Introduction
Antineoplasons are naturally-occurring peptides and
amino acid derivatives found in human blood and urine, first
described by Burzynski in 1976 (1).
Antineoplaston A10 (3-phenylacethyl-amino-2,6-piperidinedione) is
the first chemically-identified antineoplaston and it is partially
hydrolyzed in pancreatic juice to phenylacetylglutamine (PG) and
phenylacethyl isoglutamine (isoPG) when administered perorally. PG
and isoPG are further metabolized to phenylacetate (PN). The
mixture of PG and PN in the ratio of 1:4 has been formulated as
antineoplaston AS2-1. Antineoplastons have been found to control
neoplastic growth. The animal experiment and phase I clinical
toxicological studies (2) have
demonstrated minimal adverse effects of these agents. Thus, it is
postulated that combining these antineoplastons with intensive
chemotherapy may increase antitumor efficacy without an increase of
adverse effects in cancer patients. Clinical studies have described
the antitumor efficacy of antineoplastons for various tumors
including hepatocellular carcinoma, colon cancer and glioma
(3–5).
Sodium phenylbutyrate (PB) and PN (a metabolite of
PB) that is the active ingredient of antineoplaston AS2-1 induce
cytostasis, differentiation and apoptosis by several cellular
mechanisms, in glioma, neuroblastoma, leukemia cells and
adenocarcimoma cells of the breast, colon and lung (6–9). PN
activates the p53 and p21 genes through inhibition of
methyltransferase and fernesylation of the RAS protein (10). PB activates tumor suppressor genes
through inhibition of histone deacetylation (11,12).
PG that is also the active ingredient of antineoplaston AS2-1
normalizes the pattern of genome-wide methylation, stabilizing the
genes, decreasing expression of oncogenes such as AKT-2 and
c-myc (MYCC) and activating tumor suppressors proteins
phosphatase and tensin homologue (PTEN) and integrase
interactor 1 (INI1) and promotes apoptosis (13). Thus, one of the mechanisms
underlying the antitumor effect of antineoplaston AS2-1 is
considered to involve regulation of tumor suppressor gene
expression through demethylation of their promoter sequences and
modification (acetylation) of histones.
Epigenetic alterations of gene function are now well
known to contribute to the tumorigenesis and cancer progression.
Specifically, abnormal promoter region methylation which typically
occurs at CpG islands in known or candidate tumor suppressor genes
contributes to tightly heritable gene silencing and can thereby
cause the loss of gene function. Silencing of genes is a complex
process, which involves methylation of DNA, histone modification
and chromatin remodeling. Two biochemical processes play a very
important part in silencing of the genes: deacetylation of histones
and methylation of DNA (14,15).
Additional new mechanisms of methylation have been proposed
explaining two different issues of DNA methylation in cancer
progression: i) site-specific hypermethylation of promoter
sequences; and ii) genome-wide hypo-methylation is inducing genomic
instability, amplification of oncogenes and silencing of tumor
suppressor genes through RNAi mechanism (16,17).
Methylation of specific genes or methylation patterns of groups of
genes were also found to be associated with responses to
chemotherapeutics and prognosis.
The antitumor mechanisms on the epigenetic
modification of antineoplastons have not yet been clarified. In the
present study, we have investigated the epigenetic modification, in
particular in demethylation effect of the antineoplaston AS2-1 and
clarify sequential increase of transcription and translation to
protein for targeting genes in colon cancer cell lines.
Materials and methods
Antineoplaston AS2-1
Antineoplaston AS2-1 injected formulation (20 g/250
ml) which is a mixture of PG and PN in the ratio of 1:4, was a kind
gift from Dr S.R Burzynski (Burzynski Institute, Houston, TX,
USA).
Cell lines and culture
The HCT116 human colon cancer cell line (p53
wild) was obtained from the ATCC (Rockville, MD, USA). The KM12SM
human colon cancer cell line (p53 mutant) was a kind gift
from Dr Motowo Nakajima (SBI Arapromo K.K., Tokyo, Japan). Each
cell line was cultured in RPMI-1640 medium (Invitrogen, Tokyo,
Japan) supplemented with 10% fetal bovine serum (FBS) at 37°C/5%
CO2 in 75-cm2 culture dishes. The cells were
trypsinized once a week with trypsin/EDTA (0.25%/0.02) and the
medium was changed twice a week.
In vitro cell growth assay
The HCT116 and KM12SM cells were seeded at a density
of 2.5×104 into a 6-well dish containing 4 ml RPMI-1640
medium (Invitrogen, Carlsbad, CA, USA) supplement with 10% FBS and
incubated overnight. The next day, AS2-1 was added to the
subconfluent cultures at concentrations of 0.2, 0.5, 1, 2 and 5
mg/ml. The tumor cells were harvested before AS2-1 treatment as
control, and after AS2-1 treatment at 24, 48 and 72 h. The number
of viable tumor cells was determined by the trypan blue exclusion
test.
Evaluation of methylation profiles at
promoter regions by HELP assay (HpaII tiny fragment enrichment by
ligation-mediated PCR)
HpaII-MspI methylation microarray
(Methyl-Scan DNA chip; Genomictree Inc, Daejeon, South Korea) was
used to investigate the methylation status of 51 kinds of gene
promoter region in HCT116 cells and KM12SM cells before and after
treatment with 2 mg/ml of AS2-1 for 24 h. All chemical reagents
used were purchased from Sigma-Aldrich (Haverhill, MA, USA) unless
otherwise noted. HpaII and MspI restriction enzymes
were obtained from New England Biolabs (Ipswich, MA, USA).
Oligonucleotides were synthesized by Bioneer Inc. (Daejeon, South
Korea).
To avoid incomplete digestion and reduce the
background noise signals, 200 ng of genomic DNA was digested with
excessive units of HpaII and MspI (80 units each) for
6 h at 37°C with enzyme buffers recommended by the suppliers. The
digested samples were inactivated at 65°C for 20 min and then
purified with GeneClean Turbo kit (Qbiogene, Irvine, CA, USA)
according to the manufacturer’s instructions.
Multiplex PCR amplification was done with
un-digested and HpaII-, MspI-digested DNA with
primers sets to label 51 target promoter regions. The sequences of
the gene-specific primer sets used are shown in Table I. During the amplification step,
fluorescent dyes were incorporated into the amplicons; Cy3-dUTP in
MspI-digested targets, Cy5-dUTP in HpaII-digested
targets and undigested samples labeled with Cy5-dUTP by same
multiplex PCR. The amplification was carried out according to the
general guidelines: denaturating at 94°C for 5 min, followed by 30
cycles at 94°C for 30 sec, at 66°C for 45 sec, at 72°C for 45 sec
and a final extension at 72°C for 5 min. To assess PCR adequacy and
ensure scanning, the human interferon-2 gene without HpaII
site was used as a control.
 | Table IPrimer information for
HpaII-MspI-PCR assay. |
Table I
Primer information for
HpaII-MspI-PCR assay.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Amplicon size
(bp) |
|---|
| ASIC2 |
ccaggaggcgaggagagaatg |
cctcctctcctcctcctcca | 178 |
| Apaf-1 |
ccttggggcttggggtgtgt |
cctcaagtcttcgcgggtcg | 201 |
| APC |
caggcaacccagacgtccagag |
ggaagttgatggcagttgacac | 310 |
| AR |
ggacccgactcgcaaactgtt |
gctggcgtggtgcgtccct | 195 |
| BRCA1 |
gcgtgagctcgctgagacttc |
ccttcctgatcctcagcgctt | 302 |
| hCTR |
ggatcagagttggaagagtccc |
cctcccagcgccagcgact | 382 |
| CALCR |
cctgtgtttacgcggcgcttt |
gcagcagaattgatgagagcca | 229 |
| CDH13 |
ccatgcaaaacgagggagcgt |
cgcacagaacgagcggagttc | 258 |
| CDKN2A (p16) |
gcagcatggagccttcggct |
ccaggaggaggtctgtgattac | 292 |
| CDKN2B (p15) |
ccttggcccagctgaaaacg |
gcactctctccttcctaggag | 304 |
| CFTR |
cctccagcgttgccaactgg |
cgtctgggctcaagctccta | 443 |
| COMT |
cccattgctctgtgcagcct |
ggctgggtgccttgtctaag | 202 |
| DAPK1 |
ccactcactccctagctgtgtt |
cccagttgctcgaggcactgc | 224 |
| EDN1 |
ggtacacaggccatataggaac |
ccgaatccctgggcatcagg | 620 |
| EBR |
ggagggaacagcggtttccaa |
cgtaacgggaggaatacagac | 284 |
| EPHA3 |
cctgtcccatgggcgacg |
gctggtgcagagggcagtg | 295 |
| EPO |
gcagcccccatgacccaca |
gctgttatctgcatgtgtgcgt | 175 |
| ESR |
cctggatccgtctttcgcgtt |
gcagggtgcagaccgtgtcc | 510 |
| FHIT |
ctccctccctctgcctttcat |
ggcgatcccaccctgagacc | 217 |
| H19 |
gccatgtgcaaagtatgtgcag |
cctggacagttccagcacac | 228 |
| Heparanase |
gggagaggaagggatgaatact |
ggtcacgttcacttacgaaatca | 273 |
| hMLH1 |
cgggcagtacctctctcagcaac |
ggcttgtgtgcctctgctgagg | 528 |
| HTR1B |
gggagcttccttggccagga |
ctctgccctccctctcttttc | 434 |
| IL-8 |
ggaagtgtgatgactcaggttt |
ggctcttgtcctagaagcttgt | 195 |
| JunB |
ggaaacgacgccaggaaagct |
gcagcgagcggcgagctct | 174 |
| LAMA5 |
ggcacaggctgactcatgtgt |
gcttgcaggctgaccgcct | 192 |
| LDHB |
gggagtgtgcacacttgagc |
ctaccaggagagagaaggctc | 237 |
| BLT1 |
gtgagcgccatcgtgcttgc |
caccactttcagctgagggg | 332 |
| LRP2 |
cgtgtgcacgtgtgagtgtg |
cctctgctagcgaacgctcc | 485 |
| MAGE1 |
gcagagagagagtcttggctttc |
cttgactgccgaccagtcctg | 501 |
| MDR3 |
cctaggagtactcacttcagg |
cctctgcttctttgagcttgg | 230 |
| MGMT |
ccgtttgcgacttggtgagt |
ggaaaggctgggcaacacctg | 199 |
| MTHFR |
gctgcctgccccctgatgc |
ccccaggcaccaccactcc | 346 |
| MUC2 |
ggttggtcctcccagcgtaa |
cctggcaggagggtaggag | 239 |
| PGR |
ggtagggaggggctttggg |
ccagcgagcggcaagtggg | 579 |
| PIK3CG |
ccctctggggcattcattacta |
ggaagcactacagcccttcag | 139 |
| PLS3 (TLS3) |
cacccagttgatgtgacaggc |
gctccaactgaaatttctccga | 393 |
| PTGS2 |
cccatccaaggcgatcagtc |
ggtaggctttgctgtctgagg | 471 |
| RAR-b |
gtgacagaagtagtaggaagtga |
ccaggcttgctcggccaatc | 338 |
| RB1 |
ggatagggatgaggcccaca | cgtcccctgagaaaa
accgg | 341 |
| S100A2 |
ccacagttctctcattccagc |
ctcaggattctttttgcagcaac | 578 |
| SHP1 |
gctctgcttctcttcccttgc |
gggactaagcctcagatgcag | 193 |
| SKT11 (LKB1) |
cgaggacgaagttgaccctg |
ggacccagggtcctggagt | 324 |
| NIS |
cccagtccagggctgaaagg |
ctccctgggttaggaatctatg | 468 |
| HLTF |
ggagacggcgtcgacgtct |
cgctgagtgggatgacaagag | 181 |
| SRBC |
gcagtggagacctgaaacagg |
ctggctgcactacggtcagg | 286 |
| TFF1 |
ctcagatccctcagccaagata |
cgagtcagggatgagaggcc | 229 |
| TP73 |
gcctttggcgccaaagacagc |
cgaaaccgcttagtaaccaactc | 308 |
| TUSC3 (N33) |
ccttcatcatccaagaaggcatt |
cgaggacgcagagtaggaga | 295 |
| VHL |
cgagttggcctagcctcgc |
cgtcttcttcagggccgtac | 311 |
| WT1 |
gctgctgagtgaatggagcg |
gggtgaatgagtaggtgggag | 293 |
| IFN (control) |
gcagctgcagcagttccaga |
tgctcatgagttttccctggt | 221 |
After PCR amplification, all the amplicons were
mixed and purified by using QIAquick PCR cleanup kit (Qiagen K.K.,
Tokyo, Japan). The hybridization mixture (100 μl) contained the
amplified Cy5 and Cy3-labeled cDNA, 3.5X SSC, 5 μg of salmon sperm
DNA and 0.2% sodium dodecyl sulfate. It was heated for 2 min at
95°C and immediately applied onto Methyl Scan DNA chip. The arrays
were incubated at 65°C for 4 h in an eight-well platform
hybridization chamber (Genomictree). To determine the methylation
pattern, the hybridized microarray was imaged by an Axon 4000B
scanner (Axon Instruments, Inc., Union City, CA, USA). The signal
intensities were measured and analyzed by using GenePix Pro
(version 4.0) software. If the signal intensity of HpaII
amplicon is 2-fold greater than that of MspI amplicon, the
target region was considered to be methylated, while <2-fold was
considered to be unmethylated (minus). Moreover, the methylated
status was categorized into 3 degrees, high priority (three plus),
middle priority (two plus) and low priority (plus).
Real-time RT-PCR
To clarify the reinforcement of a transcription
activity of the methylate normalized gene by AS2-1, real-time
RT-PCR (relative quantitative) for p15 messenger RNA in
HCT116 cells were evaluated. The HCT116 cells were seeded at a
density of 2.5×104/ml into a 6-well dish containing 4 ml
RPMI-1640 medium (Invitrogen) supplement with 10% fetal bovine
serum (FBS) and incubated overnight. The next day, AS2-1 was added
to the subconfluent cultures at concentrations of 0.2, 1 and 2
mg/ml. The tumor cells were harvested before AS2-1 treatment as
control, after AS2-1 treatment at 12, 24 and 48 h.
Isolated RNA was controlled for quality by 2%
agarose gel separation and ethidium bromide staining. RNA was
quantified by spectrophotometry. Complementary DNA (cDNA) was
synthesized using 2 μg of total RNA. The 20 μl reverse
transcription reaction consisted of 2 μl 10X RT buffer, 0.5 mM each
dNTP, 1 μM Oligo-dT primers and 4 U Omniscript reverse
transcriptase (Qiagen K.K). The reverse transcription reaction was
incubated for 1 h at 37°C and then at 93°C for 15 min. Real-time
PCR analysis was performed using the ABI Prism 7700 sequence
detection system (Applied Biosystems, Foster City, CA, USA) as
previously described (18).
Briefly, reactions were performed in a 96-well optical reaction
plated on cDNA equivalent to 50 ng of DNase-digested RNA in a
volume of 25 μl, containing 12.5 μl of TaqMan Universal Master Mix
and optimized concentrations of carboxy fluorescein (FAM)-labeled
probe and forward and reverse primers following the manufacturer’s
protocol. All primers and FAM-labeled probes for mouse p15
(forward, 5′-TCTGCAGCTGGATCTGGTCC-3′ and reverse, 5′-TCCT
GAAAGGTAGAGGGCCC-3′) and GAPDH (forward, 5′-CA
TCTCCTCCCGTTCTGCC-3′ and reverse, 5′-GTGGTGC AGGATGCATTGC-3′) were
obtained from Applied Biosystems. The mRNA expression of p15
was normalized to the level of GAPDH mRNA. Data were
processed by the RQ Manager 1.2 software.
Western blot analysis
The cells were cultured as previously described. The
cells were directly lysed in a sample buffer (0.5 M Tris-HCl, pH
6.8, 10% glycerol, 10% SDS, 6% mercaptoethanol, 0.05% bromophenol
blue). Protein (10 μg) was separated on 4% SDS-PAGE gel at 120 V
for 1.5 h. After electrophoresis, the proteins were transferred to
a PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA) at 100 V
for 60 min. The membrane was blocked in PBS containing 0.1%
Tween-20 (FBS-Tween) with 5% skimmed milk at room temperature for
60 min, subsequently incubated with anti-p15 mouse monoclonal
antibody (Abcam, Cambridge, UK) at 4°C overnight. After washing
with PBS-Tween, the membrane was incubated with a horseradish
peroxidase-conjugated goat anti-rabbit IgG or anti-mouse IgG
secondary antibody (Santa Cruz Biotechnology, Dallas, TX, USA). The
membrane was washed with PBS-Tween, and the signal was detected
using an ECL detection kit (Amersham Pharmacia Biotech Inc.,
Piscataway, NJ, USA). A mouse monoclonal antibody against β-actin
(Sigma-Aldrich, St. Louis, MO, USA) was used to control for protein
loading. The amount of each protein was quantified as ratio to
actin. Quantification of band densities was performed using the
public domain NIH Image software.
Results
Cell growth inhibition by antineoplaston
AS2-1
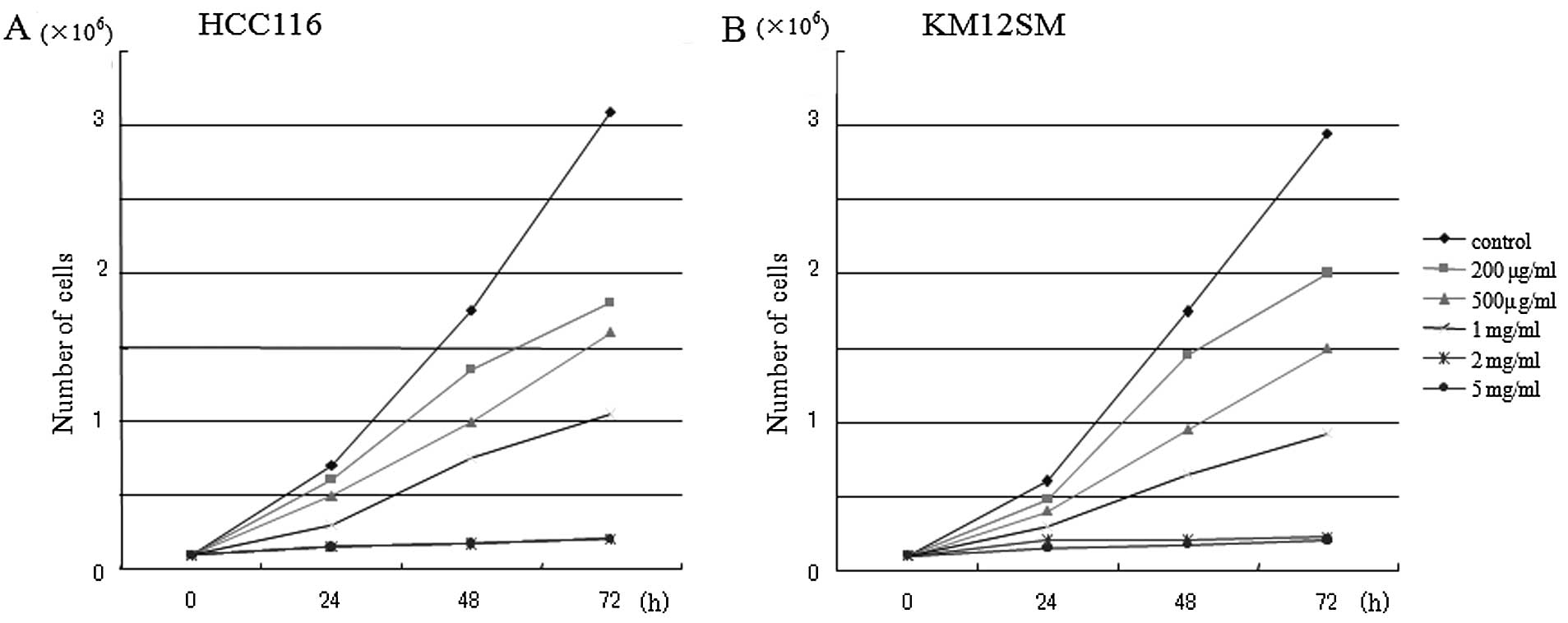
Antineoplaston AS2-1 inhibited the cell growth of
KM12SM and HCT116 in a culture in a dosa- and time-dependent manner
(Fig. 1). There was no difference
in cell growth inhibition by AS2-1 between the wild p53
HCT116 and the mutant p53 KM12SM cell lines.
Methylation status
Among the 51 genes, there were 34 methylated genes
in promoter sequence in HCT116 cells. In 19 (56%) of the 34
methylated genes the methylation status was downregulated after
treatment with 2 mg/ml of AS2-1 for 24 h, in particular, the
methylation status of cyclin-dependent kinase inhibitor p15
(INK4B) and estrogen receptor 1 (ESR1) in HCT116 cells
were dramatically downregulated from two plus to minus (Tables II and III). Among the 51 genes, there were 8
methylated genes in KM12SM cells. In 7 (88%) of the 8 methylated
genes in KM12SM cells, the methylation status was downregulated
after treatment with 2 mg/ml of AS2-1 for 24 h, in particular the
methylation status of methylenetetrahydrofolate reductase
(MTHFR) and mucin2 (MUC2) in KM12SM cells was
dramatically downregulated from two plus to minus (Tables IV and V). There was no gene with methylation
status upregulated in both HCT116 and KM12SM cells.
| Table IIMethylation status in HCT116 cells
before and after treatment with AS2-1. |
Table II
Methylation status in HCT116 cells
before and after treatment with AS2-1.
| Apaf-1 | APC | AR | ASIC2 | BLT1 | BRCA1 | CALCA |
| (+) ⇒ (−) | (+)⇒ (−) | (+)⇒ (−) | (−) ⇒ (−) | (+++)⇒ (+++) | (−) ⇒ (−) | (−) ⇒ (−) |
| CDH13 | CFTR | COMT | DAPK | EBR | EDN1 | EphA3 |
| (+)⇒ (+) | (−) ⇒ (−) | (−) ⇒ (−) | (+)⇒ (+) | (+)⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) |
| EPO | ESR1 | FHIT | hCTR | Heparanase | HLTF | hMLH1 |
| (+)⇒ (−) | (++)⇒ (−) | (+)⇒ (−) | (+)⇒ (+) | (+)⇒ (+) | (−) ⇒ (−) | (+)⇒ (−) |
| HTR1B | H19 | IL-8 | JunB | Laminin5 | LDHB | LKB |
| (+)⇒ (−) | (+)⇒ (+) | (−) ⇒ (−) | (+)⇒ (+) | (−) ⇒ (−) | (+)⇒ (−) | (+)⇒ (−) |
| LRP2 | MAGE | MDR3 | MGMT | MTHFR | MUC2 | NIS |
| (+)⇒ (−) | (+)⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (++)⇒ (++) | (++)⇒ (+) | (+)⇒ (+) |
| N33 | PGR | PIK3CG | PTGS2 | P15 | P16 | P73 |
| (+)⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (++)⇒ (−) | (−) ⇒ (−) | (+)⇒ (+) |
| RAR-β | RB1 | SHP1 | SRBC | S100A2 | TFF1 | TLS3 |
| (+)⇒ (−) | (+)⇒ (+) | (+)⇒ (−) | (+)⇒ (−) | (+)⇒ (−) | (−) ⇒ (−) | (+)⇒ (+) |
| VHL | WT1 | | | | | |
| (+)⇒ (+) | (+)⇒ (+) | | | | | |
| Table IIISummary of methylation status in
HCT116 cells. |
Table III
Summary of methylation status in
HCT116 cells.
| Before
treatment | After
treatment | |
|---|
|
| |
|---|
| Status | n | Status | n | (%) |
|---|
| (−) | 17 | (−) | 17 | 100 |
| (+) | 29 | (+) | 13 | 45 |
| | (−) | 16 | 55 |
| (++) | 4 | (++) | 1 | 25 |
| | (+) | 1 | 25 |
| | (−) | 2 | 50 |
| (+++) | 1 | (+++) | 1 | 100 |
| Table IVMethylation status in KM12SM cells
before and after treatment with AS2-1. |
Table IV
Methylation status in KM12SM cells
before and after treatment with AS2-1.
| Apaf-1 | APC | AR | ASIC2 | BLT1 | BRCA1 | CALCA |
| (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (+++)⇒ (+++) | (−) ⇒ (−) | (−) ⇒ (−) |
| CDH13 | CFTR | COMT | DAPK | EBR | EDN1 | EphA3 |
| (+)⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) |
| EPO | ESR1 | FHIT | hCTR | Heparanase | HLTF | hMLH1 |
| (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (+)⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) |
| HTR1B | H19 | IL-8 | JunB | Laminin5 | LDHB | LKB |
| (−) ⇒ (−) | (+)⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) |
| LRP2 | MAGE | MDR3 | MGMT | MTHFR | MUC2 | NIS |
| (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (++)⇒ (−) | (++)⇒ (−) | (−) ⇒ (−) |
| N33 | PGR | PIK3CG | PTGS2 | P15 | P16 | P73 |
| (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) |
| RAR-β | RB1 | SHP1 | SRBC | S100A2 | TFF1 | TLS3 |
| (−) ⇒ (−) | (+)⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) | (−) ⇒ (−) |
| VHL | WT1 | | | | | |
| (−) ⇒ (−) | (+)⇒ (−) | | | | | |
| Table VSummary of methylation status in
KM12SM cells. |
Table V
Summary of methylation status in
KM12SM cells.
| Before
treatment | After
treatment | |
|---|
|
| |
|---|
| Status | n | Status | n | (%) |
|---|
| (−) | 43 | (−) | 43 | 100 |
| (+) | 5 | (+) | 0 | |
| | (−) | 5 | 100 |
| (++) | 2 | (++) | 0 | |
| | (+) | 0 | |
| | (−) | 2 | 100 |
| (+++) | 1 | (+++) | 1 | 100 |
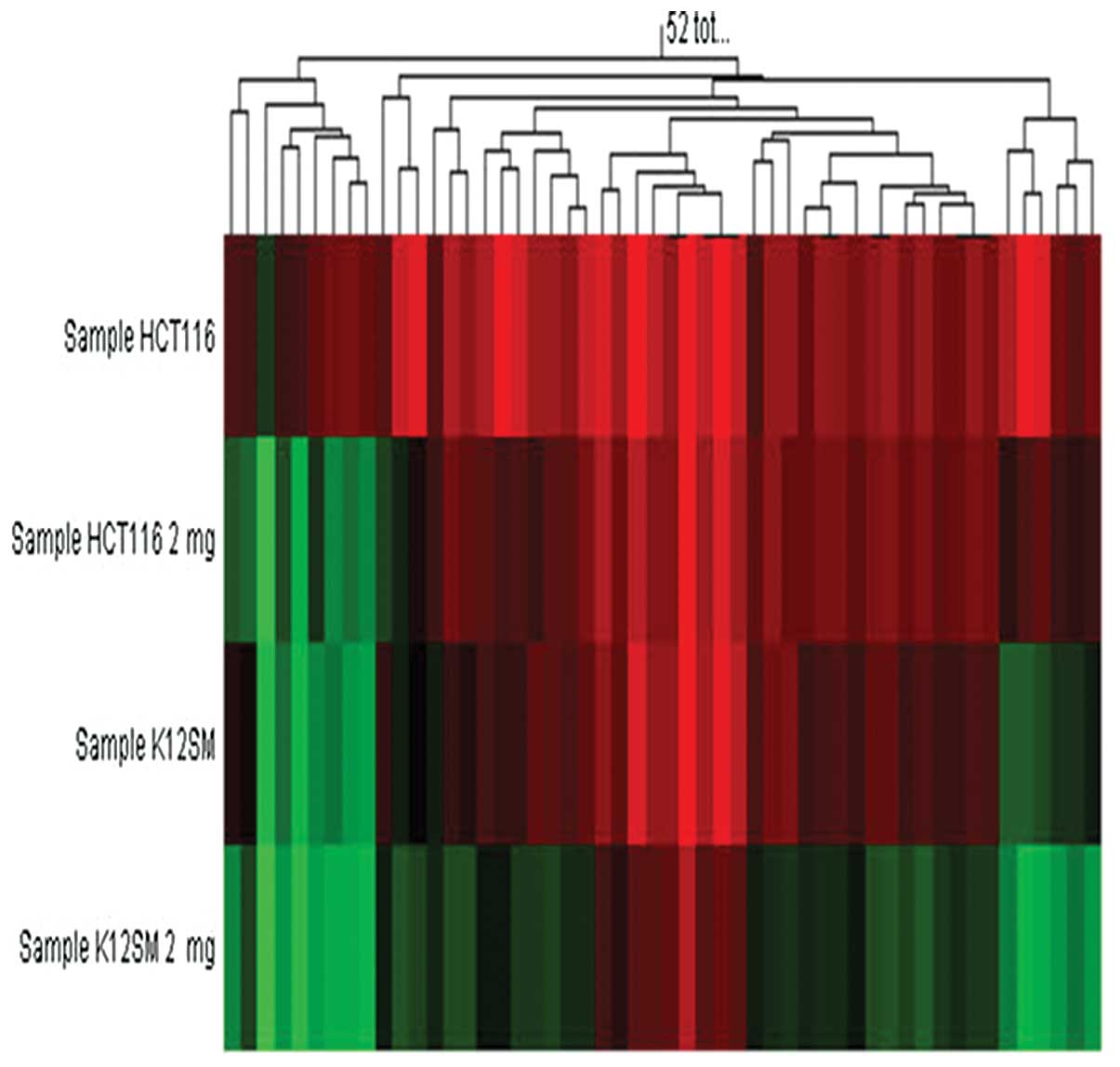
Of interest, in between the cell lines there was a
difference in demethylation effect of AS2-1 in the 8 genes which
were methylated in both HCT116 cells and KM12SM cells. Among the 8
genes, there was only one gene (MUC2) of which methylation
status was downregulated after treatment with AS2-1 in HCT116
cells, whereas in 7 of the 8 methylated genes in KM12SM cells, the
methylation status were downregulated by AS2-1 (Fig. 2).
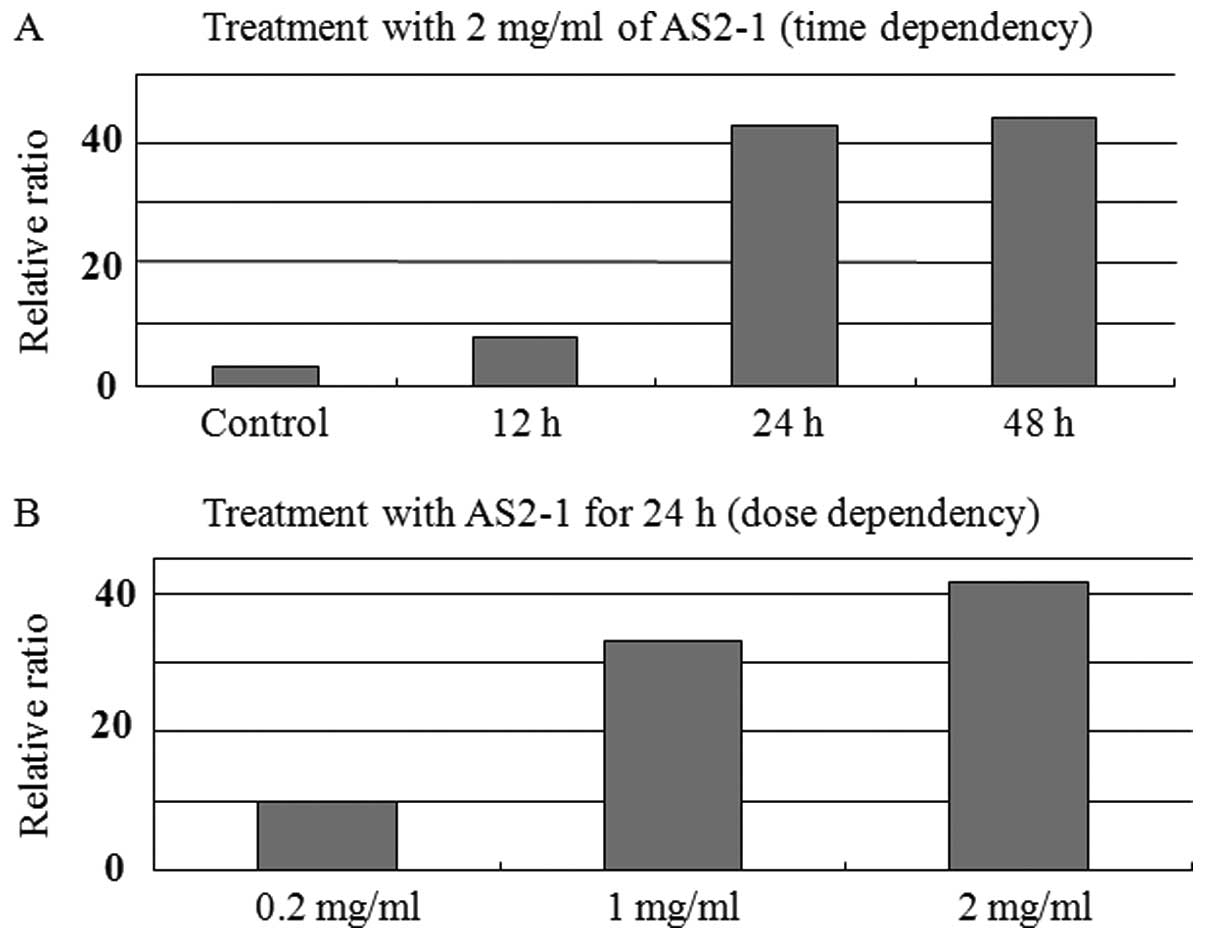
Transcription of p15 mRNA
Real-time RT-PCR analysis revealed that AS2-1
upregulated the expression of p15 mRNA in a time- and
dose-dependent manner in HCT116 cells (Fig. 3).
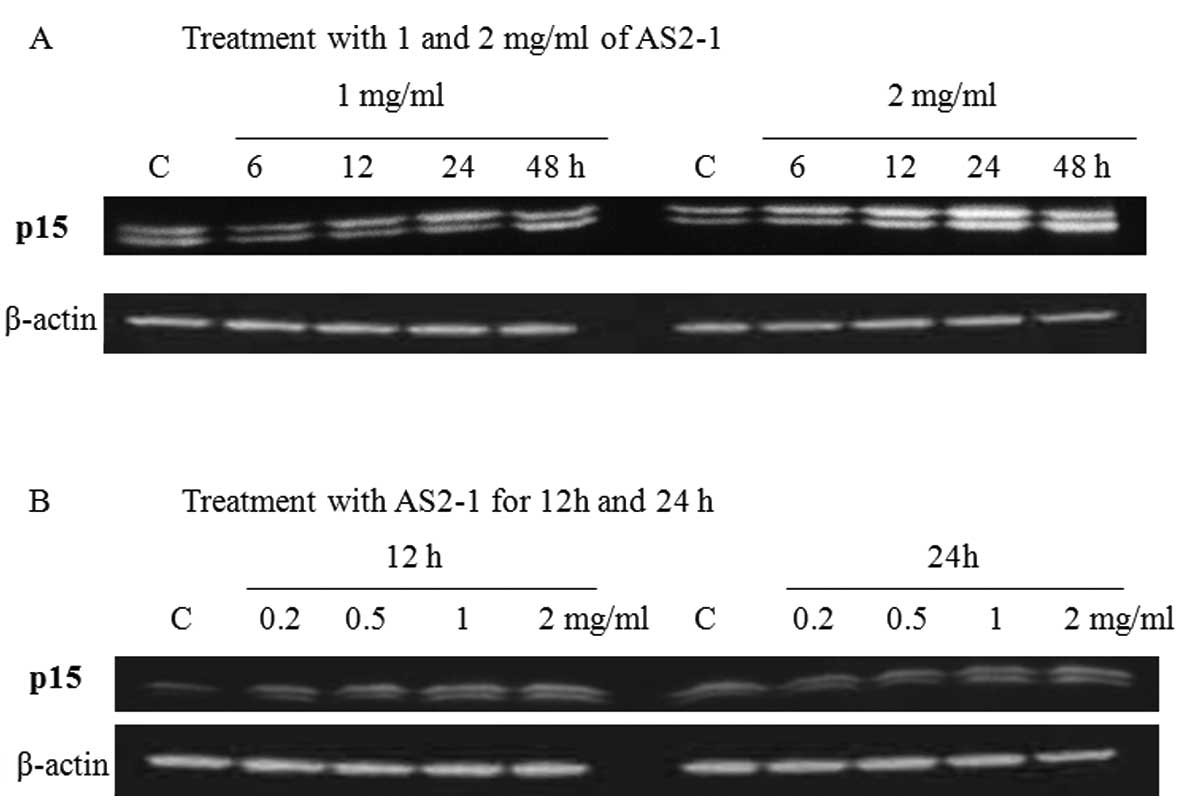
Translation of p15 protein
Western blot analysis revealed that AS2-1
upregulated the expression of p15 protein in a time- and
dose-dependent manner in HCT116 cells (Fig. 4).
Discussion
Burzynski found compounds in the blood and urine of
healthy adults that were absent in blood and urine of cancer
patients, as first described in 1976 (1). He proposed the hypothesis that these
peptide fractions could trigger normal differentiation and
apoptosis in neoplastic cells and their deficiency in cancer
patients contributed to disease progression. These peptide
fractions were termed ‘antineoplastons’.
Re-expression of abnormally silenced suppressor
genes is being investigated for potential clinical applications.
Based on animal experiments and human observations, the drugs which
can restore normal gene expression in aging should inhibit
insulin-like growth factor 1 (IGF-1)/AKT and RAS pathways and
provide proper anticancer defense through normal activity of tumor
suppressors p53 and p21(19). It has also been introduced that a
group of amino acid derivatives and organic acids activate the
tumor suppressors p53, p21, PTEN and
INI1 and decrease overexpression of RAS and
AKT-2 and MYCC oncogenes (20).
The current theory of the mechanisms of action of
antineoplastons is that they function as ‘molecular switches’,
turning on tumor suppressor genes and turning off oncogenes. It has
been suggested that AS2-1 and PN activate tumor suppressor genes
p53 and p21 through demethylation of their promoter
sequences (21). In the present
study, the methylation chip analyses revealed that AS2-1
downregulated methylation status at promoter sequences in various
genes including well-known tumor suppressor genes and the
candidates for tumor suppressor gene in both p53 wild HCT116
and p53 mutant KM12SM colon cancer cells. The results
suggest that AS2-1 has a potent effect to normalize
hypermethylation at promoter regions independently of p53
mutation. Of interest, the demethylation effect was seen more
completely in the KM12SM cells (8 methylated genes) than in the
HCT116 cells (34 methylated genes). The difference in demethylation
effect in the same genes between the two cell lines suggests that
the demethylation effect of AS2-1 may depend on the cell type not
on the genes. Cytidine analogs such as 5-azacytidine (azacitidine)
and 5-azadeoxycytidine (decitabine) are the most commonly used
demethylating agents. These compounds work by binding to DNA
methyltransferases that catalyse the methylation reaction and
titrate out DNA methyltransferases (22). If the demethylation effect of AS2-1
depends on activity of DNA methyltransferases, it is suspected that
AS2-1 may demethylate at CpG islands of promoter region more
completely in tumor cells with a low DNA methyltransferase
activity.
One of the dramatically demethylated genes after
treatment with AS2-1 in HCT116 cells was the tumor suppressor gene,
cyclin-dependent kinase inhibitor p15 (INK4B).
Sequentially the expression of p15 mRNA and p15 protein was
upregulated by treatment with AS2-1. The results indicate that
treatment with AS2-1 leads to re-expression of p15 mRNA and
p15 protein. This gene is localized to a region on chromosome 9p21
frequently deleted in human tumors. p15 gene is an important
mediator of cell cycle control, especially in pathways stimulated
by TGF-β (23), raising the issue
of whether inactivation of one or both of the genes and
cyclin-dependent kinase inhibitor p16 (INK4A) with
homozygous deletion involving the nearby p15 is necessary,
because both proteins are inhibitors of cyclin-dependent kinase 4
(INK4 family) (24,25) and are highly homologous throughout
their coding sequence (26). It has
been reported that the p15 gene is commonly inactivated in
association with promoter region hypermethylation involving
multiple sites in a 5′-CpG island in glioma, leukemia and
myelodysplastic symdromes. In other tumors, including lung, head
and neck, breast, prostate and colon cancer, inactivation of
p15 occurs rarely and with concomitant inactivation of
p16. It has been shown that aberrant methylation of
p15 is associated with transcriptional loss of this gene and
treatment with the demethylating agent decitabine leads to
reactivation of p15, inducing growth arrest and apoptosis in
myeloid cell lines (27). AS2-1 may
activate silenced tumor suppressor p15 by demethylation and
sequentially upregulates the expression of p15 protein in colon
cancer HCT116 cells.
In conclusion, antineoplaston AS2-1 may normalize
hypermethylation status at the promoter region in various tumor
suppressor genes of which expression is silenced in colon cancer.
Then, AS2-1 activates the gene transcription and protein
translation, resulting in differentiation, cell cycle arrest and
apoptosis in cancer cells.
Acknowledgements
The present study was supported by a grant-in-aid
for Scientific Research (C) (no. 13671364) and a grant-in-aid for
Young Scientist (B) (no. 14770672) from the Ministry of Education,
Culture, Sports, Science and Technology of Japan.
References
|
1
|
Burzynski SR: Antineoplastons: biochemical
defense against cancer. Physiol Chem Phys. 8:275–279.
1976.PubMed/NCBI
|
|
2
|
Tsuda H, Hara H, Eriguchi N, Nishida H,
Yoshida H, Kumabe T and Sugita Y: Toxicological study on
antineoplaston A-10 and AS2-1 in cancer patients. Kurume Med J.
42:241–246. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsuda H, Sata M, Kumabe T, Uchida M and
Hara H: The preventive effect of antineoplaston AS2-1 on HCC
recurrence. Oncol Rep. 10:391–397. 2003.PubMed/NCBI
|
|
4
|
Ogata Y, Tsuda H, Matono K, Kumabe T,
Saitsu H, Hara H, Akagi Y, Araki Y, Sata M and Shirouzu K:
Long-term survival following treatment with antineoplastons for
colon cancer with unresectable multiple liver metastasis: report of
a case. Surg Today. 33:448–453. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burzynski SR, Weaver RA, Lewy RI, Janicki
TJ, Jurida GF, Szymkowski BG, Khan MI and Bestak M: Phase II study
of antineoplaston A10 and AS2-1 in children with recurrent and
progressive multicentric glioma. a preliminary report. Drugs R D.
5:315–326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liau MC, Lee SS and Burzynski SR:
Hypomethylation of nucleic acids: a key to the induction of
terminal differentiation. Int J Exp Clin Chemother. 2:187–199.
1989.
|
|
7
|
Samid D, Shack S and Myers CE: Selective
growth arrest and phenotypic reversion of prostate cancer cells in
vitro by nontoxic pharmacological concentrations of phenylacetate.
J Clin Invest. 91:2288–2295. 1993. View Article : Google Scholar
|
|
8
|
Liu L, Shack S, Stetler-Stevenson WG,
Hudgins WR and Samid D: Differentiation of cultured human melanoma
cells induced by the aromatic fatty asid phenylacetete and
phenylbutyrate. J invest Dermatol. 103:335–340. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Melchior SW, Brown LG, Figg WD, Quinn JE,
Santucci RA, Brunner J, Thüroff JW, Lange PH and Vessella RL:
Effect of phenylbutyrate on proliferation and apotosis in human
prostate cancer cells in vitro and in vivo. Int J
Oncol. 14:501–508. 1999.PubMed/NCBI
|
|
10
|
Shack S, Chen LC, Miller AC, Danesi R and
Samid D: Increased susceptibility of ras-transformed cells to
phenylacetate is associated with inhibition of p21ras
isoprenylation and phenotypic reversion. Int J Cancer. 63:124–129.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu KH, Weng LJ, Fu S and Gore SD:
Augmentation of phenylbutyrate-induced differentiation of myeloid
leukemia cells using all-trans retinoic acid. Leukemia.
13:1258–1265. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marks PA, Richon VM and Rifkind RA:
Histone deacetylase inhibitors: inducers of differentiation or
apoptosis of transformed cells. J Natl Cancer Inst. 92:1210–1216.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Waldbillig R and Burzynski SR: Mechanism
of action, uptake, and gene array studies on the antineoplastic
agent phenylacetylglutamine (PG) in human glioma cells U-87. Neuro
Oncol. 5:3092003.
|
|
14
|
Goldberg AD, Allis CD and Bernstein E:
Epigenetics: a landscape takes shape. Cell. 128:635–638. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bernstein BE, Meissner A and Lander ES:
The mammalian epigenome. Cell. 128:669–681. 2007. View Article : Google Scholar
|
|
16
|
Sieber OM, Heinimann K and Tomlinson IP:
Genomic instability - the engine of tumorigenesis? Nat Rev Cancer.
3:701–708. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schramke V and Allshire R: Hairpin RNAs
and retrotransposon LTRs effect RNAi and chromatin-based gene
silencing. Science. 301:1069–1074. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pickett MA, Everson JS, Pead PJ and Clarke
IN: The plasmids of Chlamydia trachomatis and
Chlamydophila pneumoniae(N16): accurate determination of
copy number and the paradoxical effect of plasmid-curing agents.
Microbiology. 151:893–903. 2005.PubMed/NCBI
|
|
19
|
Vousden KH and Lu X: Live or let die: the
cell’s response to p53. Nat Rev Cancer. 2:594–604. 2002.
|
|
20
|
Burzynski SR: The present state of
antineoplaston research (1). Integr Cancer Ther. 3:47–58. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burzynski SR: Aging: gene silencing or
gene activation? Med Hypotheses. 64:201–208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Holliday R and Ho T: DNA methylation and
epigenetic inheritance. Methods. 27:179–183. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hannon GJ and Beach D:
p15INK4Bis a potential effector of TGF-β-induced cell
cycle arrest. Nature. 371:257–261. 1994.
|
|
24
|
Serrano M, Hannon GJ and Beach D: A new
regulatory motif in cell-cycle control causing specific inhibition
of cyclin D/CDK4. Nature. 366:704–707. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jen J, Harper W, Bigner SH, Bigner DD,
Papadopoulos N, Markowitz S, Willson JKV, Kinzler KW and Vogelstein
B: Deletion of p16 and p15 genes in brain tumors. Cancer Res.
54:6353–6358. 1994.PubMed/NCBI
|
|
26
|
Kamb A, Gruis NA, Weaver-Feldhaus J, Lie
Q, Harshman K, Tavtingian SV, Stockert E, Day RS, Johnson BE and
Skolnick MH: A cell cycle regulator potentially involved in genesis
of many tumor types. Science. 264:436–440. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Berg T, Guo Y, Abdelkarim M, Fliegauf M
and Lübbert M: Reversal of p15/INK4b hypermethylation in
AML1/ETO-positive and -negative myeloid leukemia cell lines. Leuk
Res. 31:497–506. 2007. View Article : Google Scholar : PubMed/NCBI
|