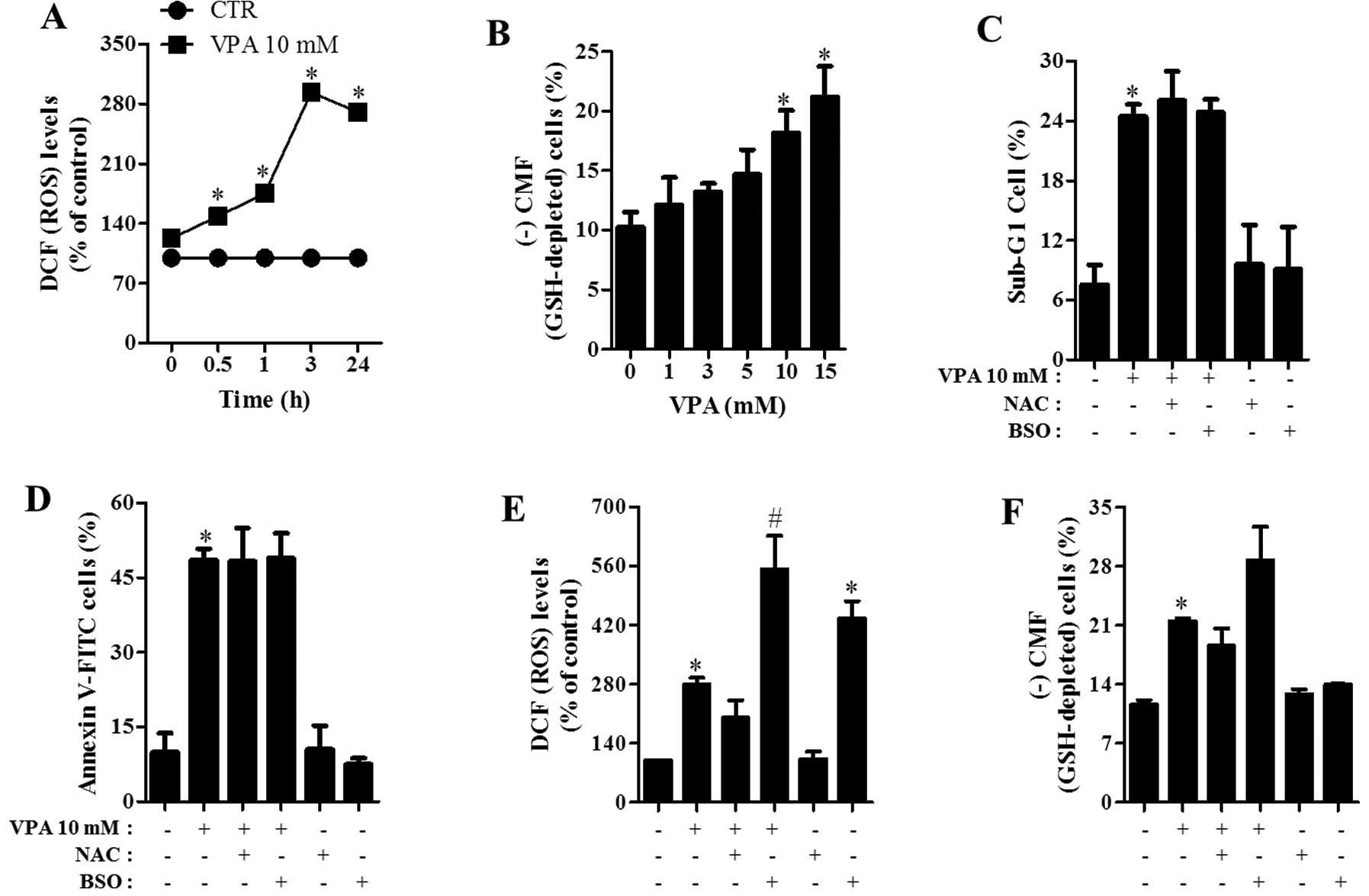
Effects of NAC and BSO on cell death, ROS
and GSH levels in VPA-treated HeLa cells
Changes in the intracellular ROS and GSH levels were
investigated in HeLa cells treated with VPA. VPA significantly
increased the intracellular ROS (DCF) level in HeLa cells from 30
min to 24 h. In relation to GSH level, VPA significantly increased
GSH-depleted cell number at 24 h in a dose-dependent manner
(Fig. 5B). Next, the effects of NAC
(an antioxidant) or BSO (an inhibitor of GSH synthesis) on cell
death were examined in VPA-treated HeLa cells. As shown in Fig. 5C and D, NAC and BSO did not affect
cell death induced by VPA. When assessing whether NAC or BSO
influences ROS level in VPA-treated HeLa cells, NAC slightly
reduced ROS levels in these cells and BSO significantly increased
ROS levels in VPA-treated and -untreated HeLa cells (Fig. 5E). Regarding GSH levels, NAC
slightly attenuated GSH depletion induced by VPA in HeLa cells
(Fig. 5F). BSO seemed to increase
GSH depletion in VPA-treated HeLa cells (Fig. 5F).
Discussion
In the present study, we assessed the effects of VPA
on HeLa cervical cancer cells in relation to cell death, ROS and
GSH levels. VPA inhibited the activities of cytosol and nuclear
HDACs in HeLa cells. These results support that VPA is a class 1
and 2 HDAC inhibitor (26). VPA
decreased the growth of HeLa cells in dose- and time-dependent
manners. When the cell cycle distributions were examined, 10 mM VPA
induced a G2/M phase arrest of the cell cycle in HeLa cells at 24
h. However, relatively lower concentrations of VPA seemed to induce
a G1 phase arrest in HeLa cells. Similarly, VPA induced a G1 phase
or a G2/M phase arrest in gastric cancer and glioblastoma cells
(27,28). Therefore, cell cycle arrest in
VPA-treated cells was an underlying mechanism to suppress the
growth of cancer cells including HeLa cells.
VPA also increased the number of sub-G1 cells and
induced apoptosis, which was accompanied by the cleavage of PARP,
caspase-3, -8 and -9 activations. Apoptosis is closely related to
the collapse of MMP (ΔΨm) (29). Our results demonstrated that VPA
triggered the loss of MMP (ΔΨm) in HeLa cells in a
dose-dependent manner. Moreover, caspase inhibitors significantly
prevented HeLa cell death caused by VPA. These data suggest that
the mitochondrial pathway as well as the cell death receptor
pathway are all together necessary for the induction of apoptosis
in VPA-treated HeLa cells. It is reported that HDAC inhibitor and
TNF-family members, especially TRAIL, synergistically induce
apoptosis in several cancer cells such as breast, liver and
lymphoma cells (10–12). According to the present study, TNF-α
synergistically enhanced cell death in VPA-treated HeLa cells.
Treatment with TRAIL or FasL did not affect cell death induced by
VPA in HeLa cells (data not shown). Although VPA induced LDH
release, TNF-α did not enhance this release in VPA-treated HeLa
cells. This result indicates that HeLa cell death caused by VPA
and/or TNF-α did not result from the necrotic pathway. In
particular, Z-IETD and Z-LEHD significantly attenuated HeLa cell
death induced by co-treatment with VPA and TNF-α. Furthermore, we
observed that VPA induced autophagy, as evidenced by the conversion
of LC3-I to LC3-II (data not shown). However, autophagy inhibitors,
hydroxychloroquinine and 3-methyladenine did not affect HeLa cell
death induced by VPA. Taken together, the main cause of HeLa cell
death induced by VPA is mediated by apoptosis rather than necrotic
or autophagic cell death.
HDAC inhibitors generate ROS in solid tumor and
leukemia cells and induce apoptosis in these cells (30). Oxidative stress might be involved in
HDAC inhibitor-induced cell death. It is reported that NAC prevents
cell death induced by HDAC inhibitors (31). Similarly, ROS levels significantly
increased in VPA-treated HeLa cells from 30 min to 24 h. However,
NAC did not attenuate cell death level in VPA-treated HeLa cells at
24 h. Since NAC decreased ROS levels in VPA-treated HeLa cells,
this agent seemed to work as an antioxidant in these cells. In
addition, although BSO increased ROS levels in VPA-treated and
-untreated HeLa cells, it did not enhance cell death. Therefore,
VPA-induced HeLa cell death was not closely related to ROS. The
increased ROS level induced by VPA seems to be a byproduct of
VPA-induced HeLa cell death.
GSH is an important intracellular antioxidant that
protects cells from damage caused by free radical and toxins. It is
able to clear away O2•− and provide electrons for
glutathione peroxidase to reduce H2O2 to
H2O. Apoptotic effects are inversely comparative to GSH
content (32–34). Similarly, VPA increased the
percentage of GSH-depleted cells in HeLa cells. NAC slightly
decreased GSH depletion whereas BSO augmented it in VPA-treated
HeLa cells. However, these agents did not affect cell death induced
by VPA in HeLa cells. Therefore, the loss of GSH content seemed to
be necessary but not sufficient to fully induce apoptosis in
VPA-treated HeLa cells.
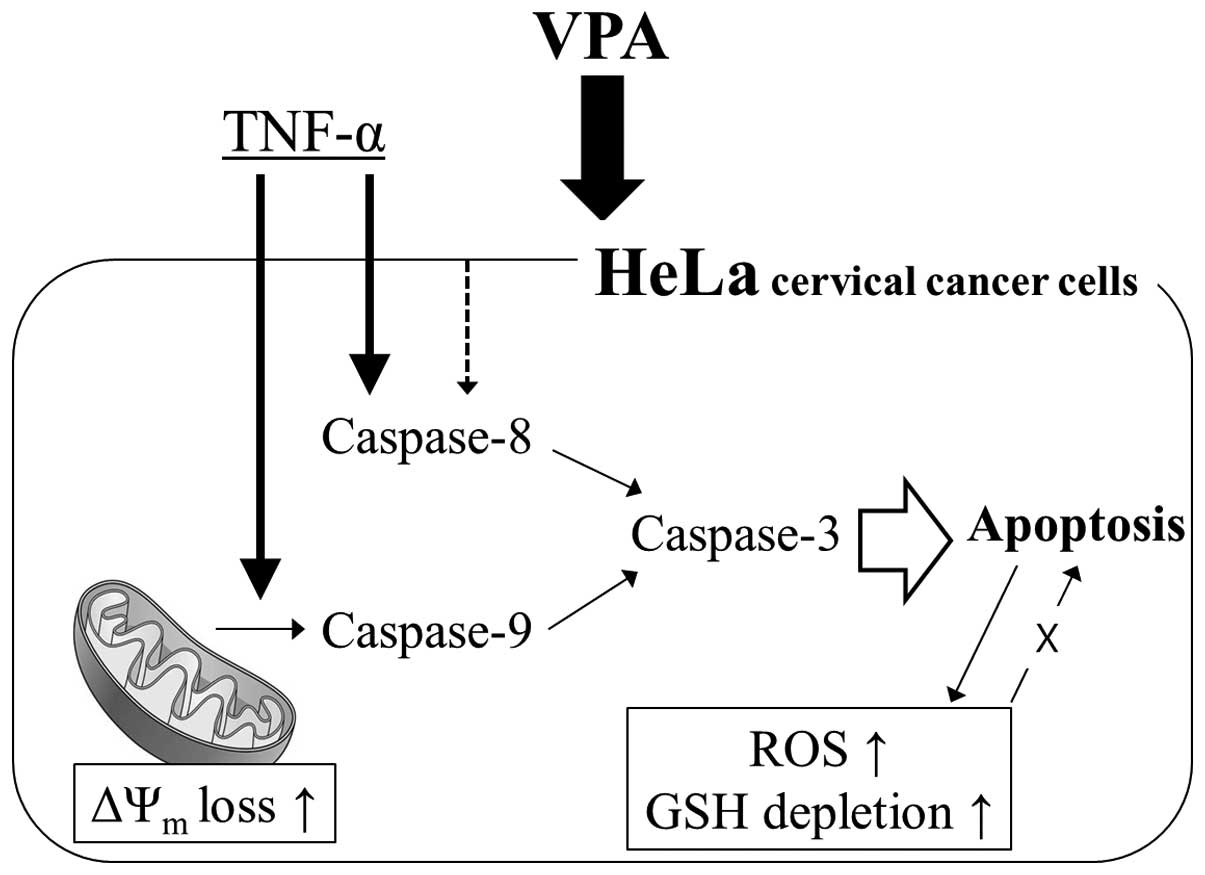
In summary, as depicted in Fig. 6, VPA inhibited the growth of HeLa
cervical cancer cells via caspase-dependent apoptosis. TNF-α
enhanced HeLa apoptotic cell death induced by VPA. The growth
inhibition was not dependent on ROS and GSH level changes.
Acknowledgements
The present study was supported by the National
Research Foundation of Korea (NRF) grant funded by the Korean
government through the Diabetes Research Center at Chonbuk National
University (2012-0009323) and the Basic Science Research Program
through the National Research Foundation of Korea (NRF) funded by
the Ministry of Education (2013006279).
Abbreviations:
|
VPA
|
valproic acid
|
|
HDAC
|
histone deacetylase
|
|
ROS
|
reactive oxygen species
|
|
GSH
|
glutathione
|
|
Z-DEVD-FMK
|
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone
|
|
Z-IETD-FMK
|
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
|
|
Z-LEHD-FMK
|
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone
|
|
TNF-α
|
tumor necrosis factor-α
|
|
LDH
|
lactate dehydrogenase
|
|
NAC
|
N-acetyl cysteine
|
|
BSO
|
L-buthionine sulfoximine
|
|
FITC
|
fluorescein isothiocyanate
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
PI
|
propidium iodide
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
References
|
1
|
Icardi L, De Bosscher K and Tavernier J:
The HAT/HDAC interplay: multilevel control of STAT signaling.
Cytokine Growth Factor Rev. 23:283–291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lu Z, Luo RZ, Peng H, et al: E2F-HDAC
complexes negatively regulate the tumor suppressor gene ARHI in
breast cancer. Oncogene. 25:230–239. 2006.PubMed/NCBI
|
|
3
|
Khan O and La Thangue NB: HDAC inhibitors
in cancer biology: emerging mechanisms and clinical applications.
Immunol Cell Biol. 90:85–94. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cebrian A, Pharaoah PD, Ahmed S, et al:
Genetic variants in epigenetic genes and breast cancer risk.
Carcinogenesis. 27:1661–1669. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang L, Zou X, Berger AD, et al: Increased
expression of histone deacetylaces (HDACs) and inhibition of
prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J
Transl Res. 1:62–71. 2009.PubMed/NCBI
|
|
6
|
Robey RW, Chakraborty AR, Basseville A, et
al: Histone deacetylase inhibitors: emerging mechanisms of
resistance. Mol Pharm. 8:2021–2031. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pettazzoni P, Pizzimenti S, Toaldo C, et
al: Induction of cell cycle arrest and DNA damage by the HDAC
inhibitor panobinostat (LBH589) and the lipid peroxidation end
product 4-hydroxynonenal in prostate cancer cells. Free Radic Biol
Med. 50:313–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frumm SM, Fan ZP, Ross KN, et al:
Selective HDAC1/HDAC2 inhibitors induce neuroblastoma
differentiation. Chem Biol. 20:713–725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rikiishi H: Autophagic and apoptotic
effects of HDAC inhibitors on cancer cells. J Biomed Biotechnol.
2011:8302602011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lauricella M, Ciraolo A, Carlisi D, et al:
SAHA/TRAIL combination induces detachment and anoikis of MDA-MB231
and MCF-7 breast cancer cells. Biochimie. 94:287–299. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carlisi D, Lauricella M, D’Anneo A, et al:
The histone deacetylase inhibitor suberoylanilide hydroxamic acid
sensitises human hepatocellular carcinoma cells to TRAIL-induced
apoptosis by TRAIL-DISC activation. Eur J Cancer. 45:2425–2438.
2009. View Article : Google Scholar
|
|
12
|
Al-Yacoub N, Fecker LF, Mobs M, et al:
Apoptosis induction by SAHA in cutaneous T-cell lymphoma cells is
related to downregulation of c-FLIP and enhanced TRAIL signaling. J
Invest Dermatol. 132:2263–2274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong K, Xie J, Yi H and Li W: CS055
(Chidamide/HBI-8000), a novel histone deacetylase inhibitor,
induces G1 arrest, ROS-dependent apoptosis and differentiation in
human leukaemia cells. Biochem J. 443:735–746. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang BH, Laban M, Leung CH, et al:
Inhibition of histone deacetylase 2 increases apoptosis and
p21Cip1/WAF1 expression, independent of histone
deacetylase 1. Cell Death Differ. 12:395–404. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Anton M, Horky M, Kuchtickova S, et al:
Immunohistochemical detection of acetylation and phosphorylation of
histone H3 in cervical smears. Ceska Gynekol. 69:3–6.
2004.PubMed/NCBI
|
|
16
|
Shan Z, Feng-Nian R, Jie G and Ting Z:
Effects of valproic acid on proliferation, apoptosis, angiogenesis
and metastasis of ovarian cancer in vitro and in vivo. Asian Pac J
Cancer Prev. 13:3977–3982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Machado MC, Bellodi-Privato M, Kubrusly
MS, et al: Valproic acid inhibits human hepatocellular cancer cells
growth in vitro and in vivo. J Exp Ther Oncol. 9:85–92.
2011.PubMed/NCBI
|
|
18
|
Han YH, Moon HJ, You BR, et al: Effects of
carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone on the growth
inhibition in human pulmonary adenocarcinoma Calu-6 cells.
Toxicology. 265:101–107. 2009. View Article : Google Scholar
|
|
19
|
You BR and Park WH: Zebularine inhibits
the growth of HeLa cervical cancer cells via cell cycle arrest and
caspase-dependent apoptosis. Mol Biol Rep. 39:9723–9731. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han YH, Moon HJ, You BR and Park WH: The
effect of MG132, a proteasome inhibitor on HeLa cells in relation
to cell growth, reactive oxygen species and GSH. Oncol Rep.
22:215–221. 2009.PubMed/NCBI
|
|
21
|
You BR and Park WH: Proteasome inhibition
by MG132 induces growth inhibition and death of human pulmonary
fibroblast cells in a caspase-independent manner. Oncol Rep.
25:1705–1712. 2011.PubMed/NCBI
|
|
22
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2(*-) generator induces apoptosis via the depletion of
intracellular GSH contents in Calu-6 cells. Lung Cancer.
63:201–209. 2009.
|
|
23
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Griffiths EJ: Mitochondria - potential
role in cell life and death. Cardiovasc Res. 46:24–27. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
You BR and Park WH: Suberoyl bishydroxamic
acid-induced apoptosis in HeLa cells via ROS-independent,
GSH-dependent manner. Mol Biol Rep. 40:3807–3816. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gurvich N, Tsygankova OM, Meinkoth JL and
Klein PS: Histone deacetylase is a target of valproic acid-mediated
cellular differentiation. Cancer Res. 64:1079–1086. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao X, Yang W, Shi C, et al: The G1 phase
arrest and apoptosis by intrinsic pathway induced by valproic acid
inhibit proliferation of BGC-823 gastric carcinoma cells. Tumour
Biol. 32:335–346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Das CM, Aguilera D, Vasquez H, et al:
Valproic acid induces p21 and topoisomerase-II (α/β) expression and
synergistically enhances etoposide cytotoxicity in human
glioblastoma cell lines. J Neurooncol. 85:159–170. 2007.PubMed/NCBI
|
|
29
|
Yang J, Liu X, Bhalla K, et al: Prevention
of apoptosis by Bcl-2: release of cytochrome c from mitochondria
blocked. Science. 275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eot-Houllier G, Fulcrand G,
Magnaghi-Jaulin L and Jaulin C: Histone deacetylase inhibitors and
genomic instability. Cancer Lett. 274:169–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ungerstedt JS, Sowa Y, Xu WS, et al: Role
of thioredoxin in the response of normal and transformed cells to
histone deacetylase inhibitors. Proc Natl Acad Sci USA.
102:673–678. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han YH, Kim SZ, Kim SH and Park WH:
Enhancement of arsenic trioxide-induced apoptosis in HeLa cells by
diethyldithiocarbamate or buthionine sulfoximine. Int J Oncol.
33:205–213. 2008.PubMed/NCBI
|
|
33
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar
|
|
34
|
Han YH, Kim SZ, Kim SH and Park WH:
Suppression of arsenic trioxide-induced apoptosis in HeLa cells by
N-acetylcysteine. Mol Cells. 26:18–25. 2008.PubMed/NCBI
|