Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of
the most lethal of all human malignancies (1). Great improvements have been made in
surgical and chemotherapeutic approaches during the past decades;
however, the survival rate of patients with PDAC has not been
significantly prolonged. Therefore, an alternative therapy for PDAC
is urgently required.
The Hedgehog (HH) signaling pathway is an essential
pathway during embryonic development (2). Sonic Hedgehog (Shh) is a secreted
mammalian ortholog of the Hedgehog family and plays multiple roles
during embryonic development (3).
Activation of the Shh pathway triggers a series of intracellular
events through the GLI transcriptional effectors Gli-1, Gli-2 and
Gli-3 (4,5).
The HH pathway is reactivated in tumor development,
and plays an important role in the tumorigenesis and maintenance of
tumors (6–8), including pancreatic cancer (9). Evidence has shown that the HH
signaling pathway is a ‘core’ signal transduction pathway in
pancreatic cancer and is abnormally expressed in almost all
pancreatic cancer cells (10).
Accumulating data suggest that the HH pathway promotes the
tumorigenesis of pancreatic cancers by enhancing cell proliferation
(11,12), increasing invasion and metastasis
(13,14) and protecting against apoptosis
(15,16).
In a phase I trial testing vismodegib (GDC-0449), an
inhibitor of Smoothened (SMO), beneficial responses were observed
in patients with advanced basal cell carcinoma (BCC) and
medulloblastoma. However, only one in eight patients with
pancreatic carcinoma experienced a stable disease as the best
response (17). The ineffectiveness
of a pathway inhibitor for pancreatic cancer can probably be
explained by the fact that there are numerous crosstalks between
the HH signaling pathway and other signaling pathways in pancreatic
cancer (18–20). Feedback from these crosstalk
pathways may reduce the therapeutic effectiveness of the HH pathway
inhibitor. Therefore, further investigation should be focused on
the mechanism of interaction between HH signaling and these
crosstalk pathways.
Autophagy was initially reported 50 years ago by
Ashford and Porter (21), and it
has recently gained considerable attention. Autophagy is a
lysosome-mediated protein and organelle degradation process that is
characterized by the formation of double-membrane vesicles,
referred to as autophagosomes (22,23).
Autophagy is involved in several pathophysiological processes and
contributes to numerous diseases, particularly to cancer (24,25).
However, the function of autophagy in cancer has yet to be fully
clarified, as it acts both as a tumor suppressor and as a tumor
promoter (26).
During the development of PDAC, autophagy initially
suppresses tumor initiation yet supports tumor growth at later
stages (27,28). Other studies have reported that
pancreatic cancers require autophagy for tumor growth (29) and that autophagy is correlated with
poor outcome in PDAC patient (30).
Many results suggest that autophagy is a cell death mechanism in
response to drugs such as gemcitabine and triptolide. It was
confirmed that autophagy is activated by gemcitabine and triptolide
in the treatment of pancreatic cancer cells and that activated
autophagy plays a role in cancer suppression (31,32).
However, it was also reported that activation of autophagy by
receptor for advanced glycation end products (RAGE) (33,34),
2-deoxy-D-glucose (2-DG) (35),
phosphatidylinositol 3-kinase (PI3K) (36) and Kangai 1 (KAI1) (37) promotes tumorigenesis and limits
apoptosis in pancreatic cancer cells. Hence, the role and mechanism
of autophagy is unclear in pancreatic cancer and requires further
investigation.
Although HH signaling is known to inhibit apoptosis,
it remains unknown whether HH signaling is able to regulate
autophagy in pancreatic cancer cells. The present study describes a
novel role of the HH signaling pathway in the regulation of
autophagy in PDAC cells. We report that inhibition of the HH
pathway induces autophagy and enhances apoptosis in PDAC cells. Our
findings suggest that the status of autophagy is a key factor that
may influence the cellular response to HH signaling-targeted
therapy.
Materials and methods
Cell cultures
Human pancreatic cancer cell line CFPAC-1 was
purchased from the Shanghai Cell Bank (Shanghai, China) and
propagated in our laboratory by culturing in Dulbecco’s modified
Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA) with 10%
fetal bovine serum (FBS) (Sigma, St. Louis, MO, USA) at 37°C with
5% CO2, supplemented with 1% penicillin/streptomycin.
BALB/c nude mice (4-week-old, female) were purchased from the Model
Animal Research Center of Nanjing University, Nanjing, China. Drug
treatment included recombinant human N-Shh (R&D Systems,
Minneapolis, MN, USA) used at 0.25/0.5 μg/ml; GANT61 (Abcam,
Cambridge, MA, USA) used at 5/10 μM soluble in DMSO (dimethyl
sulphoxide); and 3-MA (3-methyladenine) (Sigma) used at 5
mmol/l.
Reverse transcription quantitative
real-time polymerase chain reaction (RT-qPCR) for mRNA
quantitation
Total RNA of cells was isolated using TRIzol reagent
(Invitrogen). cDNA was synthesized using the PrimeScript RT reagent
kit (Takara, Dalian, China). Quantitative RT-PCR was performed
using an ABI 7500 (Applied Biosystems) with FastStart Universal
SYBR-Green Master (Rox) (Roche) for mRNA quantitation and with the
TaqMan® MicroRNA assay kit (Applied Biosystems). The
relative expression of mRNA was calculated as the inverse log of
the ΔΔCt (38) and normalized to
β-actin. Primers for mRNA qPCR were synthesized by Invitrogen
(Shanghai, China). The sequences were: Gli-1 sense, 5′-AGCC
AAGCACCAGAATCGGAC-3′ and antisense, 5′-GTTTGGTC ACATGGGCGTCAG-3′;
Gli-2 sense, 5′-GGGTCTGGGGTC AGCCTTTGGA-3′ and antisense,
5′-AATGGCGACAGGGT TGACGGT-3′; β-actin sense, 5′-AGAAAATCTGGCACCAC
ACC-3′ and antisense, 5′-TAGCACAGCCTGGATAGCAA-3′.
Cell viability assays
Cell viability was determined using an MTT assay.
Aliquots of 2×103 cells were planted in 96-well plates
with 200 μl culture medium per well accordingly, and the viability
was measured at 72 h thereafter using the following procedures.
Cells were incubated with 20 μl/well of MTT solution (5 mg/ml;
Sigma) at 37°C for 4 h. Then 150 μl of DMSO was substituted for the
supernatant, followed by oscillation for 10 min. Absorbance at 490
nm was detected by the Multiskan MK3 microplate reader (Thermo
Labsystems, Philadelphia, PA, USA). All results were normalized to
corresponding controls and are expressed in terms of
percentage.
Analysis of cell apoptosis
Cell apoptosis was assessed by flow cytometry
(Becton-Dickinson, San Jose, CA, USA). For cell apoptosis, cells
were treated with GANT61 (10 μM), 3-MA (5 mmol/l) and a combination
of GANT61 and 3-MA for 48 h. Cells were then collected, washed,
suspended in 100 μl 1X binding buffer and stained with 5 μl
fluorescein isothiocyanate (FITC)-Annexin V and 1 μl PI at room
temperature for 15 min in the dark. The stained cells were
immediately analyzed by flow cytometry.
Western blotting
For western blotting, cells were treated with 400 nM
bafilomycin for 2 h before being lysed using RIPA buffer with 1%
PMSF on ice. The concentration of total protein was determined
using a BCA kit (Nanjing KeyGen Biotech., Co., Ltd., Nanjing,
China). Equal amounts of protein (30 μg) were resolved with 10%
SDS-PAGE and transferred to polyvinylidene difluoride (PVDF)
membranes (Millipore, Bedford, MA, USA) using a Mini Trans-Blot
apparatus (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were
probed with primary antibodies for 12 h at 4°C and then incubated
with secondary antibodies for 2 h at room temperature. Primary
antibodies used were LC3 (microtubule-associated protein light
chain 3) (Novus Biologicals, Littleton, CO, USA) and tubulin (Santa
Cruz Biotechnology, Santa Cruz, CA, USA) which was used as an
internal control. The secondary antibody was purchased from
Beyotime (Santa Cruz Biotechnology). Electrochemiluminescence was
performed with a ChemiImager 5500 imaging system (Alpha Innotech
Co., San Leandro, CA, USA).
Transmission electron microscopy
For ultrastructural examination, samples (~1
mm3) were fixed with 2% OsO4 and embedded in
Araldite. Ultrathin sections were stained with uranyl acetate and
lead citrate and inspected using an electron microscope (JEOL
JEM-1010; Jeol, Tokyo, Japan).
Orthotopic xenograft tumor study
CFPAC-1 cells (2×106) suspended in 100 μl
PBS were injected undercapsule of the pancreas in 4-week-old BALB/c
nude mice. After 1 week, mice were randomized to three groups;
vehicle, GANT61 (50 mg/kg), GANT61 (50 mg/kg) in combination with
3-MA (10 mg/kg) were administered every other day, i.p. for 28
days. At the end of the drug treatment, mice were euthanized and an
autopsy was performed to obtain the primary lesions. Tumor volume
was determined as V= (L × W2)/2, where L represents the
length and W the width of the tumor. A portion of each tumor was
used for western blot analysis for LC3II.
Statistical analysis
All experiments were repeated in triplicate. All
values are expressed as the means ± standard deviation (SD).
Statistical significance was determined by a Student’s t-test using
SPSS 15.0. P-values <0.05 were considered to indicate
statistically significant differences.
Results
Effects of N-Shh and GANT61 on the
expression of Gli-1 and Gli-2 in CFPAC-1 cells
To determine N-Shh (a recombinant Shh ligand) and
GANT61 (a small-molecule inhibitor of Gli-1 and Gli-2) function in
CFPAC-1 cells, the cells were treated with N-Shh (0.25/0.5 μg/ml)
or GANT61 (5/10 μM) for 12, 24 and 48 h, respectively. The
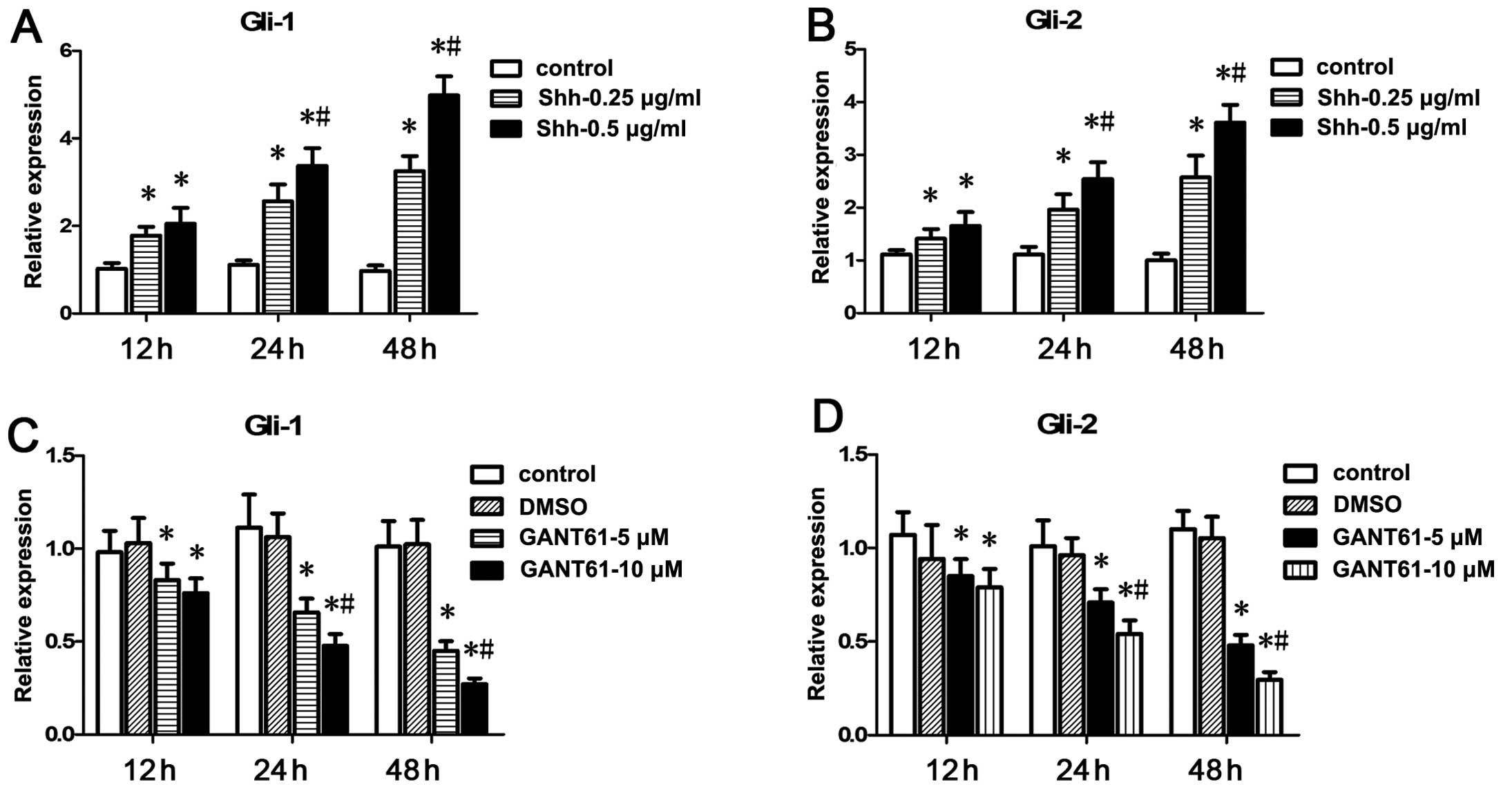
expression levels of Gli-1 and Gli-2 were confirmed by qRT-PCR. Our
data showed that N-Shh increased the mRNA levels of Gli-1 and Gli-2
while GANT61 decreased the Gli-1 and Gli-2 mRNA levels (Fig. 1). Gli-1 and Gli-2 mRNA levels were
markedly upregulated by 0.5 μg/ml N-Shh at 48 h and were markedly
downregulated by 10 μM GANT61 at 48 h, respectively. Therefore,
N-Shh was used at a concentration of 0.5 μg/ml while GANT61 was
used at 10 μM in the subsequent experiments.
HH signaling regulates autophagy in
CFPAC-1 cells
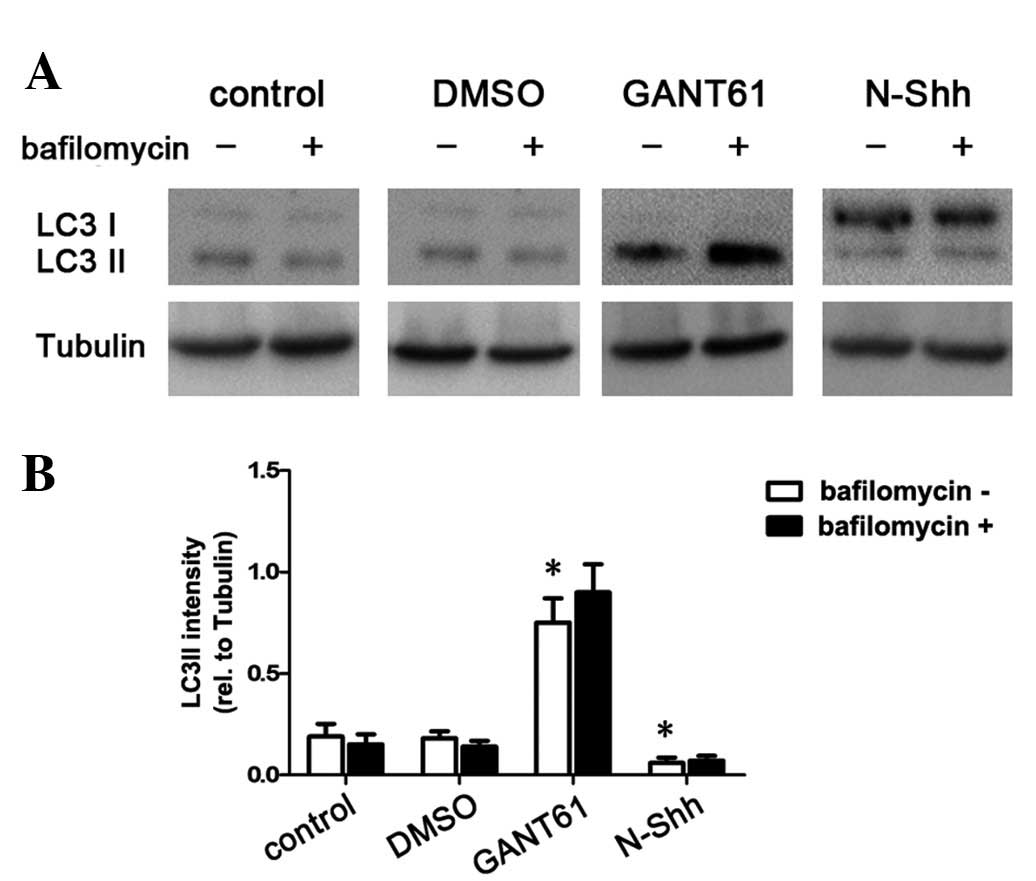
LC3 is widely used to monitor autophagy. The density
of the LC3-II band divided by the density of the tubulin band
represents the expression level of LC3-II. The ratio of the LC3-II
level/tubulin level was used as an indicator of the autophagic
level. In our experiment, CFPAC-1 cells were transfected with N-Shh
or GANT61 for 48 h. The cells were then treated with bafilomycin
(400 nM; Sigma), which significantly inhibits autophagy, for 2 h
and used as an autophagy inhibitor. Non-treated cells and the
vehicle group (DMSO) were used as controls. Western blot analysis
showed that, compared with the non-treated cells, N-Shh reduced the
ratio of the LC3-II level/tubulin level (P<0.05; Fig. 2A and B), indicating that the process
of autophagy was inhibited. Meanwhile, GANT61 significantly
elevated the ratio of the LC3-II level/tubulin level (P<0.05;
Fig. 2A and B). These data suggest
that activation of HH signaling reduces autophagy, while inhibition
of HH signaling induces autophagy. Therefore, HH signaling
regulates autophagy in pancreatic cancer CFPAC-1 cells.
GANT61-induced autophagy contributes to
induction of apoptosis
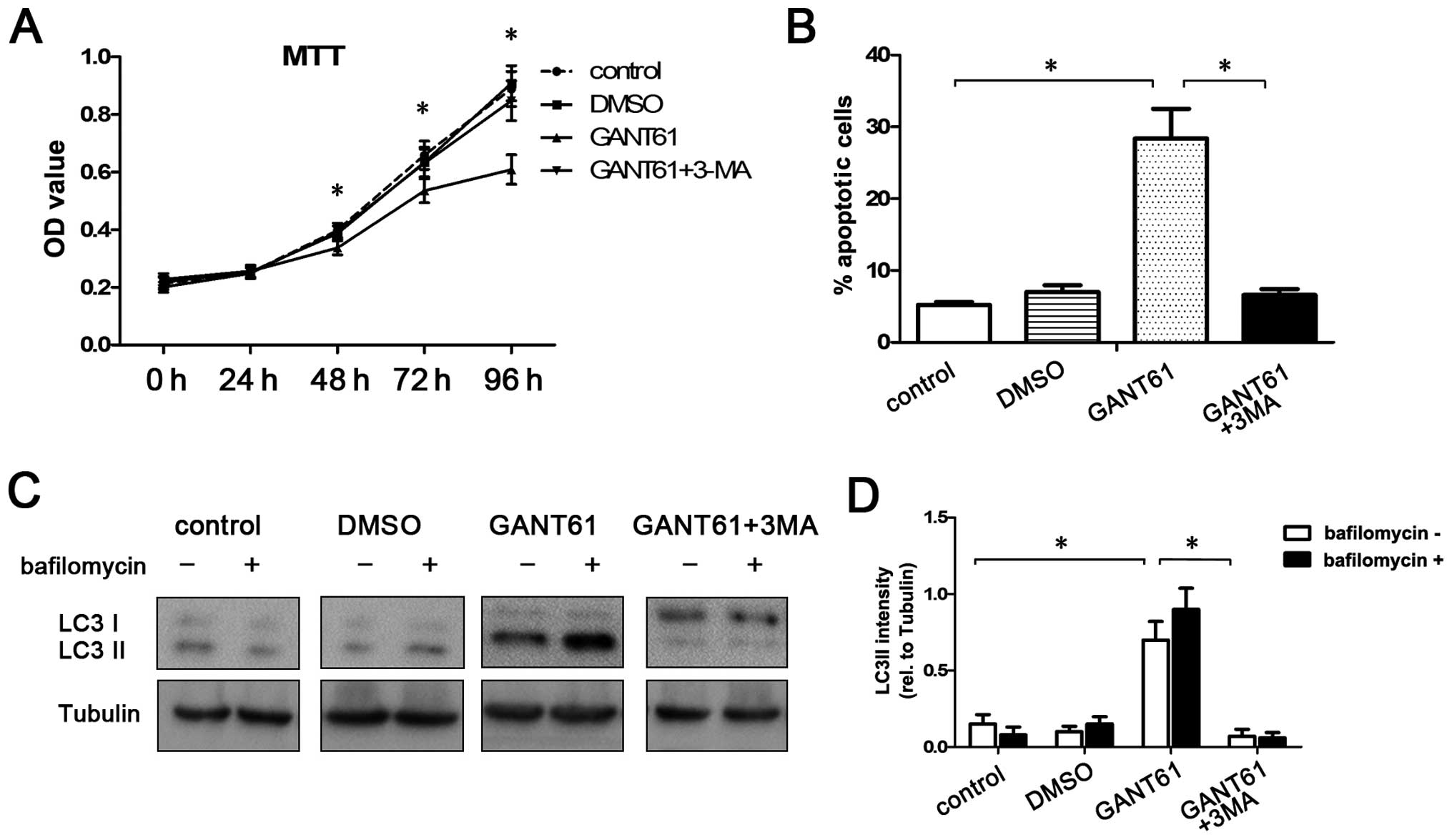
In pancreatic cancer, it remains unclear whether
autophagy acts fundamentally as a cell survival or cell death
pathway. Cell viability and apoptosis were analyzed after treatment
with 10 μM GANT61 for 48 h to investigate whether GANT61-induced
autophagy contributes to cell survival or death. MTT and flow
cytometric assay showed that GANT61 decreased the viability and
enhanced the apoptosis of CFPAC-1 cells when compared with the
control group (P<0.05; Fig. 3A and
B).
Since both HH signaling and autophagy involve
complex crosstalks with many other pathways, we used 3-MA (an
inhibitor of autophagy) to inhibit autophagy to confirm whether or
not GANT61-induced apoptosis is caused by the activation of
autophagy. Our data showed that GANT61-induced apoptosis and
reduction in cell viability in CFPAC-1 cells were reversed by
inhibition of autophagy by 3-MA (P<0.05; Fig. 3A and B). Western blot analysis
showed that, compared with the GANT61-treated group, the ratio of
the LC3-II level/tubulin level was significantly reduced in the
group treated with the combination of GANT61 and 3-MA (P<0.05;
Fig. 3C and D), suggesting the
GANT61-induced autophagy was inhibited by 3-MA. These results
suggest that GANT61-induced autophagy contributes to the viability
and apoptosis of CFPAC-1 cell.
GANT61 induces autophagy and suppresses
pancreatic cancer cell growth in vivo
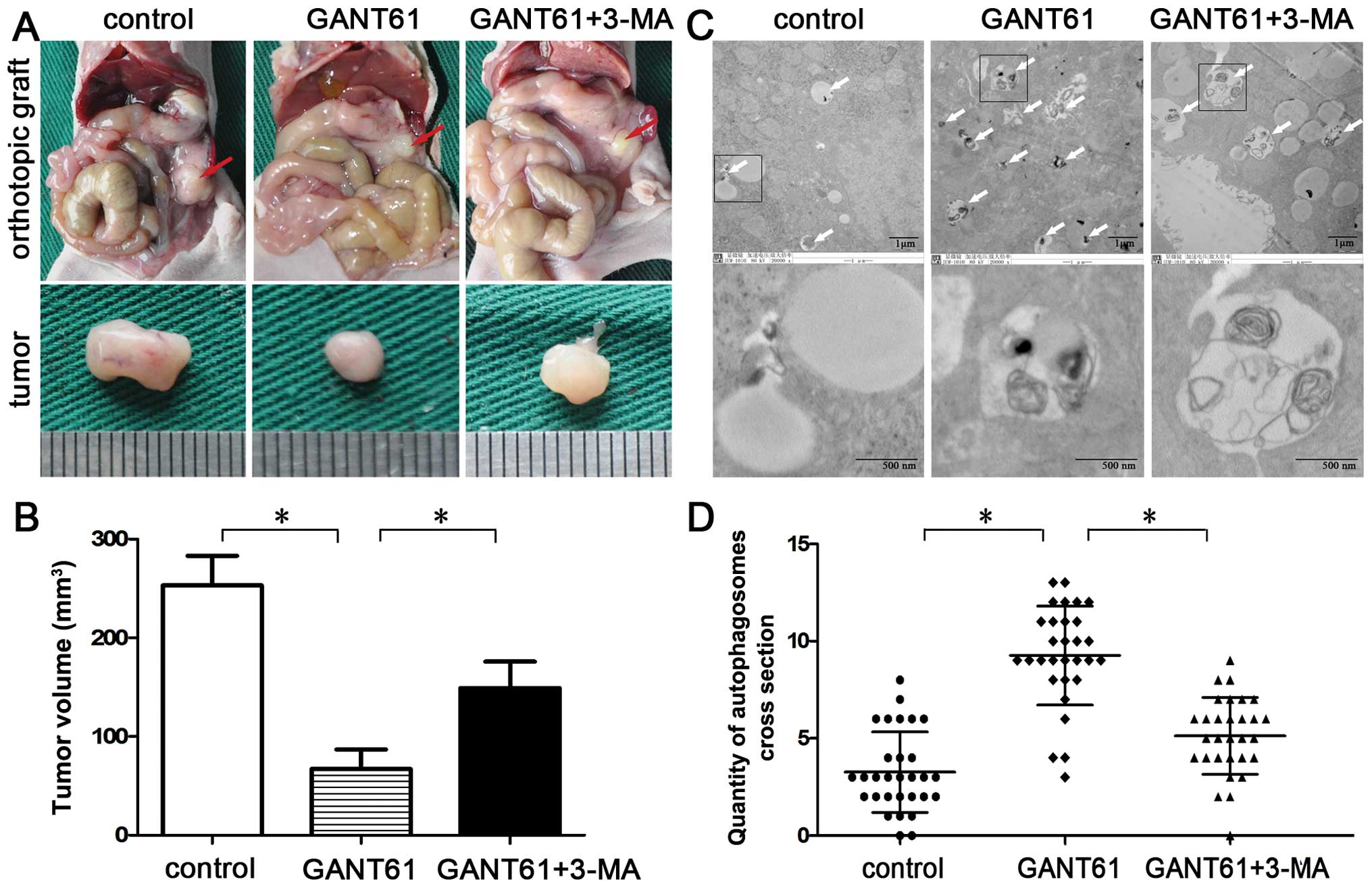
To further investigate the antitumor potential of
GANT61 and the role of autophagy in vivo, BALB/c nude mice
were injected undercapsule of the pancreas with CFPAC-1 cells.
Vehicle (DMSO), GANT61 (50 mg/kg), GANT61 (50 mg/kg) in combination
with 3-MA (10 mg/kg) were administered every other day, i.p. for 28
days (starting one week after inoculation). The data revealed that
GANT61 significantly inhibited tumor growth in CFPAC-1 cells when
compared with the control group (P<0.05; Fig. 4A and B) and the effect was reversed
by co-treatment with 3-MA, the autophagy inhibitor. Since the
formation of special double-membraned structures containing
undigested cytoplasmic contents (autophagosomes) is the most
important characteristic of autophagy, detecting these structures
by electron microscopy is considered the gold standard for
documenting autophagy. Hence, we examined the ultrastructural
changes in the orthotopic xenograft tumors. Quantification of
autophagosomes revealed that the number of autophagosomes was
significantly increased in the GANT61 treatment group (9.26±2.54);
however, the number of autophagosomes was reduced in the
combination group (5.13±1.97) (P<0.05; Fig. 4C and D). These results indicate that
GANT61-induced autophagy contributes to the viability of CFPAC-1
cells in vivo.
Discussion
In the present study, we demonstrated that the HH
signaling pathway regulates autophagy in pancreatic cancer cells.
Our data showed that activation of HH signaling by its ligand,
N-Shh, inhibits autophagy, while inhibition of the HH pathway by
GANT61 induces autophagy. In addition, GANT61-induced autophagy
contributed to reduced viability and increased apoptosis in CFPAC-1
cells and these effects were reversed by the co-treatment with
3-MA, the autophagy inhibitor.
The HH signaling pathway is a ‘core’ signal
transduction pathway in pancreatic cancer (10) that promotes the tumorigenesis of
pancreatic cancers via enhancing cell proliferation (11,12),
increasing invasion and metastasis (13,14)
and protecting against apoptosis (15,16).
Therapies targeting HH signaling were believed to be an effective
method through which to overcome pancreatic cancer. However, in a
phase I trial testing vismodegib (GDC-0449), an inhibitor of
Smoothened (SMO), no beneficial responses were observed in
pancreatic carcinoma (17). The
ineffectiveness of the pathway inhibitor for pancreatic cancer was
probably due to the fact there is much crosstalk between the HH
signaling pathway and other signaling pathways in pancreatic cancer
(18–20). In the present study, we found that
HH signaling regulated autophagy in pancreatic cancer cells. Our
data revealed that activation of HH signaling by its ligand, N-Shh,
inhibited autophagy, while inhibition of the HH pathway by GANT61
induced autophagy.
The role of autophagy in pancreatic cancer
development and progression is complex. During the development of
pancreatic cancer, autophagy initially suppresses tumor initiation
yet supports tumor growth at later stages (27,28).
Many results suggest that autophagy is a cell death mechanism in
response to drugs such as gemcitabine and triptolide. However, it
was also reported that activation of autophagy promotes
tumorigenesis and limits apoptosis in pancreatic cancer cells. In
the present study, we found that GANT61-induced autophagy
contributed to reduced viability and increased apoptosis in CFPAC-1
cells both in vivo and in vitro, and these effects
were reversed by the blockage of autophagy. Our findings were
confirmed by a recent study in which Wang et al (39) found that the HH signaling pathway
regulates autophagy in HCC, inhibition of Gli by GANT61 induces
autophagy, and blockage of autophagy attenuates GANT61-induced
apoptosis. However, in a recent study, we found that miR-101
enhanced apoptosis induced by cisplatin in HCC cells by inhibition
of autophagy (40). Although these
findings appear contradictory, these inconsistent conclusions
confirm that autophagy plays a dual role in tumor initiation and
development. The tumor-suppressor or tumor-promoter role depends on
the type of cancer and the microenvironment. In addition,
inhibition of autophagy by different approaches caused inconsistent
effects which may be related to the complex crosstalk between
autophagy and many other pathways. Different methods of autophagy
inhibition may regulate different crosstalk pathways resulting in a
different degree of effectiveness. Thus, further investigation of
the mechanism of interaction between autophagy and these crosstalk
pathways is particularly important.
In conclusion, the present study showed that HH
signaling regulates autophagy in pancreatic cancer cells.
Activation of HH signaling inhibits autophagy, while inhibition of
the HH pathway induces autophagy. Although the role of autophagy in
cell survival and apoptosis may depend on tumor type and
microenvironment, our data clearly demonstrated that GANT61-induced
autophagy contributes to reduced viability and increased apoptosis
in pancreatic cancer cells both in vivo and in vitro.
We propose that HH signaling by regulating autophagy plays an
important role in determining the cellular response to HH-targeted
therapy in pancreatic cancer and further investigation concerning
the interaction between autophagy and HH signaling is particularly
important.
Acknowledgements
The present study was supported by grants from the
Department of Public Health of Jiangsu Province (no. RC2007056) and
the National Natural Science Foundation of China (no.
81170415).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Hammerschmidt M, Brook A and McMahon AP:
The world according to hedgehog. Trends Genet. 13:14–21. 1997.
View Article : Google Scholar
|
|
3
|
Varjosalo M and Taipale J: Hedgehog:
functions and mechanisms. Genes Dev. 22:2454–2472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kalderon D: Transducing the hedgehog
signal. Cell. 103:371–374. 2000. View Article : Google Scholar
|
|
5
|
McMahon AP: More surprises in the Hedgehog
signaling pathway. Cell. 100:185–188. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Caro I and Low JA: The role of the
hedgehog signaling pathway in the development of basal cell
carcinoma and opportunities for treatment. Clin Cancer Res.
16:3335–3339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barginear MF, Leung M and Budman DR: The
hedgehog pathway as a therapeutic target for treatment of breast
cancer. Breast Cancer Res Treat. 116:239–246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Saqui-Salces M and Merchant JL: Hedgehog
signaling and gastrointestinal cancer. Biochim Biophys Acta.
1803:786–795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thayer SP, di Magliano MP, Heiser PW, et
al: Hedgehog is an early and late mediator of pancreatic cancer
tumorigenesis. Nature. 425:851–856. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jones S, Zhang X, Parsons DW, et al: Core
signaling pathways in human pancreatic cancers revealed by global
genomic analyses. Science. 321:1801–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yauch RL, Dijkgraaf GJ, Alicke B, et al:
Smoothened mutation confers resistance to a Hedgehog pathway
inhibitor in medulloblastoma. Science. 326:572–574. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morton JP, Mongeau ME, Klimstra DS, et al:
Sonic hedgehog acts at multiple stages during pancreatic
tumorigenesis. Proc Natl Acad Sci USA. 104:5103–5108. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bailey JM, Mohr AM and Hollingsworth MA:
Sonic hedgehog paracrine signaling regulates metastasis and
lymphangiogenesis in pancreatic cancer. Oncogene. 28:3513–3525.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dai J, Ai K, Du Y and Chen G: Sonic
hedgehog expression correlates with distant metastasis in
pancreatic adenocarcinoma. Pancreas. 40:233–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olive KP, Jacobetz MA, Davidson CJ, et al:
Inhibition of Hedgehog signaling enhances delivery of chemotherapy
in a mouse model of pancreatic cancer. Science. 324:1457–1461.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lonardo E, Hermann PC, Mueller MT, et al:
Nodal/activin signaling drives self-renewal and tumorigenicity of
pancreatic cancer stem cells and provides a target for combined
drug therapy. Cell Stem Cell. 9:433–446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
LoRusso PM, Rudin CM, Reddy JC, et al:
Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449)
in patients with refractory, locally advanced or metastatic solid
tumors. Clin Cancer Res. 17:2502–2511. 2011. View Article : Google Scholar
|
|
18
|
Pasca di Magliano M, Sekine S, Ermilov A,
Ferris J, Dlugosz AA and Hebrok M: Hedgehog/Ras interactions
regulate early stages of pancreatic cancer. Genes Dev.
20:3161–3173. 2006.PubMed/NCBI
|
|
19
|
Kasperczyk H, Baumann B, Debatin KM and
Fulda S: Characterization of sonic hedgehog as a novel NF-κB target
gene that promotes NF-κB-mediated apoptosis resistance and tumor
growth in vivo. FASEB J. 23:21–33. 2009.PubMed/NCBI
|
|
20
|
Eberl M, Klingler S, Mangelberger D, et
al: Hedgehog-EGFR cooperation response genes determine the
oncogenic phenotype of basal cell carcinoma and tumour-initiating
pancreatic cancer cells. EMBO Mol Med. 4:218–233. 2012. View Article : Google Scholar
|
|
21
|
Ashford TP and Porter KR: Cytoplasmic
components in hepatic cell lysosomes. J Cell Biol. 12:198–202.
1962. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lorin S, Hamai A, Mehrpour M and Codogno
P: Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eskelinen EL: The dual role of autophagy
in cancer. Curr Opin Pharmacol. 11:294–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang S and Kimmelman AC: A critical role
for autophagy in pancreatic cancer. Autophagy. 7:912–913. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aghajan M, Li N and Karin M: Obesity,
autophagy and the pathogenesis of liver and pancreatic cancers. J
Gastroenterol Hepatol. 27(Suppl 2): 10–14. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang S, Wang X, Contino G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fujii S, Mitsunaga S, Yamazaki M, et al:
Autophagy is activated in pancreatic cancer cells and correlates
with poor patient outcome. Cancer Sci. 99:1813–1819.
2008.PubMed/NCBI
|
|
31
|
Mukubou H, Tsujimura T, Sasaki R and Ku Y:
The role of autophagy in the treatment of pancreatic cancer with
gemcitabine and ionizing radiation. Int J Oncol. 37:821–828.
2010.PubMed/NCBI
|
|
32
|
Donadelli M, Dando I, Zaniboni T, et al:
Gemcitabine/cannabinoid combination triggers autophagy in
pancreatic cancer cells through a ROS-mediated mechanism. Cell
Death Dis. 2:e1522011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kang R, Tang D, Schapiro NE, et al: The
receptor for advanced glycation end products (RAGE) sustains
autophagy and limits apoptosis, promoting pancreatic tumor cell
survival. Cell Death Differ. 17:666–676. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kang R, Tang D, Lotze MT and Zeh HJ III:
AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and
bioenergetics through the IL6-pSTAT3 pathway. Autophagy. 8:989–991.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xi H, Kurtoglu M, Liu H, et al:
2-Deoxy-D-glucose activates autophagy via endoplasmic reticulum
stress rather than ATP depletion. Cancer Chemother Pharmacol.
67:899–910. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mirzoeva OK, Hann B, Hom YK, et al:
Autophagy suppression promotes apoptotic cell death in response to
inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J
Mol Med (Berl). 89:877–889. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu CY, Yan J, Yang YF, et al:
Overexpression of KAI1 induces autophagy and increases MiaPaCa-2
cell survival through the phosphorylation of extracellular
signal-regulated kinases. Biochem Biophys Res Commun. 404:802–808.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
39
|
Wang Y, Han C, Lu L, Magliato S and Wu T:
Hedgehog signaling pathway regulates autophagy in human
hepatocellular carcinoma cells. Hepatology. 58:995–1010. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu Y, An Y, Wang Y, et al: miR-101
inhibits autophagy and enhances cisplatin-induced apoptosis in
hepatocellular carcinoma cells. Oncol Rep. 29:2019–2024.
2013.PubMed/NCBI
|