Introduction
Lung cancer treatment continues to present
difficulties and survival rates remain low, despite research
efforts. Chemotherapy is the primary form of treatment for lung
cancer; however, its response rate is low, and complete recovery is
rare (1,2). Thus, the development of a more
efficacious therapy for lung cancer is urgently required. Autophagy
is a type of programmed cell death that occurs in both
physiological or pathophysiological environments (3,4).
Autophagy is the mechanism by which protein conversion and the
removal of aging or damaged organelles occurs; this helps maintain
cellular homeostasis (5,6). Autophagy is also known to be involved
in cell survival. However, excessive autophagy or improper
activation can lead to apoptosis. Autophagy is believed to play an
important role in the incidence of tumors; it is also considered to
have a role in primary tumorigenesis. Additionally, it is known to
maintain cell survival in tumors via survival pathways during
periods of stress, such as during tumor progression or chemotherapy
(7–9). The protein p53 is an ‘intracellular
gatekeeper’ that protects the cell from various stress signals, and
p53 is a mutant gene in most cancer cells that is activated by DNA
damage, cancer gene activation, hypoxia and other stresses
(10,11). It is involved in cell cycle
inhibition, apoptosis, aging, metabolism, differentiation,
inhibition of blood vessel formation, and autophagy regulation
(12–14). Cisplatin is the most widely used
chemotherapy drug for lung cancer and has powerful anticancer
effects. However, it has many side-effects. Therefore, in an
attempt to reduce these side-effects, we confirmed the anticancer
effect of low-dose cisplatin, and examined the effects of these
low-doses on autophagy and apoptosis. To ascertain whether p53
influences autophagy and apoptosis, we compared the role of p53 in
lung cancer cell lines of wild-type p53 and null-type p53.
Materials and methods
Cell lines
The wild-type p53 NCI-H460 and null-type p53
NCI-H1299 lung cancer cell lines were purchased from the American
Type Culture Collection (ATCC, Manassas, VA, USA).
Reagents
RPMI-1640, antibiotics, trypsin and fetal bovine
serum (FBS) were obtained from Gibco-BRL (Grand Island, NY, USA).
24- and 48-well plates, along with 6 and 10-cm diameter dishes,
were purchased from Nunc (Thermo Fisher Scientific, Roskilde,
Denmark). Cisplatin (0.5 mg/ml) was obtained from Ildong
Pharmaceutical Co., Ltd. (Seoul, Korea). Propidium iodide (PI),
3-methyladenine (3-MA),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
and acridine orange (AO) were purchased from Assay Designs (Ann
Arbor, MI, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
antibody and p53 were obtained from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Secondary antibody was obtained from
Amersham (Buckinghamshire, UK). Polyvinylidene fluoride (PVDF)
membrane, and enhanced chemiluminescent (ECL) kit were purchased
from Millipore Co. (Billerica, MA, USA).
Cell culture
NCI-H460 and NCI-H1299 cells were cultured with RPMI
medium supplemented with 10% FBS, 1% antibiotics, and
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES). Cells
were incubated in 5% CO2 at 37ºC. The medium was
replaced every 24 h. Experiments were conducted with cells in the
log phase. The viability of the cultured cells was measured after
24 h of treatment with cisplatin at 5 μM (LD20) and 20 μM
(LD50).
Cell viability
Cell viability was determined with MTT assays. Cells
were seeded in 24-well plates at a density of 1×104
cells and incubated for 24 h. After treatment with cisplatin (5 μM)
in the presence or absence of 3-MA (10 mM), MTT (5 mg/ml) was added
to each well and incubated in 5% CO2 at 37ºC for 4 h.
Crystals were dissolved in 200 μl dimethyl sulfoxide (DMSO). The
absorbance of the solution was measured spectrophotometrically at
570 nm with a microplate ELISA reader (Thermo Scientific). The
absorbance of formazan formed in control cells was considered as
showing 100% cell viability, and the positively stained cells with
MTT were expressed as the percentage of control cells.
Morphological observations
To observe the cell morphology after cisplatin
treatment, H460 and H1299 cells were cultured in a 24-well plate.
They were then treated with 5 and 20 μM cisplatin for 24 and 48 h.
After the treatment, the medium was removed and washed twice with
phosphate buffered saline (PBS) (pH 7.4). It was then treated with
300 μl crystal violet solution (0.05% crystal violet, 3.7%
paraformaldehyde) for 5 min at room temperature and then washed
twice with PBS. The nucleus and the cytoplasm were then observed
under a microscope (Olympus, Japan).
Assay for autophagy detection
Autophagy was detected by measuring the expression
levels of LC3-II protein and by using AO stain to detect acidic
vesicular organelles (AVOs) within the cytoplasm. After adding AO
(1 μg/ml), the cells were incubated in 5% CO2 at 37ºC
for 15 min. Next, the cells were washed with PBS. The number of
AO-stained cells was observed under a microscope (Olympus,
Japan).
LC3 protein was detected by western blot analysis.
The cells were treated with cisplatin (5 μM) in the presence or
absence of 3-MA (10 mM) for 24 h. After treatment, each cell was
harvested and washed twice with ice-cold PBS and lysed in lysis
buffers (50 mM HEPES, pH 7.4, 150 mM NaCl, 1% deoxycholate, 1 mM
EDTA, 1 mM PMSF, 1 μg/ml aprotinin). After incubation for 1 h on
ice, the cells were centrifuged at 10,000 rpm for 30 min at 4ºC,
and the supernatants were collected. The protein concentration was
determined using the Bradford method. For western blot analysis,
equal amounts of total protein were loaded onto 10 or 15% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
then transferred onto a PVDF membrane. The PVDF membrane was
blocked with 5% skimmed milk in PBS for 20–90 min. After washing in
PBS, immunoblots were analyzed using specific primary antibodies.
The membrane was then washed with PBS and treated with secondary
antibodies for 1 h. Proteins were then visualized using an ECL
kit.
Quantitative analysis of apoptosis and
autophagy
To quantitatively analyze apoptosis and autophagy,
the cells were plated in 6-well plates and incubated for 24 h. They
were then treated with cisplatin (5 μM) in the presence or absence
of 3-MA (10 mM). After 24 h of treatment, the cells were harvested
using trypsin and were then washed twice in PBS. Subsequently, they
were treated with Annexin V/fluorescein isothiocyanate (FITC) (0.5
μg/ml final concentration) combination, PI (2 μg/ml final
concentration), and apoptosis detection kit (Assay Designs), for 10
min at room temperature. The cells were then immediately examined
using a flow cytometer (FACSVantage flow cytometer;
Becton-Dickinson Immunocytometry System, San Jose, CA, USA)
following the addition of 250 μl binding buffer. Analysis was
performed using CellQuest software (Becton-Dickinson, Franklin
Lakes, NJ, USA). To quantitatively analyze autophagy, the cells
were plated in 6-well plates and incubated for 24 h. The cells were
then treated with cisplatin (5 μM) in the presence or absence of
3-MA (10 mM). After 24 h of treatment, the cells were stained with
AO (1 μg/ml) for 15 min at 37ºC, harvested with trypsin and
subsequently washed with PBS. We then added 500 μl FACS buffer (1%
FBS in PBS). The cells were immediately counted by flow cytometry
(FACSVantage flow cytometer) after addition of 250 μl binding
buffer. Analysis was performed using CellQuest software.
Statistical analysis
The experiment was performed thrice independently,
with the means ± standard deviation (SD) recorded. Results were
analyzed using the Student’s t-test. p<0.05 was considered to
indicate a statistically significant difference.
Results
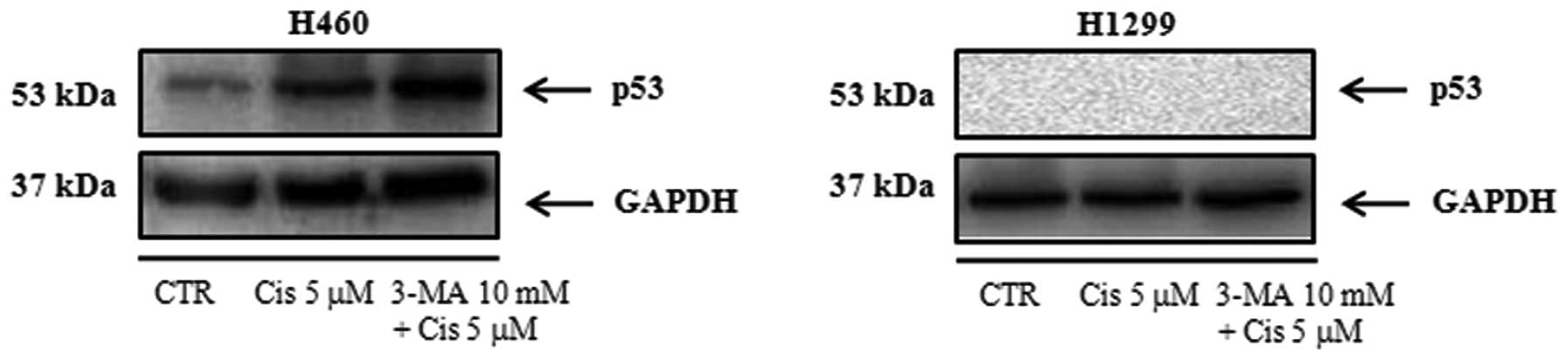
Detection of wild-type p53 expression in
the NCI-H460 and NCI-H1299 cell lines
We confirmed wild-type p53 protein expression after
treatment of NCI-H460 cell lines with 5 μM cisplatin in the
presence or absence of 3-MA (10 mM), an autophagy-specific
inhibitor. We found that wild-type p53 was not expressed after
treatment with 5 μM cisplatin in the presence or absence of 3-MA in
NCI-H1299 cell lines (Fig. 1).
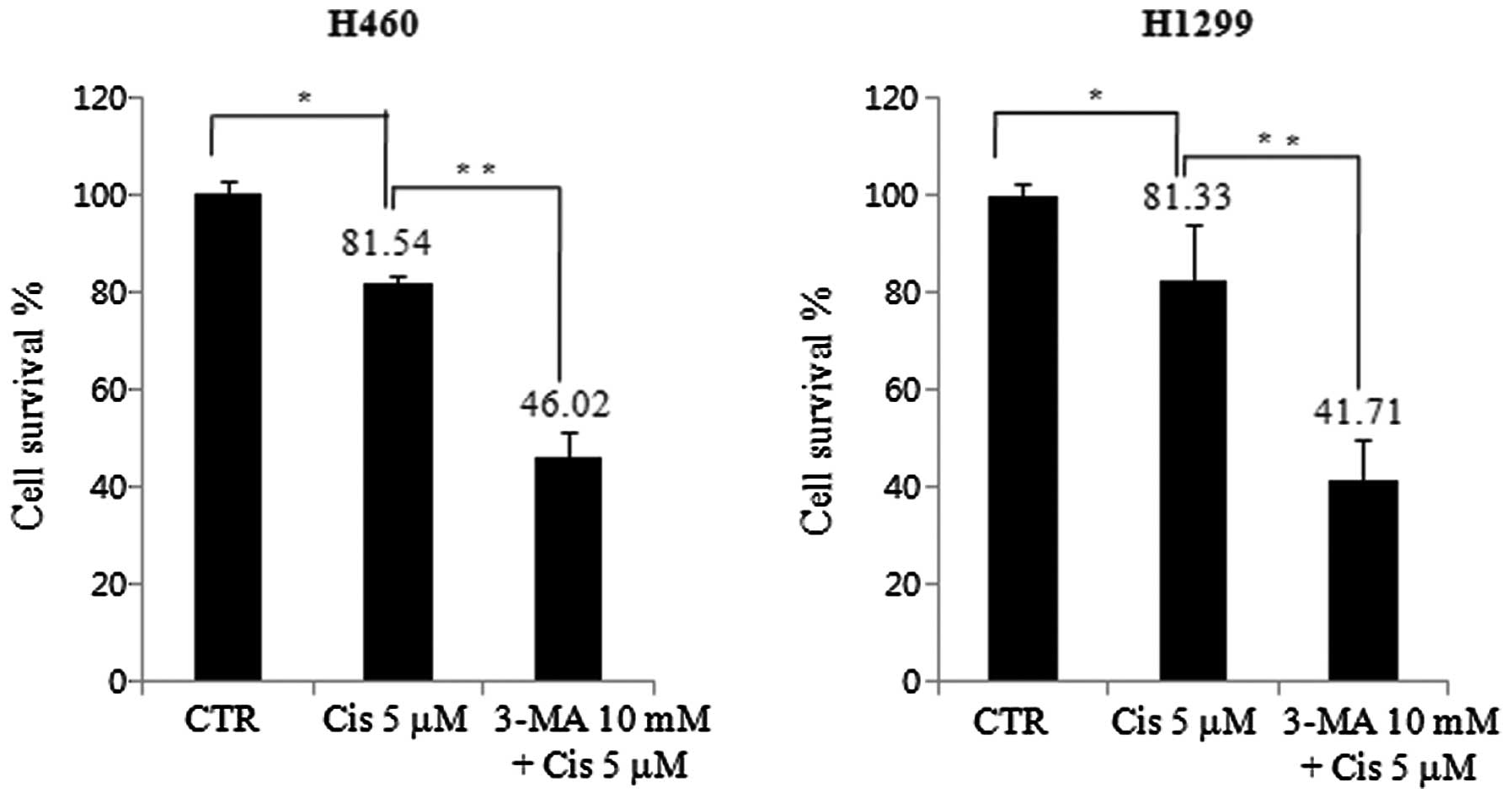
Cell viability decreases by low-dose
cisplatin in H460 and H1299 cell lines
We investigated cell death by treating both cell
lines with 5 μM cisplatin in the presence or absence of 10 mM 3-MA.
The cell viability was measured by an MTT assay. For the H460 cell
line, cell viability in the group treated with 5 μM cisplatin was
81.54%, lower than that in the control group. This further
decreased to 46.02% in the presence of 10 mM 3-MA. Cell viability
was further decreased by autophagy inhibition. In the H1299 cell
line, cell viability in the group treated with 5 μM cisplatin was
81.33%, lower than that in the control group, and it further
decreased to 41.71% in the presence of 10 mM 3-MA. However, there
was no difference in cell viability between cisplatin and 3-MA
pretreatment in H460 and H1299 cell lines (Fig. 2).
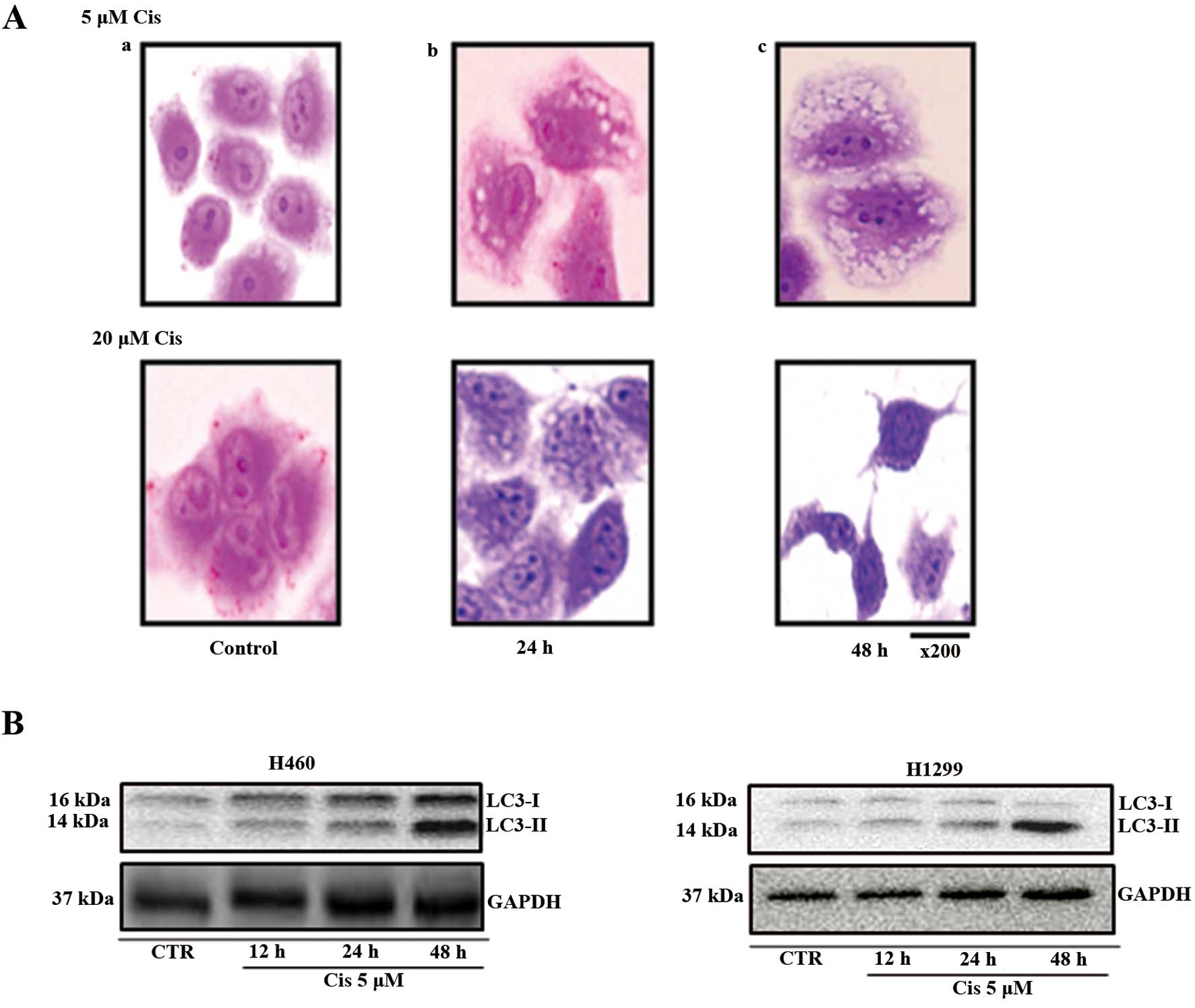
Observation of morphological
characteristics and biodynamics analysis of autophagy
After 24 h, autophagosomes, the morphological
characteristics of autophagy, were observed in cells treated with 5
μM cisplatin. After 48 h, the number of autophagosomes observed
increased. Cells treated with 20 μM cisplatin showed low
autophagosome formation after 24 h compared with those treated with
5 μM cisplatin. After 48 h, no autophagosomes were observed in the
cells treated with 20 μM cisplatin.
LC3-I is present in the cytoplasm, and LC3-II, which
is formed during autophagy, is present in the autophagosome. LC3-I
is converted to LC3-II during the initiation of autophagy. Thus,
LC3 is a marker of autophagosome formation. In the H460 cell lines
treated with 5 μM cisplatin, autophagosome-incorporated LC3-II
protein expression increased from 12 to 24 and 48 h, compared with
the control. Therefore, autophagy increased during this period.
However, in the H1299 cell lines, LC3-II protein expression did not
change compared with the control. Therefore, in the H1299 cell
lines, no change in autophagy was observed (Fig. 3).
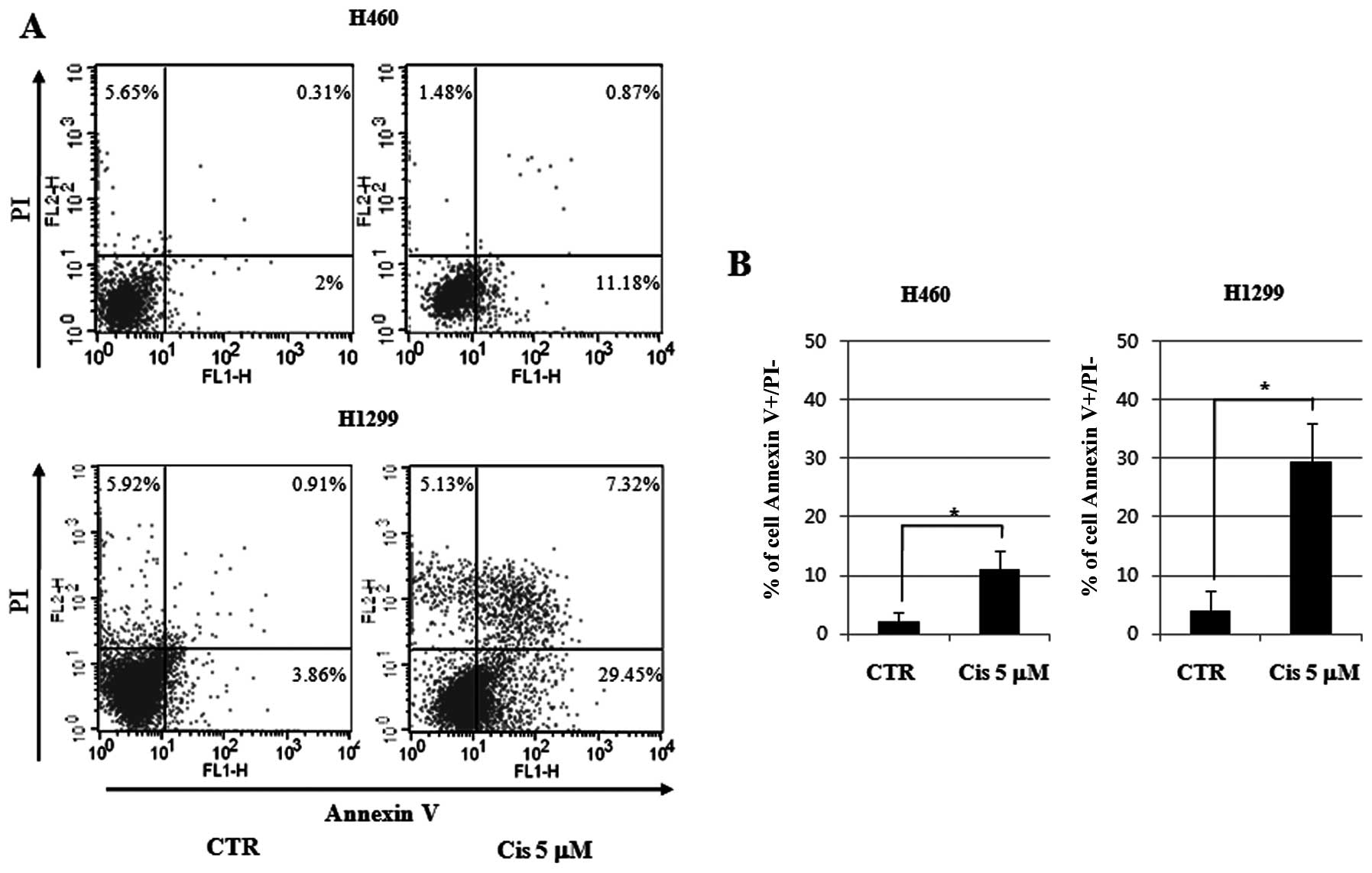
Quantitative measurements of apoptosis
and autophagy in cisplatin-treated NCI-H460 and NCI-H1299 cell
lines
For quantitative measurement of apoptosis, we
stained the cell lines with Annexin V and PI after treatment with 5
μM cisplatin for 24 h, followed by analysis using flow cytometry. A
5.9-fold increase in apoptosis was observed in the H460 cell lines
of wild-type p53. In the control group, 2% more apoptosis was
observed and 11.18% more apoptosis was observed in the group
treated with 5 μM cisplatin. In the H1299 cell lines with null-type
p53, apoptosis increased by 7.62-fold. In the control group, a
3.86% increase in apoptosis was observed along with a 29.45%
increase in the 5 μM cisplatin group. Thus, more apoptosis was
observed in the p53 null cell line groups than in the p53 wild-type
cell line groups (Fig. 4).
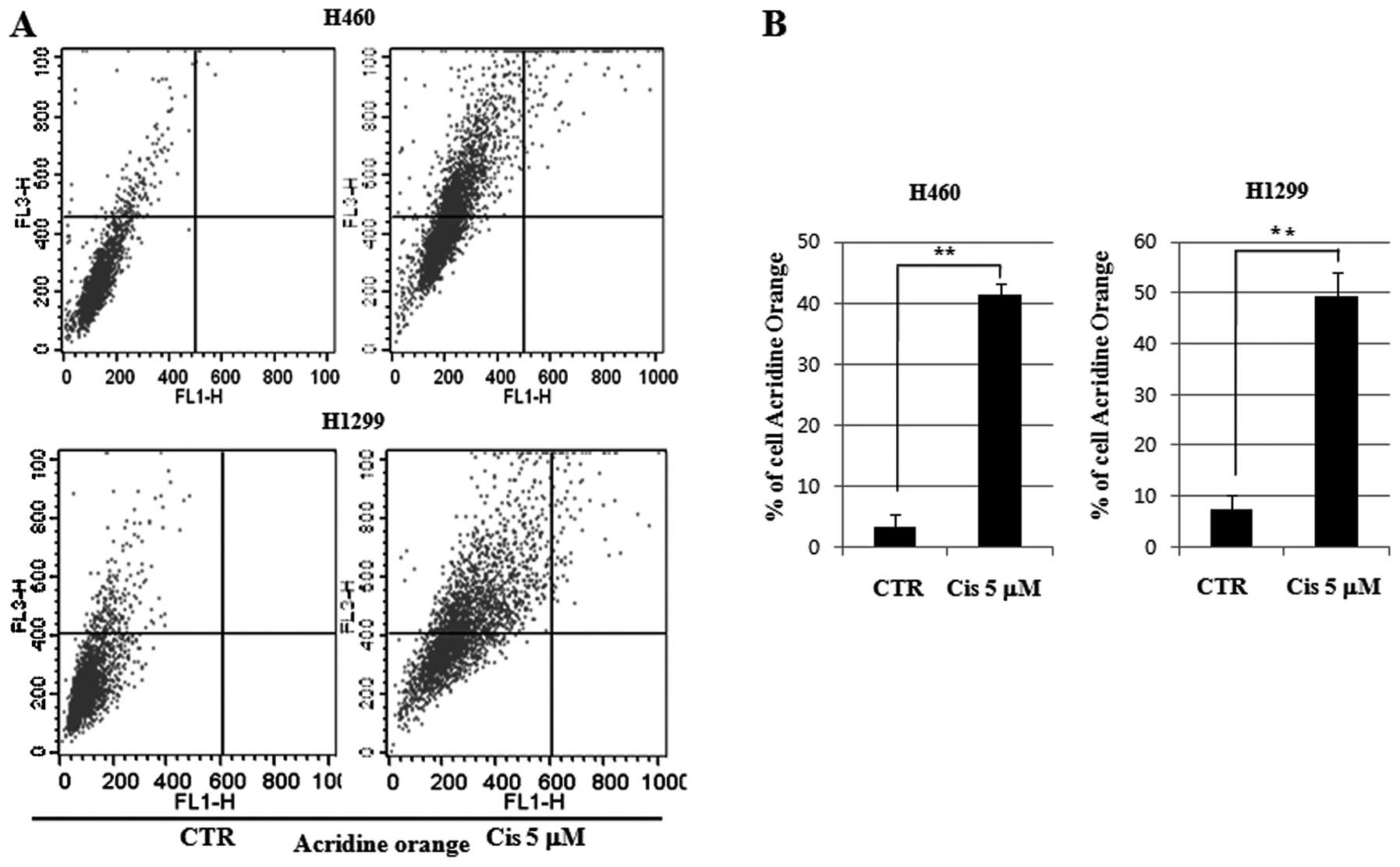
For quantitative measurement of autophagy, we
analyzed AVOs by flow cytometry in the cell lines treated with 5 μM
cisplatin for 24 h. A 12.49-fold increase in autophagy was observed
in the H460 cell lines of wild-type p53. In the control group,
autophagy increased by 3.3%, and in the 5 μM cisplatin group, it
increased by 41.22%. A 6.67-fold increase was observed in the H1299
cell lines of null-type p53. In the control group, autophagy
increased by 7.4%, and in the 5 μM cisplatin group, it increased by
49.39%. Therefore, autophagy was induced more in the p53 wild-type
cell lines than in the p53 null-type cell lines (Fig. 5). These results suggest that
cisplatin-treated p53 wild-type cells play a role in inducing
autophagy and inhibiting apoptosis.
Quantitative measurements of
cisplatin-induced apoptosis and autophagy after 3-MA treatment in
H460 and H1299 cell lines
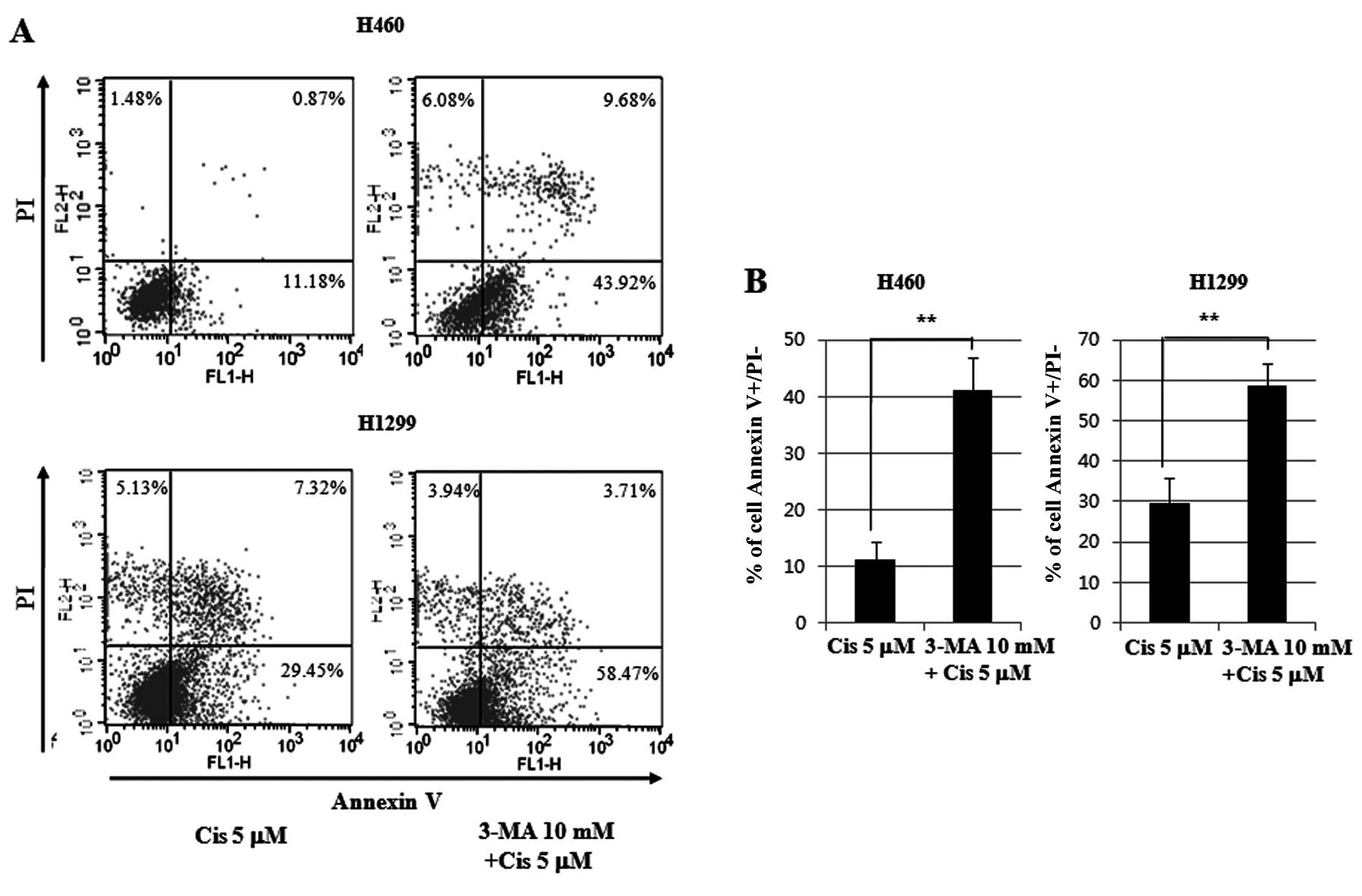
The 5 μM cisplatin-treated cell lines with 3-MA
pretreatment for 24 h were stained with Annexin V and PI, and
subjected to flow cytometry. A 3.72-fold increase in apoptosis in
the p53 wild-type H460 cell line was observed. An 11.18% increase
was observed in the 5 μM cisplatin group, and a 43.92% increase was
observed in the 5 μM cisplatin group that was pretreated with 3-MA.
In the H1299 cell line of p53 null-type, a 1.98-fold increase in
apoptosis was observed. A 29.45% increase was observed in the 5 μM
cisplatin group, and a 58.47% increase was observed in the 5 μM
cisplatin group that was pretreated with 3-MA. The p53 wild-type
cell lines induced twice as much apoptosis as the p53 null-type
cell lines (Fig. 6).
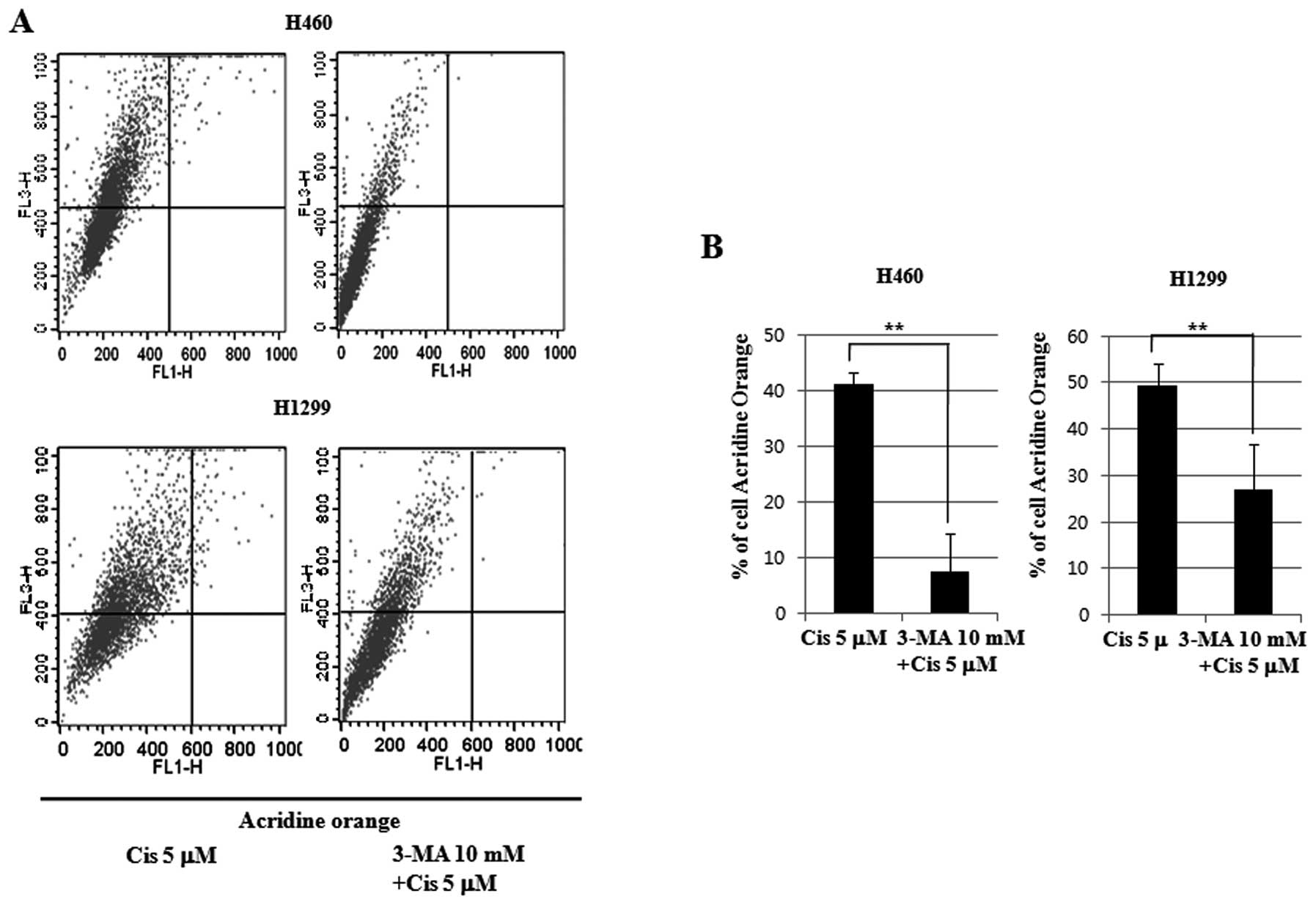
To measure autophagy, the 5 μM cisplatin-treated
cell lines with 3-MA pretreatment for 24 h were again stained with
Annexin V and PI, and subjected to flow cytometry. A 5.57-fold
decrease in autophagy in the p53 wild-type H460 cell line was
observed. A 41.22% decrease was observed in the 5 μM cisplatin
group, and a 7.4% decrease was observed in the 5 μM cisplatin group
that was pretreated with 3-MA. In the H1299 cell line of p53
null-type, a 1.83-fold decrease in autophagy was observed. A 49.39%
decrease was observed in the 5 μM cisplatin group, and a 26.88%
decrease was observed in the 5 μM cisplatin group that was
pretreated with 3-MA. The p53 wild-type cell lines reduced
autophagy 3-fold more than the p53 null-type cell lines (Fig. 7). Therefore, we concluded that p53
inhibited low-dose cisplatin-induced autophagy and induced
apoptosis.
Discussion
At the time of diagnosis, many cases of lung cancer
are considered inoperable due to infiltration of the surrounding
tissue by cancer cells. Treatment of lung cancer by chemotherapy
and radiation therapy presents difficulties, and, therefore,
prognosis for lung cancer is often bleak. However, the discovery
and subsequent adoption of cisplatin in the 1980s helped improve
survival rates of patients with lung cancer. New second-generation
anticancer drugs were developed in the 1990s, but they have yet to
produce satisfactory results.
Research has recently started to focus on targeted
therapies. However, anticancer treatment is still restricted to
cisplatin for local progression of lung cancer, despite the fact
that the use of cisplatin causes suffering and often results in
treatment being stopped due to toxicity, low white blood cell
counts, thrombocytopenia symptoms, vomiting and neurological
toxicity, among other side-effects.
Therefore, some patients choose to undergo
anticancer treatment using non-cisplatin based chemotherapy for
local lung cancer progression. However, these are often less
effective than cisplatin treatments. Furthermore, targeted
therapies often only show significant results in some patients.
Many clinicians regulate the dose intensity of cisplatin so as to
reduce the effects of toxicity. However, low-dose treatments
sometimes have minor effects.
Research has shown that autophagy affects signal
transduction. Autophagy and apoptosis have already prompted much
research in cancer. However, the role of autophagy in cell survival
or apoptosis remains to be clarified. Autophagy assists cell
survival by helping cancer cells resist radiation therapy,
chemotherapy and low-nutrient environments. However, apoptosis
helps contribute to cancer cell suicide and death on exposure to
chemotherapy or radiation. These paradoxical features are a point
of debate as to whether autophagy is a friend or foe (15–17).
Without autophagy, genome damage caused by metabolic stress is
mostly unhindered. Furthermore, autophagic defects are associated
with increased tumorigenesis. However, it is not possible to
conclude that cancer cell extirpation occurs via autophagy
induction or inhibition (18). The
number of discrepancies related to the role of autophagy in cell
death and cancer presents difficulties. Animal experiments have
shown that autophagy increases tumor cell survival during periods
of metabolic stress. However, it has also been shown to prevent
tumors, necrosis and inflammation. In this case, the
tumor-suppressing effects of autophagy had a greater effect than
the cell survival mechanisms promoted by autophagy. In other animal
experiments, treatment-induced apoptosis and tumor extinction
increased when chloroquine was used to inhibit autophagy. The
combination therapy of chloroquine and the histone deacetylase
inhibitor SAHA has a multiplier effect in killing
imatinib-refractory chronic myeloid leukemia cells. This protective
function supports the therapeutic use of autophagic inhibitors for
cancer treatment (19–22). Autophagosomes in the cytoplasm were
observed at 24 and 48 h in cell lines treated with low-dose (5 μM)
cisplatin in a previous study. The conversion of LC3-I into LC3-II
within the cytoplasm increases over time. In this study, we
investigated the role of p53 in autophagy induction in wild-type
p53 NCI-H460 cell lines and null-type p53NCI-H1299 cell lines
treated with low-dose cisplatin. No difference was observed in the
viability of H460 and H1299 cells treated with cisplatin and
pretreated with 3-MA. However, a difference between apoptosis and
autophagy was observed. After treatment with 5 μM cisplatin,
apoptosis increased by 5.59-fold in the wild-type p53 H460 cell
line compared to the control group, and apoptosis in the null-type
p53 H1299 cell lines increased by 7.62-fold.
For autophagy, a 12.49-fold increase after 5 μM
cisplatin treatment was observed in the wild-type p53H460 cell line
compared to the control group, and autophagy in the null-type p53
H1299 cell line increased by 6.69-fold. Autophagic activity in the
null-type p53 H1299 cell lines was twice that of the wild-type p53
H460 cell lines. On the basis of this result, we conclude that
wild-type p53 has a role in autophagic induction and apoptosis
inhibition when treated with low-dose cisplatin. In addition, after
3-MA pretreatment and 5 μM cisplatin treatment, apoptosis increased
by 3.92-fold in the wild-type p53 H460 cell line compared to the
control group. In the null-type p53 H1299 cell line, apoptosis
increased by 1.98-fold. Apoptotic activity in the null-type p53
H1299 cell lines was twice that of the wild-type p53 H460 cell
lines. After 3-MA pretreatment and 5 μM cisplatin treatment,
autophagy decreased 5.57-fold in the wild-type p53H460 cell lines
and decreased by 1.83-fold in the null-type p53 H1299 cell lines.
Autophagic activity decreased thrice as much in the null-type than
in the wild-type p53 cell line. This result suggests that
p53-induced apoptosis also inhibited autophagy induced by low-dose
cisplatin. However, this result does not clarify the role of p53.
Therefore, further studies are required to ascertain how
3-MA-pretreated and cisplatin-treated cells affect apoptosis and
autophagy.
Acknowledgements
This study was supported by a grant from the Korea
Health 21 R&D Project (Ministry of Health, Welfare and Family
Affairs, Republic of Korea, A010251).
References
|
1
|
Ferreira CG, Span SW, Peters GJ, et al:
Chemotherapy triggers apoptosis in a caspase-8-dependent and
mitochondria-controlled manner in the non-small cell lung cancer
cell line NCI-H460. Cancer Res. 60:7133–7141. 2000.PubMed/NCBI
|
|
2
|
Fukuoka M, Yano S, Giaccone G, et al:
Multi-institutional randomized phase II trial of gefitinib for
previously treated patients with advanced non-small-cell lung
cancer. J Clin Oncol. 21:2237–2246. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kundu M and Thompson CB: Autophagy: basic
principles and relevance to disease. Annu Rev Pathol. 3:427–455.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hara T, Nakamura K, Matsui M, et al:
Suppression of basal autophagy in neural cells causes
neurodegenerative disease in mice. Nature. 441:885–889. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakai A, Yamaguchi O, Takeda T, et al: The
role of autophagy in cardiomyocytes in the basal state and in
response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roy S and Debnath J: Autophagy and
tumorigenesis. Semin Immunopathol. 32:383–396. 2010. View Article : Google Scholar
|
|
8
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosenfeldt MT and Ryan KM: The role of
autophagy in tumour development and cancer therapy. Expert Rev Mol
Med. 11:e362009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Giaccia AJ and Kastan MB: The complexity
of p53 modulation: emerging patterns from divergent signals. Genes
Dev. 12:2973–2983. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levine B and Abrams J: p53: the Janus of
autophagy? Nat Cell Biol. 10:637–639. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zilfou JT and Lowe SW: Tumor suppressive
functions of p53. Cold Spring Harb Perspect Biol. 1:a0018832009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Green DR and Kroemer G: Cytoplasmic
functions of the tumour suppressor p53. Nature. 458:1127–1130.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karantza-Wadsworth V and White E: Role of
autophagy in breast cancer. Autophagy. 3:610–613. 2007. View Article : Google Scholar
|
|
16
|
Levine B: Unraveling the role of autophagy
in cancer. Autophagy. 2:65–66. 2006. View Article : Google Scholar
|
|
17
|
Morselli E, Galluzzi L, Kepp O, et al:
Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta.
1793:1524–1532. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maiuri MC, Tasdemir E, Criollo A, et al:
Control of autophagy by oncogenes and tumor suppressor genes. Cell
Death Differ. 16:87–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar
|
|
21
|
Feng Z, Zhang H, Levine AJ and Jin S: The
coordinate regulation of the p53 and mTOR pathways in cells. Proc
Natl Acad Sci USA. 102:8204–8209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tasdemir E, Maiuri MC and Galluzzi L:
Regulation of autophagy by cytoplasmic p53. Nat Cell Biol.
10:676–687. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maiuri MC, Galluzzi L, Morselli E, et al:
Autophagy regulation by p53. Curr Opin Cell Biol. 22:181–185. 2010.
View Article : Google Scholar
|