Introduction
Epidemiological studies have revealed that chronic
inflammation predisposes individuals to colorectal carcinoma
(1) and increases the risk of
carcinogenesis of colorectal tissue (2). Inflammatory cytokines (for example,
IL-6 and INF-α) influence the behavior of colorectal cells in the
inflammatory microenvironment through the JAK-STAT pathway
(3). Signal transducer and
activator of transcription (STAT) proteins play a key role in
determining whether immune responses in the tumor microenvironment
promote or inhibit tumorigenesis. Continuous activation of STAT3
signaling increases tumor cell proliferation, survival and invasion
while the antitumor immunosystem is suppressed by the inflammatory
microenviroment. The persistent activation of STAT3 also mediates
tumor-promoting inflammation (4).
Consequently, STAT3 has an important role in tumorigenesis and may
be a promising target for cancer therapy.
Suppressor of cytokine signaling 3 (SOCS3) is one of
the negative regulators of cytokine signaling that functions via
the JAK/STAT pathway (5). SOCS3
binds the cytokine receptor with high affinity with its Src
homology 2 protein domains and attenuates the activity of JAKs via
its kinase inhibitory region in order to abolish STAT3
phosphorylation (6). In addition,
it has been revealed that SOCS3 is frequently silenced in several
types of tumor cells (7–9).
Our previous studies demonstrated that oncolytic
adenoviral vectors carrying apoptosis-inducing genes induce strong
antitumor activity (10–12). As we deleted the CR2 region of the
vector genome which promotes the native E1A by hTERT, the oncolytic
adenoviral vector was selectively amplified in most tumor cells.
Meanwhile, endogenous major late promoter (MLP) in the adenoviral
genome was found to promote cpp-SCOS3 expression, and the promoter
contributed to the tumor-specific viral replication of adenoviral
vectors and more effectively attenuated tumor cell malignancy
(10).
In the present study, we treated colorectal
carcinoma (CRC) cells with an oncolytic adenovirus that expressed
the SOCS3 gene and found that CRC cell growth was suppressed
by overexpression of SOCS3. Finally, the data suggested that the
recombinant adenovector has high efficacy in inhibiting CRC cell
proliferation and promoting apoptosis in vitro and in
vivo.
Materials and methods
Cell culture
The HEK293 cell line, the control cell line L02, and
CRC cell lines HCT-116, HT-29 and SW620 were purchased from the
Shanghai Cell Collection (Shanghai, China) and were cultured in
RPMI-1640 or DMEM containing 10% fetal bovine serum (FBS;
Gibco-Life Technologies, Grand Island, NY, USA) at 37°C in 5%
CO2. All cells were infected by the adenoviral vectors
which expressed the genes of interest for further examination.
Recombinant adenoviruses
The vectors AdCN305-cppSOCS3, Ad-WT and
AdCN305-HcRed were constructed as previously reported (10–12).
Briefly, human SOCS3 CDS and fibroblast growth factor 4 cpp
sequences were obtained from the cell line by PCR and inserted into
pBS/IRES, and then the pBS/IRES-cppSOCS3 plasmid was constructed.
Subsequently, the IRES-cppSOCS3 fragment was cloned into the pCZ305
plasmid, and the recombinant plasmid pCN305-cppSOCS3 was generated
by the recombination of pCZ305-IRES-cppSOCS3 and pCN103 plasmid in
E coli. CN305-HcRed was obtained followed the above
protocol. The recombinant adenoviruses were amplified in HEK293
cells and purified by cesium chloride gradient ultracentrifugation.
The recombinant adenoviruses were titrated by a plaque assay in
HEK293 cells.
Cell viability assay
Cells were seeded on 96-well plates at a density of
1×104/well in 100 μl complete medium. Twenty-four hours
later, they were infected with a wild-type adenovirus (Ad-wt),
adenovirus CN305 expressing HcRed fluorescence protein
(AdCN305-HcRed) and AdCN305-cpp-SOCS3 at a multiplicity of
infection (MOI) of 10, respectively. Twenty microliters of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma Chemical Co., St. Louis, MO, USA) solution (5 mg/ml) was
added to each well at 24, 48, 72 and 96 h after infection. Plates
were incubated at 37°C for 4 h, and then 150 μl of DMSO was added
to each well and shaken for 10 min. Finally, the absorbance was
read at 595 nm with a DNA-Expert (Tecan).
Total RNA isolation and qRT-PCR
HCT-116, HT-29 and SW620 cell lines were grown to
80% confluence in 6-well plates and then infected by the three
vectors (Ad-WT, AdCN305-HcRed and AdCN305-cppSOCS3). The culture
medium was then replaced by RPMI-1640 medium for 48 h. RNA samples
were then prepared, followed by complimentary DNA (cDNA) synthesis
and qRT-PCR analysis which was performed on an ABI-7400 instrument.
The following primers were employed: SOCS3-F
(5′-gcacaagcacaaaaatccagc-3′) and SOCS3-R
(5′-agaagccaatctgcccctg-3′) for the SOCS3 gene; survivin-F
(5′-ggcatgggtgccccgacgttg-3′) and survivin-R
(5′-cagaggcctcaatcccatggca-3′); for the survivin gene; Bcl-2-F
(5′-tgtggccttctttgagttcg-3′) and Bcl-2-R
(5′-cacttgtggctcagatagg-3′) for the Bcl-2 gene; c-Myc-F
(5′-tggtcttcccctaccctctcaac-3′) and c-Myc-R
(5′-gatccagactctgaccttttgcc-3′) for the c-Myc gene; cyclin D1-F
(5′-ctgtgctgcgaagtggaaaccat-3′) and cyclin D1-R
(5′-ttcatggccaggcgggaagacctc-3′) for the cyclin D1 gene and GAPDH-F
(5′-gacatcaagaaggtggtgaagc-3′) and GAPDH-R
(5′-gtccaccaccctgttgctgtag-3′) for the GAPDH gene. Transcript
abundance was first normalized to the level of GAPDH mRNA and then
compared to each other.
Protein preparation and western blot
analyses
Total cellular proteins were prepared from the cells
under different culture conditions using a previously described
method (9). For western blot
analyses, the sample proteins (50 μg/well) were separated by
electrophoresis in 10% sodium dodecylsulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), and the separated protein bands were
transferred to a polyvinylidene difluoride membrane (Amersham,
Buckinghamshire, UK). After the membrane was blocked with 5%
skimmed milk in TBS-T (10 mM Tris-HCl, pH 8.0, 150 mM NaCl and 0.5%
Tween-20) it was maintained at 4°C overnight, and then rinsed three
times (10 min each time) with TBST, followed by a 3-h incubation at
room temperature with the primary antibodies at appropriate
concentrations. Incubation with HRP-conjugated anti-mouse or rabbit
IgG was carried out for 1 h (Zymed Laboratories Inc., San
Francisco, CA, USA). The bound antibody was detected using the
enhanced chemiluminescence system (Roche GmbH, Mannheim, Germany).
After removing the labeling signal by incubation with stripping
buffer (62.5 mM Tris-HCl, pH 6.7, 100 mM 2-mercaptoethanol, 2% SDS)
at 55°C for 30 min, the membrane was re-detected with other
antibodies one by one following the same experimental procedure
until all of the parameters were examined.
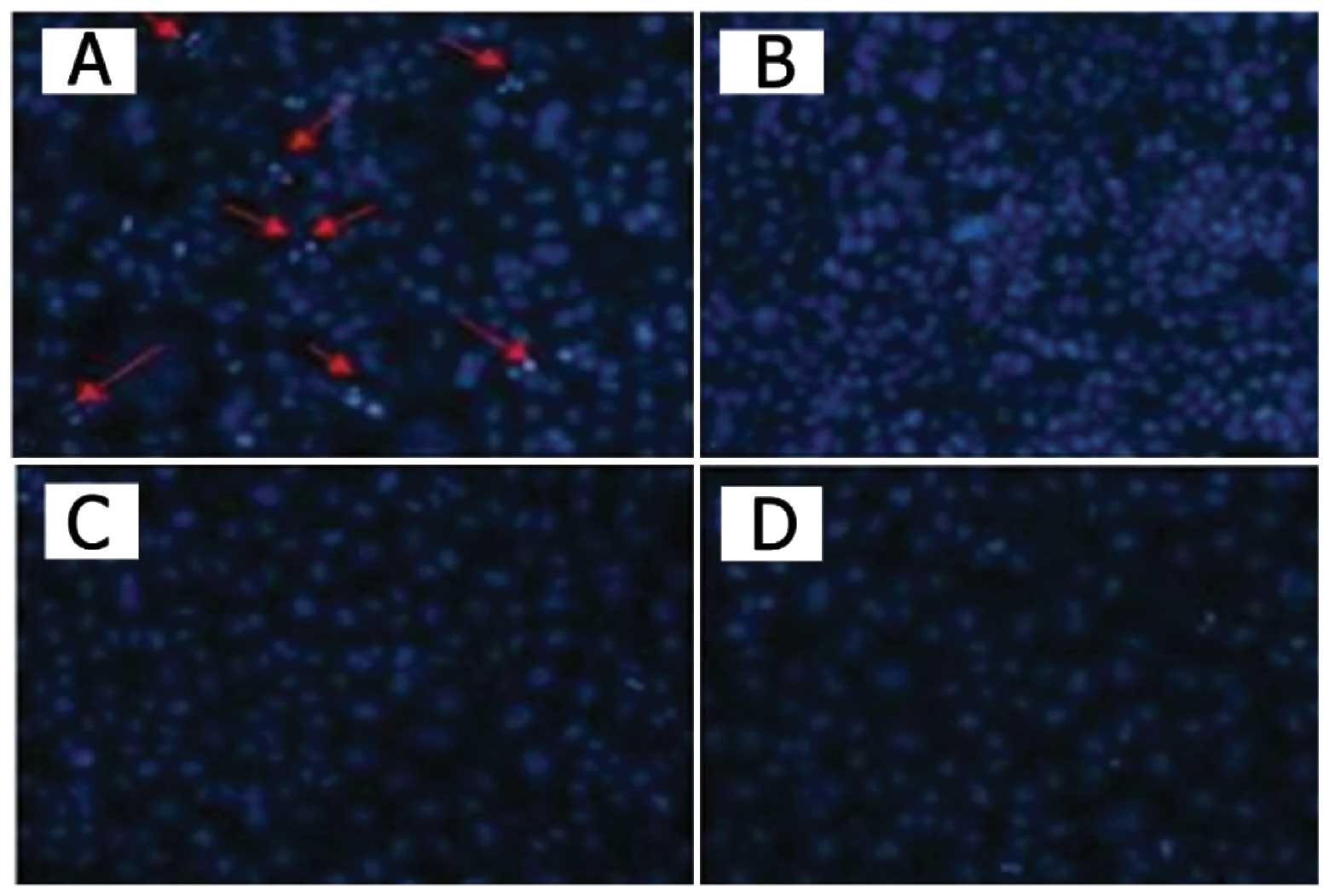
Apoptotic cell staining
The cells seeded in chamber slides were infected
with the recombinant adenoviruses or phosphate-buffered saline
(PBS). After a 48-h infection, the cells were incubated with
Hoechst 33258 (Molecular Probes, Eugene, OR, USA) for 10 min,
washed with PBS twice and observed under a fluorescence microscope
and images were captured.
Tumor xenografts in nude mice
All animals used in these experiments were
maintained at institutional facilities and received humane care
according to the criteria outlined in the Guide for the Care and
Use of Laboratory Animals. Female BALB/c nude mice (4–5 weeks of
age) were obtained from the Animal Research Committee of the
Institute of Biochemistry and Cell Biology (Shanghai, China). Mice
were inoculated subcutaneously with SW620 cells (2×106
for each mouse). When the tumor volume reached 100–150
mm3, the inoculated mice were randomly divided into five
groups. An intratumoral injection of each of the adenoviruses
(5×108 PFU/dose) with 50 μl of PBS was performed once
every other day for a total of 4 times. After therapy, the tumor
size was measured with a vernier caliper every 7 days. The tumor
volume (mm3) was calculated as follows: (length *
width2)/2.
Immunocytochemical staining
Sections from the frozen tumor samples (on day 6
after treatment) were stained with antibodies. Immunohistochemical
staining was performed on the coverslips obtained from each of the
experimental groups. The antibodies against STAT3,
phosphorylated-STAT3 (p-STAT3), Bcl-2 were purchased from Cell
Signaling Technology (Beverly, MA, USA) and were used according to
the manufacturer’s instructions. Briefly, the coverslips were
washed with phosphate-buffered solution (PBS, pH 7.4), incubated
for 10 min in 3% H2O2 and then diluted with
the appropriate primary antibody at 37°C for 60 min in a humidity
chamber, followed by the biotinylated peroxidase-conjugated
streptavidin system (BioGenex Laboratories, San Ramon, CA, USA) and
at last photographed (DP70 digital camera; Olympus, Tokyo, Japan)
under a microscope (BX51; Olympus).
Statistical analysis
The statistical significance of the data was
calculated with an analysis of variance and a one-sided Student’s
t-test with Microsoft Excel. Data were considered to indicate a
statistically significant result at P<0.05.
Results
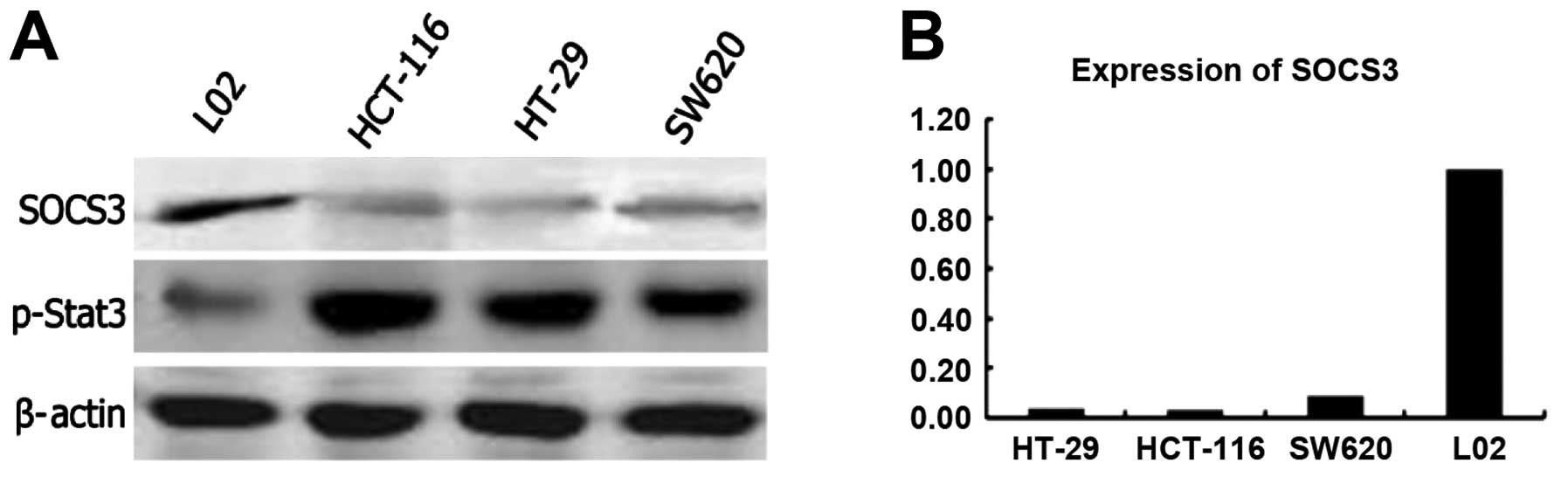
Expression of SOCS3 in the CRC cell
lines
To elucidate the the effect of SOCS3 expression on
CRC cells, we analyzed expression of endogenous SOCS3 in different
CRC cell lines and in a normal cell line (L02) by western blot
analysis (Fig. 1A) and qRT-PCR
(Fig. 1B). The expression of the
SOCS3 transcripts was markedly reduced in the CRC cell lines HT-29,
HCT-116 and SW620. In contrast, a high level of SOCS3 expression
was noted in the L02 normal cell line (Fig. 1A).
Expression and the phosphorylation status
of STAT3 in the CRC cell lines
SOCS3 is a negative regulatory factor of the
JAK/STAT3 pathway in inflammation signaling. We examined the level
of STAT3 expression and the phosphorylation status of STAT3. We
found that the level of STAT3 expression was similar in all of the
tested cell lines (CRC cell lines and the normal cell line).
However, phosphorylation of STAT3 was much higher in the tumor
cells than that in the normal cells (Fig. 1A). These data suggest that loss of
SOCS3 expression correlates with persistent STAT3 phosphorylation
and promotes the growth of tumor cells consistent with previous
observations (13,14).
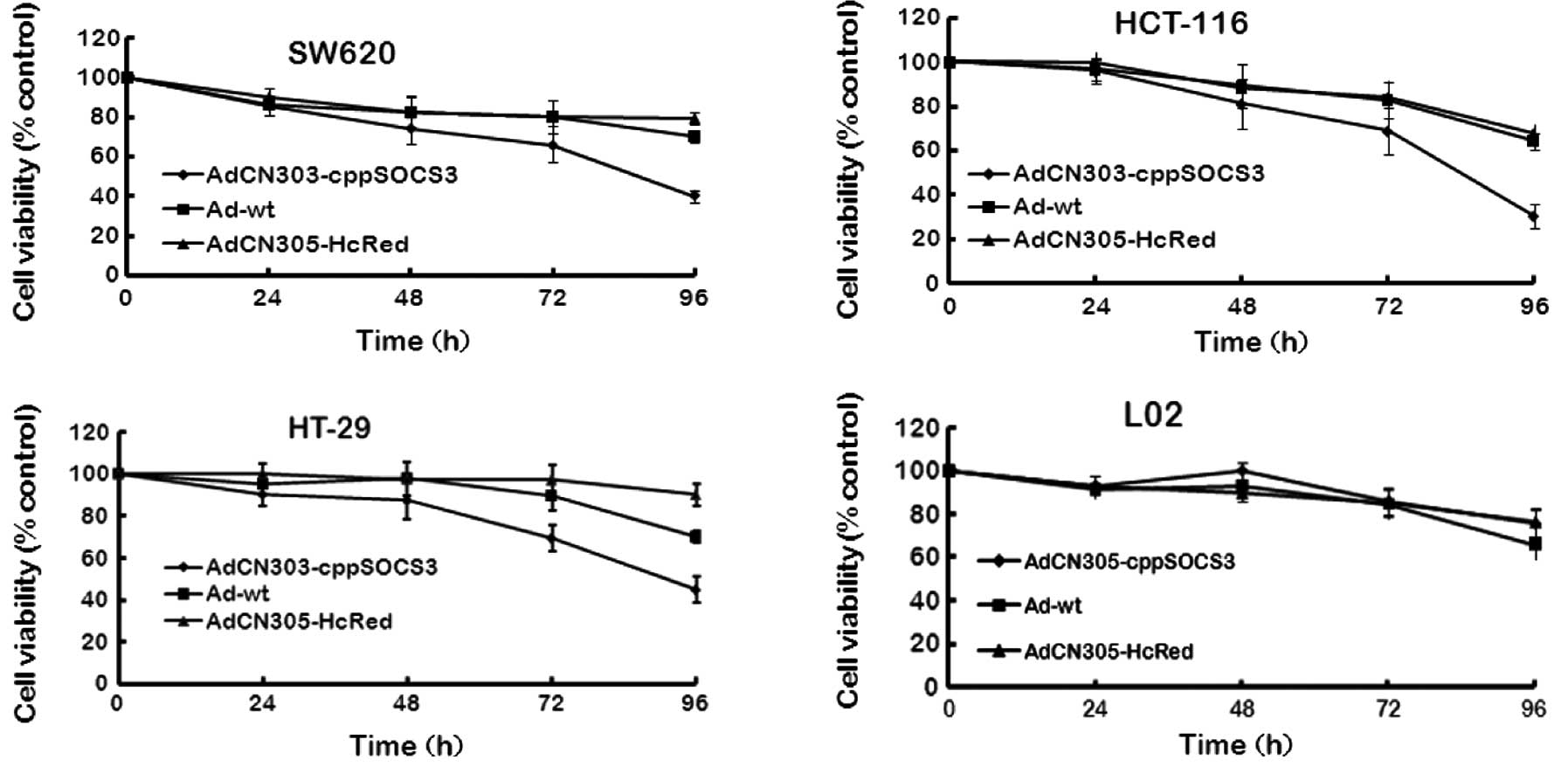
Cytotoxicity induced by the oncolytic
adenoviral vectors in CRC cells
To analyze the antitumor efficacy of recombinant
adenoviruses, three CRC cell lines (HCT-116, HT-29 and SW620) and a
normal cell line (L02) were infected with AdCN305-cppSOCS3,
AdCN305-HcRed or Ad-wt at an MOI of 10 with PBS as a negative
control. Cytotoxicity was determined by MTT assay and Hoechst 33258
staining. As shown in Figs. 2 and
3, control vector AdCN305-HcRed
induced a cytotoxic effect in the tumor cells similar to that
induced by Ad-wt. However, AdCN305-cppSOCS3 markedly reduced the
viability in all three CRC cell lines. Furthermore,
AdCN305-cppSOCS3 induced even higher cytotoxicity in the tumor
cells when compared with that induced by Ad-wt and AdCN305-HcRed.
In addition, AdCN305-cppSOCS3 and AdCN305-HcRed did not induce
obvious cytotoxicity to the normal cells due to the selective
replication ability of the adenoviral vector (Figs. 2 and 3).
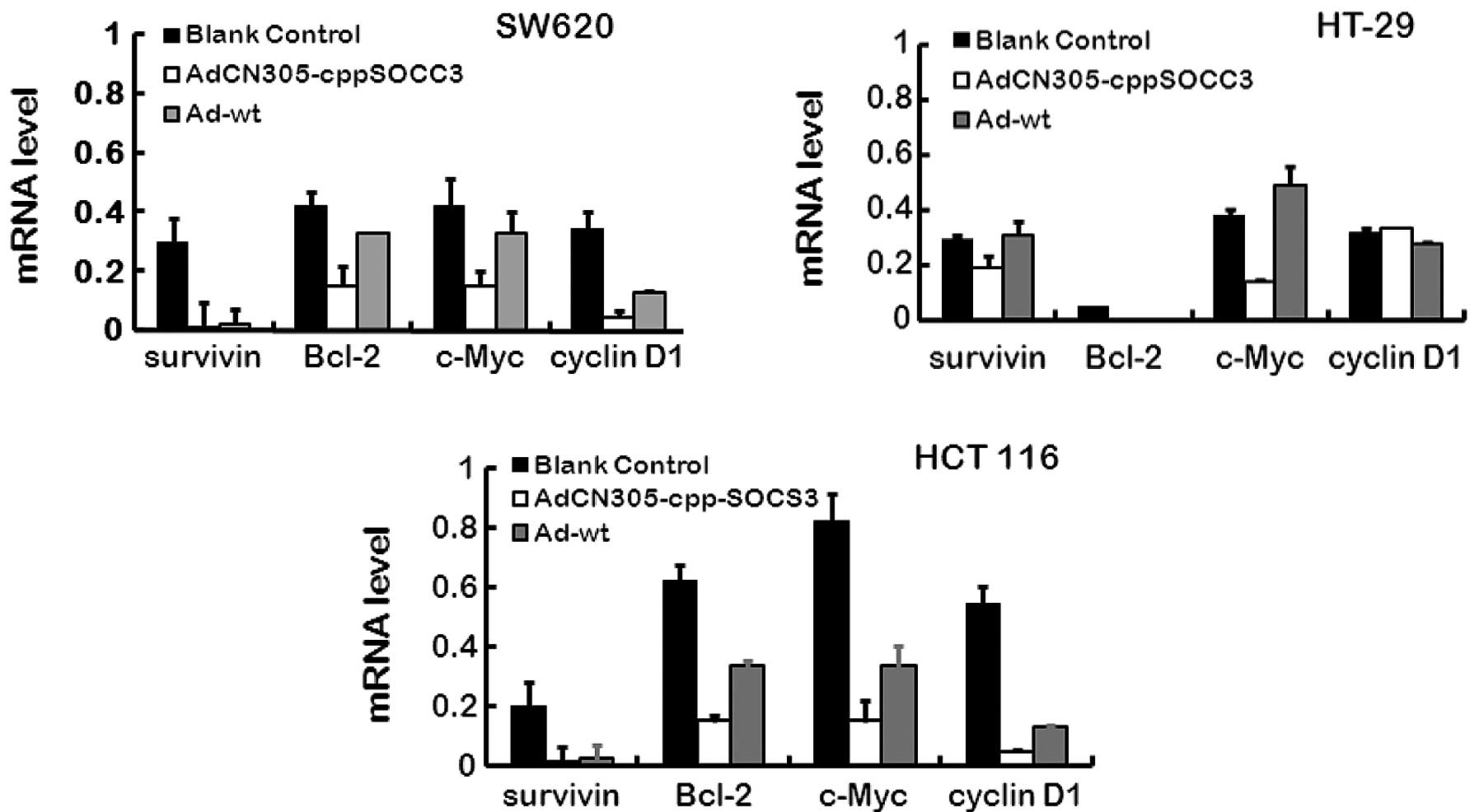
Inhibition of STAT3 phosphorylation and
its downstream targets by overexpression of SOCS3
As previously stated, recombinant oncolytic
adenoviruses harboring the gene cppSOCS3 suppress the growth of
tumor cells in vitro, while the mechanisms of STAT3 activity
which are negatively regulated by SOCS3 are not clear. In our
experiment, in the three CRC cell lines transfected with
AdCN305-cppSOCS3 or Ad-wt, several downstream factors of the
JAK-STAT3 pathway were detection by qRT-PCR. Our results showed
that AdCN305-cppSOCS3 inhibited STAT3 phosphorylation and induced
apoptosis. The factors, survivin, cyclin D1, Bcl-2 and c-Myc are
involved in promoting cancer cell proliferation and apoptosis, and
all of these factors are regulated by the JAK-STAT3 pathway
directly or indirectly. As shown in Fig. 4, all of the above factors exhibited
a decreased expression level as revealed using qRT-PCR, and the
results indicated that expression of SOCS3, the upregulated
negative feedback factor of the JAK-STAT3 pathway, efficiently
downregulated cancer proliferation-associated factors in CRC cells.
These results demonstrated that the SOCS3 protein was a potent
negative regulator of CRC cell growth and induced CRC cell death by
reducing the expression of cancer proliferation-associated
genes.
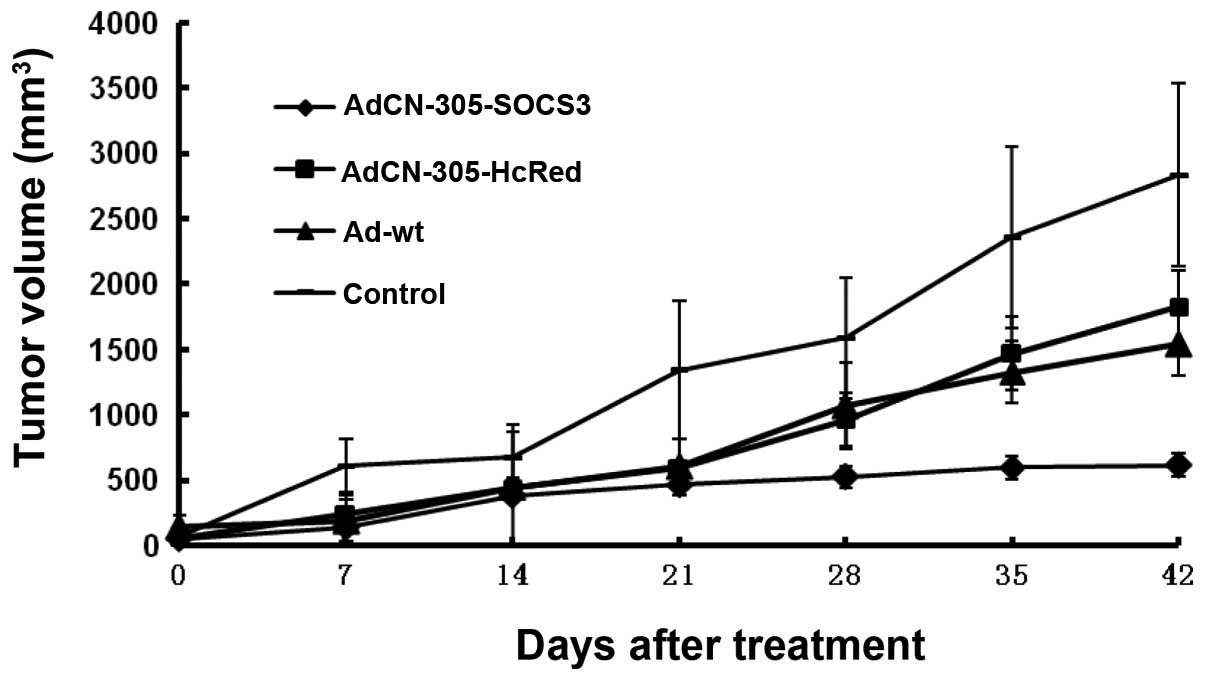
Antitumor activity of the oncolytic
adenoviral vectors in an established tumor animal model
To evaluate the antitumor activity of the
recombinant adenoviruses in vivo, a transplanted CRC mouse
model was established based on implantation of human CRC cells
(SW620) in nude mice. When the tumors reached 100–150
mm3, the animals were treated with an intratumor
injection of AdCN305-cppSOCS3, AdCN305-HcRed, Ad-wt or PBS.
In the control animals that received PBS, the tumors
grew progressively during the course of the experiment. In
contrast, the animals treated with control vector AdCN305-HcRed or
Ad-wt exhibited a significant suppression of tumor development
(P<0.01). Treatment with AdCN305-cppSOCS3 resulted in
significant inhibition of tumor growth in comparison with animals
treated with AdCN305-HcRed, Ad-wt or PBS (P<0.01) (Fig. 5).
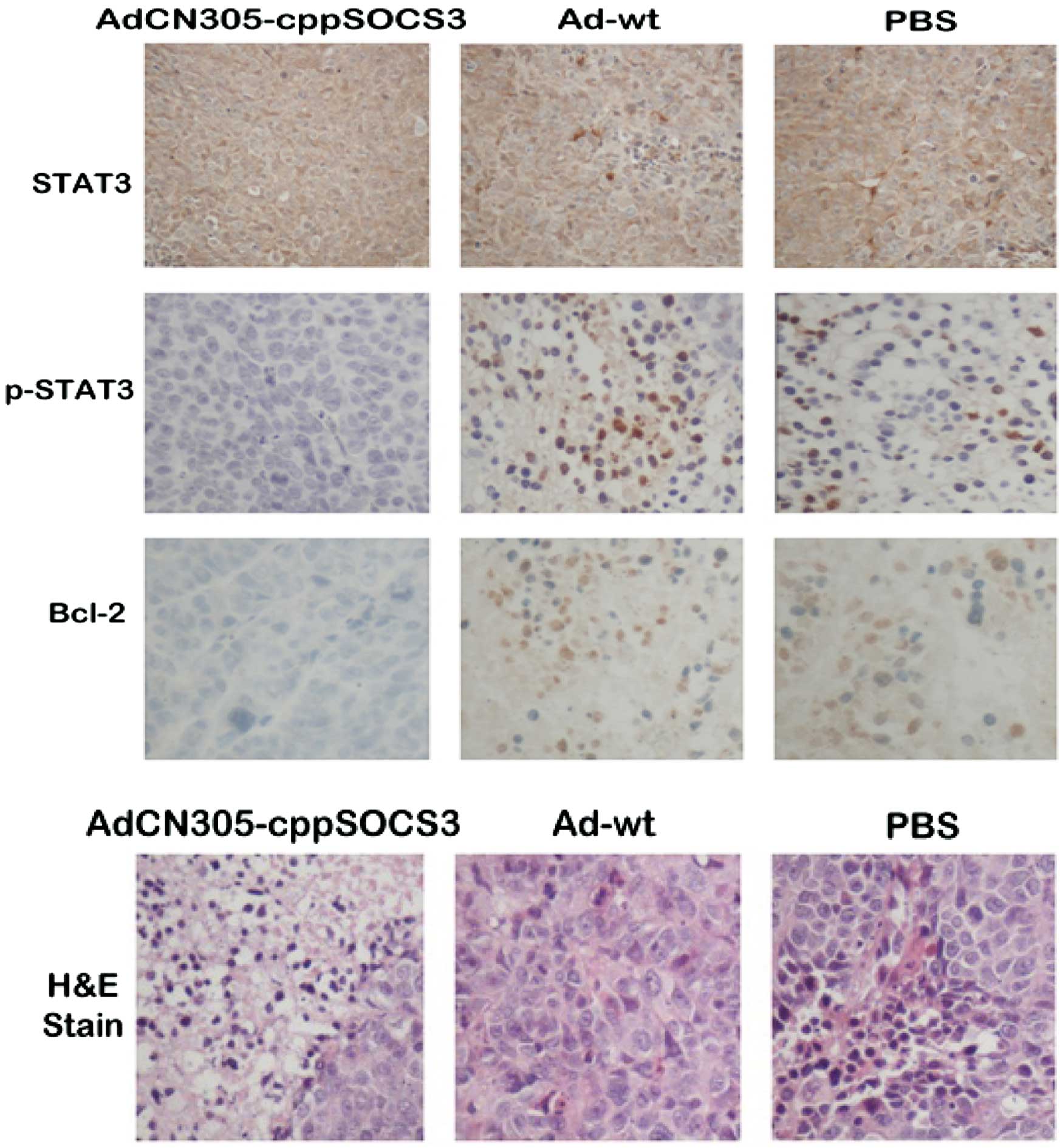
In vivo STAT3 phosphorylation is further
analyzed in transplanted tumor tissues by immunohistochemistry
As shown in Fig. 6,
treatment with AdCN305-cppSOCS3 resulted in marked inhibition of
STAT3 phosphorylation. In addition, histopathological analysis of
tumor tissue sections revealed that treatment with
AdCN305-cpp-SOCS3 resulted in a higher extent of necrosis than that
induced by AdCN305-HcRed, Ad-wt and the PBS control.
Discussion
STAT3, a member of the STAT family, has been proven
to act as a tumorigenesis driver, an oncogene. Researchers have
previously assessed the efficicacy of STAT3 as a therapy for
malignancies. For example, they performed repression of
upstream-related receptors of the pathway (15), dimerization of STAT3 (16), transcription activation by nuclear
entry of protein STAT3 and binding to target cis elements of
the associated genes (17). Various
factors which usually inhibit cancer cell apoptosis and promote
progression of the cell cycle such as c-Myc, survivin and cyclin D1
(7,10,14)
are commonly overexpressed as a results of STAT3 overactivity. Due
to important role that this pathway plays in cancer cell
progression, more and more researchers have focused on JAK/STAT3
signaling. SOCS3, a member of the SOCS family that has a negative
effect on the JAK/STAT pathway, indirectly suppresses the activity
of JAKs and plays an important role in the immune response and
embryonic development (18–21).
In the present study, we employed a selectively
replicating adenoviral vector (AdCN305) expressing SOCS3 which
exhibits strong activity against hepatocarcinoma cells (10). Replication of the AdCN305 vector
system depends on overexpression of hTERT and dysfunction of the
retinoblastoma tumor-suppressor gene in vector-infected cells.
Simultaneously, the SOCS3 gene was fused with the cpp gene
(cell-penetrating peptide), which has the capacity to deliver
peptides, proteins, and oligonucleotides into intact cells
(22,23). In addition, in the backbone of the
adenovector, expression of the SOCS gene was controlled by an
endogenous adenoviral major late promoter (MLP) which restricts
SOCS3 expression within the tumor microenvironment due to
tumor-specific viral replication (24). A previous study showed that this
recombination manipulation of the oncolytic vector did not affect
replication of the virus (10). As
a result, we aimed to ascertain whether overexpression of SOCS3
affects CRC which is believed to be initiated partially by local
inflammation.
Our data suggest that the recombinant vector
effectively suppressed proliferation of CRC cells. In contrast to
the control vectors, AdCN305-cppSOCS3 exhibited strong cytotoxicity
in CRC cells, and this effect was even higher than that induced by
Ad-wt. More importantly, the vectors did not induce irrelevant
cytotoxicity in normal cells as did AdCN305-HcRed (red fluorescence
protein).
The positive effect induced by AdCN305-cppSOCS3 may
be partially due to the cppSOCS3 fusion protein which can enter the
surrounding tumor cells, which are not infected by oncolytic
adenoviral vectors directly, with the help of the cpp peptide. The
application of cpp for intercellular traffic of proteins has been
successfully explored in vitro and in vivo(10,23).
To decipher the mechanism of the inhibition of CRC
growth by the vector, we analyzed the expression of several factors
downstream of the STAT3 pathway with qRT-PCR. The results showed
that AdCN305-cppSOCS3 resulted in high expression of SOCS3, and a
normal level of SOCS3 effectively suppressed hyperphosphorylation
of the STAT3 protein and blocked the proliferation of CRC cells. We
also found that several oncogenes (c-Myc, cyclin D1 and survivin)
and an anti-apoptosis gene (Bcl-2) were expressed at a high level
in the CRC cells, while the oncolytic vector carrying the SOCS3
gene decreased the expression of these two types of genes and
facilitated apoptosis of the tumor cells.
To further examine the therapeutic potential of the
AdCN305-cppSOCS3 vectors on CRC in vivo, we treated the
established tumors in an SCID mouse model with the vector.
Consistent with the in vitro results, a dramatic inhibition
of STAT3 phosphorylation was noted in the xenograft tumor cells
that were infected with AdCN305-cppSOCS3. The infection of tumor
cells with AdCN305-cppSOCS3 also resulted in a reduction in the
expression of Bcl-2 and induced apoptosis of tumor cells. These
data indicate that transfer of the SOCS3 gene into the tumors
suppressed tumor growth and inhibited activation of STAT3.
In summary, we found that overexpression of SOCS3
inhibited JAK/STAT3 signaling and induced programmed apoptosis in
CRC cells. At the same time, various downstream genes of STAT3
pathway, such as survivin, cyclin D1 and c-Myc were also
downregulated. These results suggest that hyperphosphorylation of
STAT3 and overactivity of STAT3 signaling may have an important
role in the malignancy maintaining of colorectal cells.
In colorectal carcinoma cells, decreased SOCS3
expression definitely exists, accompanied by hypophosphorylation of
STAT3. As a result, the associated cell signals of the inflammatory
pathway promote and maintain CRC cell growth. Recombinant oncolytic
adenovirus AdCN-305-cppSOCS3 was found to selectively replicate in
CRC cells in vitro and is relative safe when used in
vivo. Restoration of SOCS3 was obviously achieved by expression
of the adenoviral vector in cells, and a simultaneous antitumor
effect was achieved by inhibiting constitutive STAT3
phosphorylation in vitro and in vivo. The present
study provides a method for developing a novel cancer gene
therapeutic strategy.
Acknowledgements
The authors would like to thank Yaojun Wang and
Qiuping Lu for their helpful support of this work. This research
was supported by the Zhejiang Provincial Natural Science Foundation
of China (No. Y2100891), Zhejiang Provincial Top Key Discipline of
Biology and the National High Technology Research and Development
Program (No. 2012ZX09102301-009).
References
|
1
|
Flossmann E and Rothwell PM: Effect of
aspirin on long-term risk of colorectal cancer: consistent evidence
from randomised and observational studies. Lancet. 369:1603–1613.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu S, Rhee KJ, Albesiano E, et al: A human
colonic commensal promotes colon tumorigenesis via activation of T
helper type 17 T cell responses. Nat Med. 15:1016–1022. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar
|
|
4
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: a leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yasukawa H, Sasaki A and Yoshimura A:
Negative regulation of cytokine signaling pathways. Annu Rev
Immunol. 18:143–164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yoshimura A, Nishinakamura H, Matsumura Y
and Hanada T: Negative regulation of cytokine signaling and immune
responses by SOCS proteins. Arthritis Res Ther. 7:100–110. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu LJ, Wu ML, Li H, et al: Inhibition of
STAT3 expression and signaling in resveratrol-differentiated
medulloblastoma cells. Neoplasia. 10:736–744. 2008.PubMed/NCBI
|
|
8
|
Calvisi DF, Ladu S, Gorden A, et al:
Ubiquitous activation of Ras and Jak/Stat pathways in human HCC.
Gastroenterology. 130:1117–1128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Niwa Y, Kanda H, Shikauchi Y, et al:
Methylation silencing of SOCS-3 promotes cell growth and migration
by enhancing JAK/STAT and FAK signalings in human hepatocellular
carcinoma. Oncogene. 24:6406–6417. 2005.PubMed/NCBI
|
|
10
|
Cui Q, Jiang W, Wang Y, et al: Transfer of
suppressor of cytokine signaling 3 by an oncolytic adenovirus
induces potential antitumor activities in hepatocellular carcinoma.
Hepatology. 47:105–112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao L, Gu J, Dong A, et al: Potent
antitumor activity of oncolytic adenovirus expressing mda-7/IL-24
for colorectal cancer. Hum Gene Ther. 16:845–858. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pei Z, Chu L, Zou W, et al: An oncolytic
adenoviral vector of Smac increases antitumor activity of TRAIL
against HCC in human cells and in mice. Hepatology. 39:1371–1381.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsareva SA, Moriggl R, Corvinus FM, et al:
Signal transducer and activator of transcription 3 activation
promotes invasive growth of colon carcinomas through matrix
metalloproteinase induction. Neoplasia. 9:279–291. 2007. View Article : Google Scholar
|
|
14
|
Corvinus FM, Orth C, Moriggl R, et al:
Persistent STAT3 activation in colon cancer is associated with
enhanced cell proliferation and tumor growth. Neoplasia. 7:545–555.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karim R, Tse G, Putti T, Scolyer R and Lee
S: The significance of the Wnt pathway in the pathology of human
cancers. Pathology. 36:120–128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Calvert PM and Frucht H: The genetics of
colorectal cancer. Ann Intern Med. 137:603–612. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Allgayer H: Molecular regulation of
urokinase-receptor gene expression as one potential concept for
molecular staging and therapy. Recent Results Cancer Res.
162:15–30. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takeda K, Kaisho T, Yoshida N, Takeda J,
Kishimoto T and Akira S: Stat3 activation is responsible for
IL-6-dependent T cell proliferation through preventing apoptosis:
generation and characterization of T cell-specific Stat3-deficient
mice. J Immunol. 161:4652–4660. 1998.PubMed/NCBI
|
|
19
|
Lutticken C, Wegenka UM, Yuan J, et al:
Association of transcription factor APRF and protein kinase Jak1
with the interleukin-6 signal transducer gp130. Science. 263:89–92.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Akira S: Roles of STAT3 defined by
tissue-specific gene targeting. Oncogene. 19:2607–2611. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ihle JN: The Stat family in cytokine
signaling. Curr Opin Cell Biol. 13:211–217. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Joliot A and Prochiantz A: Transduction
peptides: from technology to physiology. Nat Cell Biol. 6:189–196.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mae M and Langel U: Cell-penetrating
peptides as vectors for peptide, protein and oligonucleotide
delivery. Curr Opin Pharmacol. 6:509–514. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rivera AA, Wang M, Suzuki K, et al: Mode
of transgene expression after fusion to early or late viral genes
of a conditionally replicating adenovirus via an optimized internal
ribosome entry site in vitro and in vivo. Virology. 320:121–134.
2004. View Article : Google Scholar
|