Introduction
Osteosarcoma (OS), the most common tumor of the
bone, is a rare malignant neoplasm affecting mostly children and
adolescents. Although long-term survival in high-grade OS has
markedly improved in the last decades, owing to neoadjuvant
chemotherapy (1), data emerging
from clinical studies show that 35–45% of OS patients have a
natural or acquired drug-resistance (2).
The possibility of identifying tumor molecular
background and signaling pathway key end-points may provide new
targets for planning tailored therapies combined with conventional
therapeutic modalities (3).
Metformin (1,1-dimethylbiguanide hydrochloride)
belongs to the biguanide class of oral hypoglycemic agents and is
widely used as an antidiabetic drug (4,5) by
regulating glucose homeostasis and reducing insulin resistance.
Recent evidence indicates that metformin may reduce
the risk of cancer and improve prognosis and that in patients with
type 2 diabetes it reduces the risk of cancer (6–8).
In vitro and in vivo data (2,9–11)
emphasized the role of 5′-monophosphate-activated protein kinase
(AMPK) in action mechanism of metformin and demonstrated that the
reduction of tumor cell proliferation and survival is mediated by
inactivation of mTOR in both insulin-dependent and -independent
pathways (12).
AMPK is a heterotrimeric serine/threonine kinase
composed of a catalytic α subunit, and two regulatory subunits, β
and γ (13,14). Activation of AMPK requires an
allosteric change induced by AMP, as well as phosphorylation at
Thr172, that inhibits the downstream target mTOR implicated in
protein synthesis and proliferation (15) and promotes vascular endothelial
growth factor expression and angiogenesis (16–20).
In vitro and in vivo studies
demonstrated that metformin inhibits tumor cell growth and survival
in numerous tumors (8,21–23),
emphasizing its role as an antineoplastic agent through a variety
of responses including inhibition of growth factor signaling
pathway, and/or cell arrest in G1 phase (8,24,25).
The present study investigated the antitumor effects
of metformin on OS cell lines alone and in combination with
cisplatin (CDDP), a DNA-damaging chemotherapeutic drug frequently
used in OS patients.
Findings of the present study indicated that
metformin may sensitize OS cells to CDDP through inactivation of
critical intracellular end-points and lengthening of cell cycle
phases.
Materials and methods
Reagents
Anti-cyclin D1 (HD11) and anti-p-p53 (hSer20) were
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Anti-phospho AMPKα (Thr172), anti-AMPKα, anti-phospho-p70S6K (S6K1)
(Thr389), anti-IGF-1Rβ, anti-phospho Chk1 (Ser345) were purchased
from Cell Signaling Technology (Beverly, MA, USA). Anti-cyclin A
and anti-cyclin E were obtained from Calbiochem, Merck KGaA,
(Darmstadt, Germany). Anti-actin was from Sigma Chemical Co., (St.
Louis, MO, USA). Horseradish peroxidase-conjugated anti-rabbit IgG,
anti-mouse IgG were purchased from GE Healthcare. Enhanced
chemiluminescent substrate LiteAblot Plus was obtained from
EuroClone S.p.A (Pero, Milan, Italy).
Metformin was obtained from Sigma-Aldrich
Biotechnology (St. Louis, MO, USA), and diluted in PBS 1X to make a
1 M stock solution that was stored at −20°C. It was used across a
range of concentrations at 0, 5, 10, 20 and 40 mM diluted in
media.
Cisplatin was purchased by Teva Pharmaceuticals B.V.
(Utrecht, The Netherlands) and was stored at 4°C; it was used
across a range of concentrations at 0.01, 0.1, 1.0, 10 and 100
ng/ml diluted in media.
Cell lines and culture conditions
Human OS cell lines U2OS (pRB+/+, p53+/+), 143B
(pRB+/+, p53+/+) and MG63 (pRB+/+, p53−/−) were obtained from the
American Type Culture Collection (ATCC, Rockville, MD, USA) and
cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented
with 10% fetal bovine serum (FBS), L-glutamine (2 mM), 100 U/ml
penicillin and 100 μg/ml streptomycin (Invitrogen) at 37°C in a 5%
CO2 humidified incubator. Cells were routinely passed
when they reached ~80% confluence.
Cell growth and sensitivity study
The number of adherent, viable cells was assessed
microscopically using an improved Neubauer haemocytometer and
proliferation was assessed as the percentage of cells that excluded
0.2% trypan blue. Cells were seeded at 100,000/well in 6-well
plates and incubated in medium containing 10% FBS. Twenty-four
hours after seeding, cells were treated either with or without
increasing doses of metformin (0, 5, 10, 20 and 40 mM) for 24, 48
and 72 h. After 24, 48 and 72 h, cells were washed once with
Dulbecco’s phosphate-buffered saline (PBS) 1X, harvested by
trypsinization and cell number was determined using trypan
blue.
IC50 and IC30 values, defined
as the concentration of drugs inhibiting cell growth by 50 and 30%,
respectively, were calculated for experiments with 72 h of
treatment.
Cells were also treated with increasing doses of
CDDP (0.01, 0.1, 1.0, 10 and 100 ng/ml) and cytotoxicity was
evaluated as cell viability up to 72 h.
For combined treatment, cells were treated at the
same time in combination with metformin IC30 and CDDP at
different concentrations for 72 h; cells were also treated in
sequential manner with metformin IC30 for 72 h, followed
by 24 h of CDDP treatment at different concentrations.
Flow cytometry for apoptosis
OS cells were seeded at 100,000/well in 6-well
plates, allowed to attach overnight, and incubated with or without
an IC50 dose of metformin for 48 and 72 h. According to
the protocol kit (MEBCYTO Apoptosis kit; MBL International, Woburn,
MA, USA), the adherent cells were trypsinized, detached, and
combined with floating cells from the original growth medium,
centrifuged and washed twice with PBS 1X. Cells were re-suspended
in 500 μl of staining solution containing FITC-conjugated Annexin V
antibody and propidium iodide (PI) for 30 min and analyzed by flow
cytometry.
The number of viable (Annexin−/PI−), apoptotic
(Annexin+/PI−) and necrotic (Annexin+/PI+) cells were determined
with the CellQuest Software (BD Biosciences, San Jose, CA, USA),
using a peak fluorescence gate to exclude cell aggregates during
cell cycle analysis in a FACSCalibur flow cytometer
(Becton-Dickinson, San Jose, CA, USA).
Cell cycle analysis by FACS
OS cells were plated in 6-well plates (100,000
cells/well), allowed to attach overnight, and incubated with
IC50 doses of metformin. After 48 and 72 h they were
harvested by trypsinization, fixed with 70% ethanol and washed with
appropriate buffer (PAT) several times. After α-bromodeoxyuridine
incorporation and α-mouse FITC incubation as secondary antibody,
cells were stained for total DNA content with a solution containing
PI (1:5 in PAT). Cell cycle distribution was then analyzed with a
FACScan flow cytometer (Becton-Dickinson).
Protein extraction and western blot
analysis
Expression levels of proteins were determined by
western blot analysis. After 48 h of IC50 metformin
incubation, cells were washed three times with PBS and lysed in
100–400 μl lysis buffer [20 mM Tris-HCl (pH 7.5)], 150 mM NaCl, 2.5
mM sodium pyrophosphate, 1 mM β-glycerol phosphate, 1 mM
Na3VO4, 1 mM EGTA, 1% Triton and complete
protease inhibitor mixture inhibitors from Roche Diagnostics
(Laval, QC, Canada). Cellular debris was removed by centrifugation
at 14,000 × g for 20 min at 4°C. Following assay for total protein
(Bio-Rad Laboratories, Mississauga, ON, Canada), clarified protein
lysates (50 μg) were boiled for 5 min and analyzed by 8.0–15%
SDS-polyacrylamide gel, followed by blotting at 40 V for 1 h and
100 V for 2 h. Blots were probed with anti-p-p53 (Ser20) (1:200),
anti-IGF-IRβ (1:200), anti-phospho-AMPKα (Thr172) (1:1,000),
anti-AMPKα (1:1,000), anti-phospho-p70S6K (S6K1) (Thr389)
(1:1,000), anti-phospho Chk1 (Ser345) (1:1,000), anti-cyclin A
(1:300), anti-cyclin E (1:200), and anti-cyclin D1 (1:200).
Horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG
were used as secondary antibodies. The signal was visualized by
enhanced chemiluminescent substrate LiteAblot Plus (EuroClone
S.p.A.) and quantified using GS-800 imaging densitometer (Bio-Rad
Laboratories, Hercules, CA, USA). A rabbit anti-actin antibody was
used as control.
Statistical analysis
All experiments were performed three times and
results are expressed as means ± SD. Significance was analyzed by
the Student’s t-test and a probability value of P≤0.05 was
considered to indicate a statistically significant difference.
Results
Susceptibility of OS cell lines to
metformin
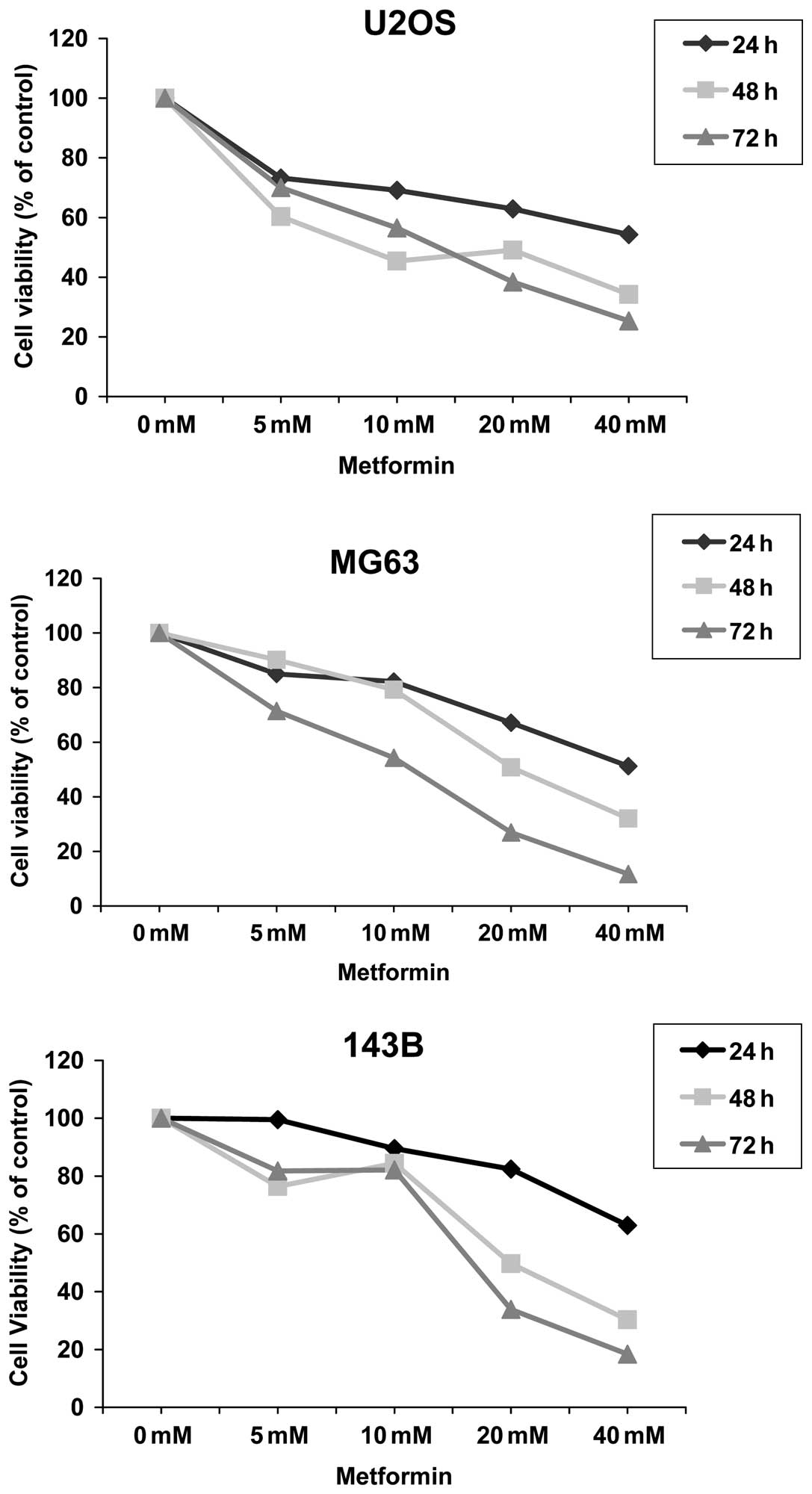
When OS cell lines were exposed to increasing doses
of metformin (0–40 mM), a progressive loss of proliferation up to
72 h was observed when cell growth decreased by 75% for U2OS, 89%
for MG63 and 82% for 143B (Fig.
1).
Cell sensitivity evaluation indicated that U2OS,
MG63 and 143B were sensitive to metformin with IC50 mean
values at 72 h of 9.13±0.3, 8.72±0.4 and 7.29±0.7, respectively,
and IC30 mean values of 4.11±0.7, 6.2±1.1 and 3.2±0.4,
respectively.
Effect of metformin on cell cycle and
apoptosis
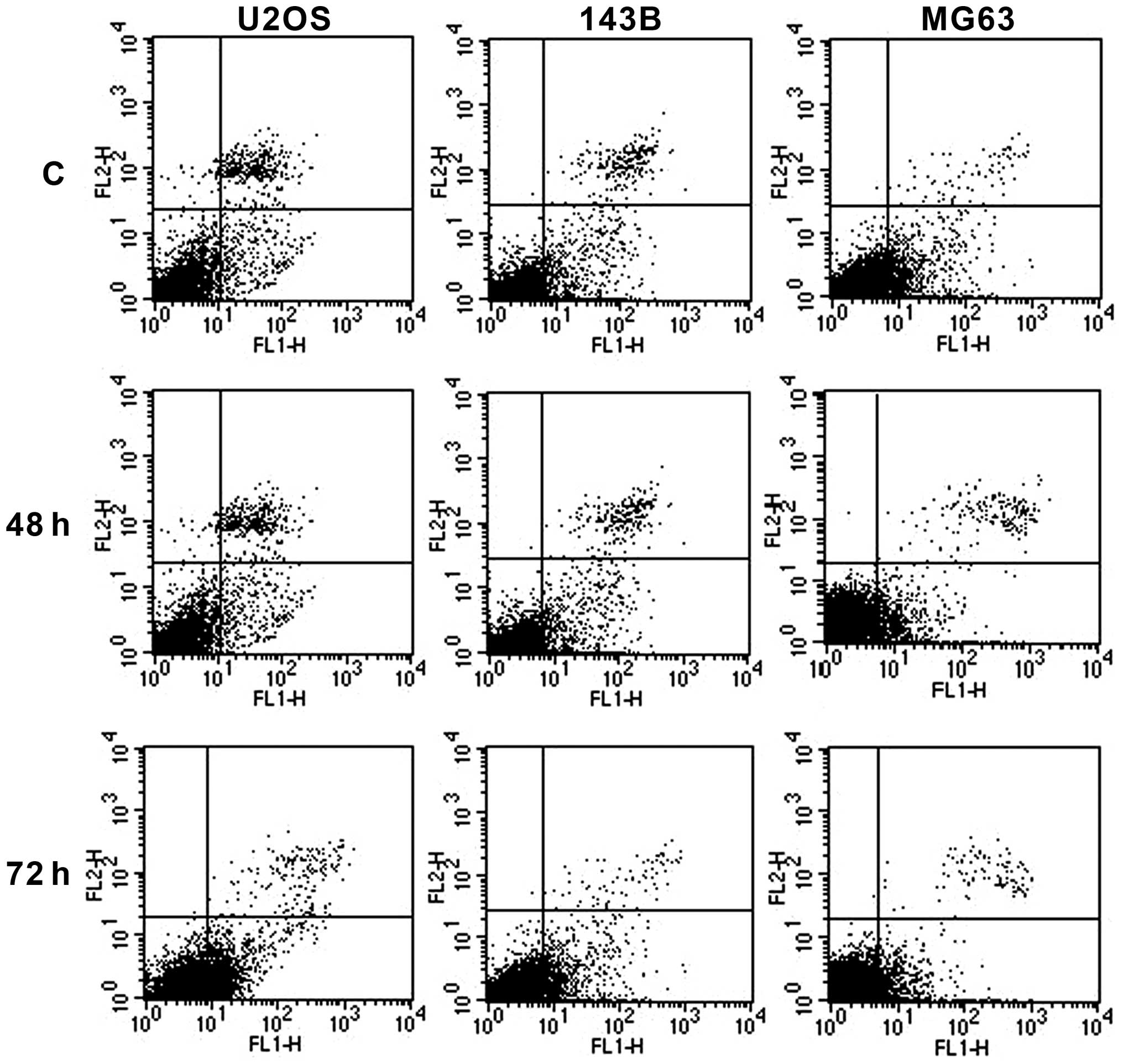
Following exposure of U2OS to IC50 dose
of metformin, cell cycle analysis revealed a transient arrest in G2
phase at 48 h, while a longer exposure (72 h) caused accumulation
of cells in S phase (Fig. 2) with a
significant time-dependent induction of apoptosis (from 4.6% in
non-stimulated cells to 17.2 and 21.7%, respectively, in stimulated
cells) (Fig. 3).
Conversely, 143B responded to the IC50
doses of metformin with relevant arrest of cells in G1 at 48 h
associated with a decrease of number of cells in S and G2 phase.
The following 72 h treatment resulted in lengthening of S phase,
concomitant with a significant decrease of G2 phase (Fig. 2) and a moderate induction of
apoptosis when compared to non-treated cells (8.10% non-treated
cells, 8.86% at 48 h and 11.20% at 72 h) (Fig. 3).
In MG63, metformin treatment was effective only at
the 72 h with accumulation of cells in G1 and G2 phases concomitant
with strong decrease in S phase (Fig.
2). No cases showed apoptotic induction by Annexin V-FITC assay
(7.6% in non-treated cells, 6.79% at 48 h, 8% at 72 h) (Fig. 3), suggesting a predominant
cytostatic effect of metformin exposure.
Protein analysis
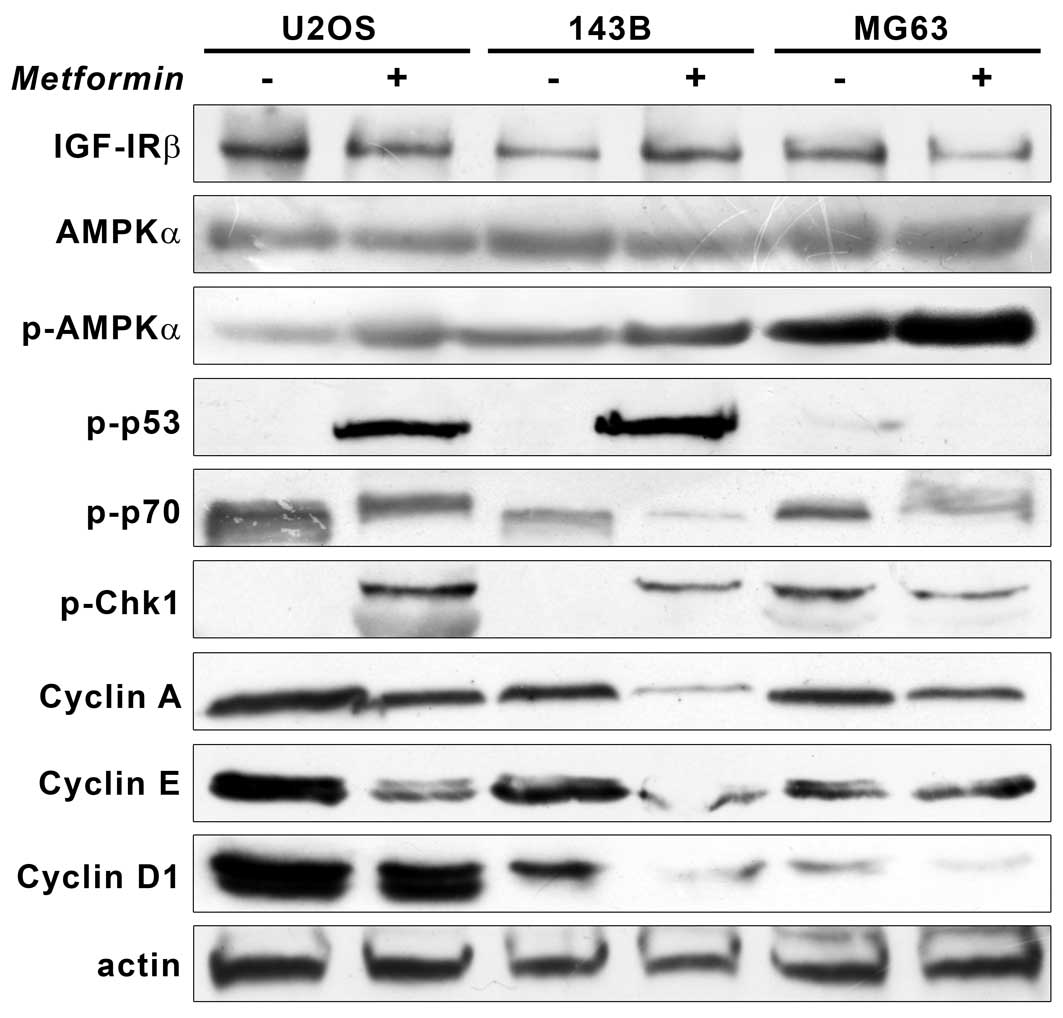
All OS cell lines were positive to IGF-IRβ and total
AMPKα without showing changes in expression levels after metformin
exposure. However, at 48 h of IC50 treatment,
phosphorylation level of p-AMPKα Thr172 increased in all cell lines
and accumulation of p53 (Ser20) was seen in wild-type-p53 U2OS and
143B.
p70S6K phosphorylated at Thr389, substrate of mTOR
activity, markedly decreased after treatment (Fig. 4).
When proteins involved in cell cycle control were
analyzed, both wild-type U2OS and 143B cells showed increased
expression of Chk1 (Ser345) associated with downregulation of
active cyclin A and cyclin E. No significant changes in the volume
of electrophoretic bands were seen for MG63. In parallel, we
observed a loss of cyclin D1 expression in 143B and MG63 and to a
lesser extent in U2OS (Fig. 4).
Susceptibility of OS cell lines to
CDDP
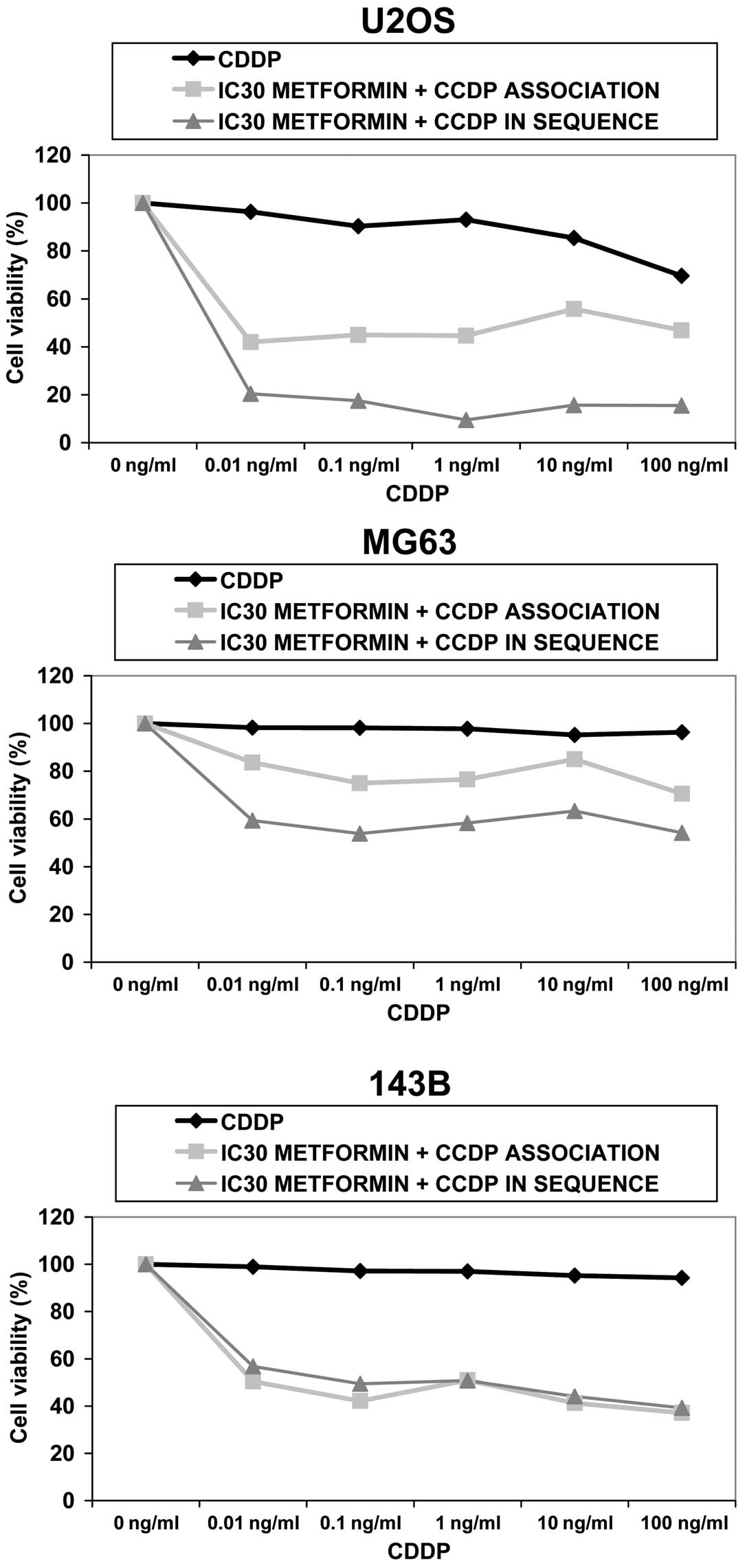
All OS cell lines were exposed to increasing doses
of CDDP up to 100 ng/ml for 72 h; MG63 and 143B did not show cell
growth inhibition, while U2OS had a slight reduction of 30% with
the maximum dose of CDDP (Fig.
5).
Metformin sensitizes OS cells to
CDDP
First U2OS, 143B and MG63 were exposed to increasing
concentrations of cisplatin (0.01–100 ng/ml) combined with
sub-toxic doses of metformin (IC30) for 72 h.
Data demonstrate that U2OS and 143B responded to
simultaneous treatment with reduction of cell proliferation of 33%
(P<0.01) and 60% (P<0.001), respectively, when compared with
CDDP alone showing a synergistic effect up to 1.0 ng/ml of CDDP for
U2OS and 100 ng/ml for 143B. MG63 responded to a lesser extent by
reduction of cell proliferation of 27% (P<0.05 at maximum dose
of CDDP). An antagonistic effect was observed between the two drugs
at any dose.
Subsequently, we evaluated whether pre-treatment
with metformin better sensitizes OS cells to CDDP treatment by
administering the drugs in sequence. OS cells were exposed to
IC30 metformin for 72 h, followed by increasing doses of
CDDP for 24 h.
In U2OS and MG63, cell proliferation dropped by 78%
(P<0.001) and 44% (P<0.01), respectively, with respect to
CDDP alone, while 143B responded with a percentage of decrease
equal to that of simultaneous treatment (60%) (P<0.01) (Fig. 5).
When CDDP was administered after metformin, a
synergistic interaction was seen in all cell lines.
Discussion
The first choice in OS treatment consists of
combined chemotherapeutic treatments often associated with serious
problems, such as frequent acquisition of drug-resistant phenotypes
and toxic side-effects that impair the quality and expectancy of
life in sarcoma patients.
Identification of critical end-points implicated in
the control of tumor cell survival (26) may provide the rationale for new
combined regimens able to overcome conventional treatment
failure.
Several experimental approaches have demonstrated
the therapeutic potential of mTOR inhibitors (27) and the strengthening of cell response
to anticancer agents through checkpoint activation and arrest of
cell cycle (28,29).
Evidence shows that metformin may inhibit tumor cell
growth (30) and enhance the effect
of chemotherapy through different anticancer mechanisms including
insulin-dependent and/or -independent activity (12,31).
Our data show that OS cell lines differing in
proliferation, transmigration and genetic background (32) respond to metformin by decreasing
cell proliferation through cell cycle lengthening associated or not
with apoptosis induction.
This effect appears to be correlated with increased
expression of AMPKα phosphorylated at Tyr172 and inhibition of mTOR
downstream signaling pathway measured by dephosphorylation of
p70S6K, resulting in an inhibition of protein synthesis and cell
growth (33).
Some reports support the hypothesis that inhibition
of cell proliferation by AMPKα activation is determined other than
by mTOR signaling inhibition, by arresting cell cycle through
activation of phospho-p53 and downregulation of cyclin-dependent
kinase (CDK) activity (34,35).
It is well known that cell cycle is regulated by
phosphorylation and dephosphorylation events controlled by
CDK/cyclin complexes and CDK inhibitors that arrest cell growth at
G1/S and/or G2/M checkpoints (28,36).
Our data showed that in wt-p53 U2OS and 143B cell
lines, metformin treatment induced accumulation of p53 (Ser20)
associated with apoptosis induction and prevalent lengthening of S
phase after long-term exposure (72 h). This delay in cell cycle
progression resulted from activation of phospho-Chk1 at Ser345 that
activates S and G2 checkpoints through downregulation of cyclin A
and cyclin E. Evidence that Chk1 contributes to cell cycle
checkpoints in human cells comes from studies showing that Chk1 is
an important regulator of S phase arrest and its disruption
abrogates S and G2 checkpoints (37,38).
These events may contribute to sensitize our wt-p53 OS cell lines
to CDDP showing a synergistic effect with metformin both in
combined and sequence treatments. Null-p53 MG63 where no activation
of phospho-Chk1 was seen, responded to long-term exposure of
metformin with prevalent accumulation of cells in G1 associated
with downregulation of cyclin D1 without apoptosis induction,
suggesting cytostatic rather than cytotoxic effect. Ben Sahra et
al(8) demonstrated that in
prostate cancer, the block of cell cycle in G1 by metformin is not
mediated by the AMPK pathway. By contrast, in breast cancer,
inhibitors of AMPK induced downregulation of D1 and G1 arrest even
in mut-p53 cells (35).
Moreover, CDDP in sequence with metformin was more
effective in decreasing MG63 cell proliferation than in
simultaneous treatment, where the two agents presented antagonistic
effects.
These results show that treatment with metformin
induces significant growth inhibition of OS cell lines through
arrest of cell cycle and decrease of S6K activity mediated by AMPKα
phosphorylation. In addition, metformin may sensitize OS cells
otherwise resistant to CDDP in a p53-independent manner through
synergistic drug-drug interaction.
Our data may have clinical relevance for novel
therapeutic strategies for the treatment of OS.
Acknowledgements
The authors thank Cristina Ghinelli and Alba
Balladelli for their help in editing the manuscript. Chiara Novello
was supported by the Fondazione Italiana per la Ricerca sul Cancro
(FIRC); triennial fellowship ‘Mario e Valeria Rindi’ 2013–2015
(research project no. 13748). This study was supported by the
Italian Ministry of Public Health and 5‰ donation (Italy).
References
|
1
|
Picci P, Mercuri M, Ferrari S, Alberghini
M, Briccoli A, Ferrari C, Pignotti E and Bacci G: Survival in
high-grade osteosarcoma: improvement over 21 years at a single
institution. Ann Oncol. 21:1366–1373. 2010.PubMed/NCBI
|
|
2
|
Hattinger CM, Pasello M, Ferrari S, Picci
P and Serra M: Emerging drugs for high-grade osteosarcoma. Expert
Opin Emerg Drugs. 15:615–634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liotta LA and Petricoin E: Cancer
biomarkers: closer to delivering on their promise. Cancer Cell.
20:279–280. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stumvoll M, Nurjhan N, Perriello G, Dailey
G and Gerich JE: Metabolic effects of metformin in
non-insulin-dependent diabetes mellitus. N Engl J Med. 333:550–554.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N,
Hirshman MF, Goodyear LJ and Moller DE: Role of AMP-activated
protein kinase in mechanism of metformin action. J Clin Invest.
108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bowker SL, Majumdar SR, Veugelers P and
Johnson JA: Increased cancer related mortality for patients with
type 2 diabetes who use sulfonylureas or insulin. Diabetes Care.
29:254–258. 2006. View Article : Google Scholar
|
|
8
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586.
2008.PubMed/NCBI
|
|
9
|
Hawley SA, Gadalla AE, Olsen GS and Hardie
DG: The antidiabetic drug metformin activates the AMP-activated
protein kinase cascade via an adenine nucleotide-independent
mechanism. Diabetes. 51:2420–2425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Towler MC and Hardie DG: AMP-activated
protein kinase in metabolic control and insulin signaling. Circ
Res. 100:328–341. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zou MH, Kirkpatrick SS, Davis BJ, Nelson
JS, Wiles WG IV, Schlattner U, Neumann D, Brownlee M, Freeman MB
and Goldman MH: Activation of the AMP-activated protein kinase by
the anti-diabetic drug metformin in vivo. Role of mitochondrial
reactive nitrogen species. J Biol Chem. 279:43940–43951. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dowling RJ, Goodwin PJ and Stambolic V:
Understanding the benefit of metformin use in cancer treatment. BMC
Med. 6:332003.
|
|
13
|
Kahn BB, Alquier T, Carling D and Hardie
DG: AMP-activated protein kinase: ancient energy gauge provides
clues to modern understanding of metabolism. Cell Metab. 1:15–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carling D: The AMP-activated protein
kinase cascade - a unifying system for energy control. Trends
Biochem Sci. 29:18–24. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stein SC, Woods A, Jones NA, Davison MD
and Carling D: The regulation of AMP-activated protein kinase by
phosphorylation. Biochem J. 345:437–443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yun H, Lee M, Kim SS and Joohun HA:
Glucose deprivation increases mRNA stability of vascular
endothelial growth factor through activation of AMP-activated
protein kinase in DU145 prostate carcinoma. Biol Chem.
280:9963–9972. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Neurath KM, Keough MP, Mikkelsen T and
Claffey KP: AMP-dependent protein kinase alpha 2 isoform promotes
hypoxia-induced VEGF expression in human glioblastoma. Glia.
53:733–743. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee M, Hwang JT, Lee HJ, Kang I, Kim SS
and Ha J: AMP-activated protein kinase activity is critical for
hypoxia-inducible factor-1 transcriptional activity and its target
gene expression under hypoxic conditions in DU145 cells. J Biol
Chem. 278:39653–39661. 2003. View Article : Google Scholar
|
|
19
|
Ouchi N, Shibata R and Walsh K:
AMP-activated protein kinase signaling stimulates VEGF expression
and angiogenesis in skeletal muscle. Circ Res. 96:838–846. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nagata D, Mogi M and Walsh K:
AMP-activated protein kinase (AMPK) signalling in endothelial cells
is essential for angiogenesis in response to hypoxic stress. J Biol
Chem. 278:31000–31006. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Buzzai M, Jones RG, Amaravadi RK, Lum JJ,
DeBerardinis RJ, Zhao F, Viollet B and Thompson CB: Systemic
treatment with the antidiabetic drug metformin selectively impairs
p53-deficient tumor cell growth. Cancer Res. 67:6745–6752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Phoenix KN, Vumbaca F and Claffey KP:
Therapeutic metformin/AMPK activation promotes the angiogenic
phenotype in the ERα negative MDA-MB-435 breast cancer model.
Breast Cancer Res Treat. 113:101–111. 2009.
|
|
23
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tomimoto A, Endo H, Sugiyama M, Fujisawa
T, Hosono K, Takahashi H, Nakajima N, Nagashima Y, Wada K, Nakagama
H and Nakajima A: Metformin suppresses intestinal polyp growth in
ApcMin/+ mice. Cancer Sci. 99:2136–2141. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luo Q, Hu D, Hu S, Yan M, Sun Z and Chen
F: In vitro and in vivo anti-tumor effect of metformin as a novel
therapeutic agent in human oral squamous cell carcinoma. BMC
Cancer. 12:5172012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Novello C, Pazzaglia L, Cingolani C, Conti
A, Quattrini I, Manara MC, Tognon M, Picci P and Benassi MS: miRNA
expression profile in human osteosarcoma: Role of miR-1 and
miR-133b in proliferation and cell cycle control. Int J Oncol.
42:667–675. 2013.PubMed/NCBI
|
|
27
|
Hidalgo M and Rowinsky EK: The
rapamycin-sensitive signal transduction pathway as a target for
cancer therapy. Oncogene. 19:6680–6686. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Collins I and Garrett MD: Targeting the
cell division cycle in cancer: CDK and cell cycle checkpoint kinase
inhibitors. Curr Opin Pharmacol. 5:366–373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Merry C, Fu K and Wang J, Merry C, Fu K
and Wang J: Targeting the checkpoint kinase Chk1 in cancer therapy.
Cell Cycle. 9:279–283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alimova IN, Liu B, Fan Z, Edgerton SM,
Dillon T, Lind SE and Thor AD: Metformin inhibits breast cancer
cell growth, colony formation and induces cell cycle arrest in
vitro. Cell Cycle. 8:909–915. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jalving M, Gietema JA, Lefrandt JD, de
Jong S, Reyners AK, Gans RO and de Vries EG: Metformin: taking away
the candy for cancer? Eur J Cancer. 46:2369–2380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Montanini L, Lasagna L, Barili V, Jonstrup
SP, Murgia A, Pazzaglia L, Conti A, Novello C, Kjems J, Perris R
and Benassi MS: MicroRNA cloning and sequencing in osteosarcoma
cell lines: differential role of miR-93. Cell Oncol. 35:29–41.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rocha GZ, Dias MM, Ropelle ER,
Osório-Costa F, Rossato FA, Vercesi AE, Saad MJ and Carvalheira JB:
Metformin amplifies chemotherapy-induced AMPK activation and
antitumoral growth. Clin Cancer Res. 17:3993–4005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Motoshima H, Goldstein BJ, Igata M and
Araki E: AMPK and cell proliferation - AMPK as a therapeutic target
for atherosclerosis and cancer. J Physiol. 574:63–71. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhuang Y and Miskimins WK: Cell cycle
arrest in Metformin treated breast cancer cells involves activation
of AMPK, downregulation of cyclin D1, and requires
p27Kip1 or p21Cip1. J Mol Signal. 3:182008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Eastman A: Cell cycle checkpoints and
their impact on anticancer therapeutic strategies. J Cell Biochem.
91:223–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tu YS, Kang XL, Zhou JG, Lv XF, Tang YB
and Guan YY: Involvement of Chk1-Cdc25A-cyclin A/CDK2 pathway in
simvastatin induced S-phase cell cycle arrest and apoptosis in
multiple myeloma cells. Eur J Pharmacol. 670:356–364. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao H, Watkins JI and Worms H: Disruption
of the checkpoint kinase 1/cell division cycle 25A pathway
abrogates ionizing radiation-induced S and G2 checkpoints. Proc
Natl Acad Sci USA. 99:14795–14800. 2002. View Article : Google Scholar : PubMed/NCBI
|