Introduction
Pancreatic cancer is the fourth most common cause of
cancer-related mortality in the western world, with an estimated
35,000 deaths in 2009 in the United States (1). Less than 20% of pancreatic cancer
patients are diagnosed with resectable and potentially curable
disease, whereas most patients have advanced disease at the time of
diagnosis and hence a dismal prognosis (2). This poor prognosis has been related to
the difficulty of detection at the early stages of development,
resulting in advanced disease at the time of presentation of first
symptoms. With regard to patients with pancreatic cancer, for all
stages, the 1-year survival rate is 23% and the 5-year overall
survival rate from diagnosis is 4%. Median survival for patients
with locally advanced disease is 9–12 months, and for patients with
metastatic disease, the median survival is 3–6 months. The 5-year
survival after curative resection is only 15–25% (3). In recent years, few agents have
demonstrated significant benefit for patients with metastatic
disease. Thus, effective new cytotoxic chemotherapy is needed for
these diseases.
Oridonin, an ent-kaurane diterpenoid isolated from
Rabdosia rubescens, is an important traditional Chinese
herbal remedy, which has multiple biological activities, such as
anti-inflammatory, antibacterial and antitumor effects (4). Existing research has confirmed that
oridonin confers an inhibitive effect on the development of various
types of cancers, and is a potential, effective, low-toxic
antitumor medicine (5–13). However, mechanisms underlying the
antitumor activity of oridonin and whether or not oridonin has
anti-pancreatic cancer activity remain largely unknown.
In the present study, we investigated the
involvement of MAPK signaling pathways in the anticancer effects of
oridonin in pancreatic cancer cells, and we demonstrated that the
MAPK pathway participated in oridonin-induced pancreatic cancer
cell apoptosis. Importantly, a p38 inhibitor but neither an ERK
inhibitor nor a JNK inhibitor, blocked the phosphorylation of p38
and also reduced the activation of p53, p21 and caspase-9 and -3.
Taken together, the p38-MAPK pathway is required in
oridonin-induced pancreatic cancer cell apoptosis, and which is
dependent on its downstream target p53 and caspase activation.
Materials and methods
Chemicals and reagents
Oridonin was obtained from the Beijing Institute of
Biological Products (Beijing, China). The purity of the oridonin
was measured by HPLC and determined to be 99.4%. Oridonin was
dissolved in dimethyl sulfoxide (DMSO) to make a stock solution at
a 10 mmol/l concentration and stored at −20°C. The DMSO
concentration was kept below 0.1% in all the cell cultures and did
not exert any detectable effect on cell growth or cell death. Fetal
bovine serum (FBS), trypsin containing EDTA, Roswell Park Memorial
Institute-1640 (RPMI-1640) and the Cell Counting Kit-8 (CCK-8) were
obtained from Gibco (USA). Annexin V-FITC/PI Apoptosis Detection
Kit I was obtained from BD Bioscience. The p53 inhibitor pifithrin
α (PFT α) was obtained from Biomol International (Plymouth Meeting,
PA, USA). The ERK inhibitor PD98059, p38 inhibitor SB203580 and JNK
inhibitor SP600125 were obtained from Calbiochem (San Diego, CA,
USA). RNA extraction kit was purchased from Life Technologies Co.,
cDNA First Strand Synthesis kit was purchased from Fermentas, and
2X Taq PCR Master Mix was purchased from Tiangen. Ribonuclease A
(RNase A), propidium iodide (PI), Hoechst 33258 and DMSO were
obtained from Sigma. Antibodies against p53, phospho-p53, p38,
phospho-p38, p21, caspase-9, caspase-3, β-actin and horseradish
peroxidase (HRP)-conjugated secondary antibodies (goat anti-rabbit
and goat anti-mouse) were purchased from Sigma.
Cell culture
The SW1990 pancreatic cancer cell line was obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). The human normal pancreas cell line HPDE6c7 was obtained from
Guangzhou Jennio Biotech Co., Ltd. (Guangzhou, China). The cells
were cultured in RPMI-1640 supplemented with 10% FBS, 100 U/ml
penicillin, and 100 μg/ml streptomycin. Cells were maintained at
37°C in a humidified atmosphere of 5% CO2. The medium
was changed every second to third day, and cells were subcultured
when confluency reached 70–80% by 0.25% trypsin at 37°C.
Cytotoxicity assay
The cytotoxic effect of oridonin on SW1990 and
HPDE6c7 cells was determined using the CCK-8. Briefly, the
logarithmic phase cells were plated in 96-well culture plates
(5×103 cells per well). After 24 h of incubation, the
cells were treated with vehicle alone (0.1% DMSO) and various
concentrations (10, 20, 40, 80, 160 μM) of oridonin, followed by a
48-h cell culture. In addition, in experiments to determine the
effects of MAPK inhibitors (PD98059, SP600125 and SB203580) and the
p53 inhibitor (PFT α) on cell cytotoxicity, cells were pretreated
with the inhibitors for 1 h at the given concentrations and then
incubated with the specified concentration of oridonin for 48 h.
Each group had 6 wells, and CCK-8 (100 μl) was added to each well 1
h before the end of incubation. The absorbance at 450 nm was read
using Bio-Tek, ELX800. Experiments were repeated three times. The
cytotoxic effect was expressed as a relative percentage of cell
death calculated as follows: Cell death (%) = 1 - (dosing
absorbance - blank absorbance)/(control absorbance - blank
absorbance) ×100.
Observation of apoptotic cell
morphology
Apoptotic morphology was monitored in DAPI- and
Hoechst 33258-stained cells. Cells (4×105) were grown
for 48 h on coverslips on a 6-well plate in the presence or absence
of 40 μM oridonin. After 48 h of incubation, the coverslips were
carefully washed with PBS, fixed with 4% paraformaldehyde at room
temperature for 30 min and respectively incubated with 10 μg/ml
DAPI and 10 μg/ml Hoechst 33258 at room temperature for 30 min.
Thereafter, cells were again washed and resuspended in PBS for
morphological observation under a fluorescence microscope (Nikon,
Japan).
Flow cytometric analysis of cell cycle
distribution and apoptosis
Analysis of cell cycle distribution was measured by
staining DNA with PI according to the manufacturer’s protocol.
SW1990 cells were seeded into 6-well plates at a density of
~5×105 cells per well, cultured overnight, and then
treated with various concentrations of oridonin (20, 40, 80 μM).
Control cells were treated with 0.1% DMSO only. Cells were
incubated for 48 h. Both detached and adherent cells were collected
and centrifuged at 1000 × g for 5 min at 4°C. Pellets were rinsed
with ice-cold phosphate-buffered saline (PBS) and fixed with 70%
ethanol for 24 h at 4°C. Cells were then stained with staining
buffer (PBS containing 50 μg/ml of PI, 10 μg/ml RNase A, 0.1%
sodium citrate and 0.1% Triton X-100) for at least 15 min at 37°C
in the dark. Samples were analyzed by a flow cytometer (BD
Bioscience).
Apoptosis in SW1990 cells was evaluated using
Annexin V-FITC Apoptosis Detection Kit I, which was performed
according to the manufacturer’s protocol. The SW1990 cells were
cultured as above, then treated with various concentrations of
oridonin (20, 40, 80 μM). Control cells were treated with 0.1% DMSO
only. Cells were collected after a 48-h incubation, washed with
PBS, resuspended in binding buffer, and incubated with FITC and PI
staining solution following the manufacturer’s instructions.
Samples of 10,000 stained cells were analyzed by flow cytometry (BD
Bioscience).
Western blot analysis
SW1990 cells were treated with the desired
concentration of oridonin in the absence or presence of inhibitors
for 48 h. Both adherent and floating cells were collected, and then
western blot analysis was performed. The cells were washed with
ice-cold PBS, and lysed on ice for 40 min in a solution containing
50 mM Tris, 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 2 mM
Na3VO4, 2 mM EGTA, 12 mM β-glycerolphosphate,
10 mM NaF, 16 μg/ml benzamidine hydrochloride, 10 μg/ml
phenanthroline, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml
pepstatin, and 1 mM phenylmethylsulfonyl fluoride, and then lysed
at 4°C for 60 min. The cell lysate was centrifuged at 14,000 × g
for 15 min, and the supernatant fraction was collected for western
blotting. Protein content in the supernatant fraction was
determined by bicinchoninic acid (BCA) assay kit (Sigma) according
to the manufacturer’s instructions. The protein lysates (20
μg/lane) were separated by electrophoresis on 12% SDS
polyacrylamide gel and then transferred to a nitrocellulose
membrane. After blocking for 1 h in 10% nonfat dry milk in
Tris-buffered saline, the membrane was incubated with the desired
primary antibody for 1 h. The membrane was then treated with the
appropriate peroxidase-conjugated secondary antibody, and the
immunoreactive proteins were detected using an enhanced
chemiluminescence kit (NEN Life Science Products, Boston, MA, USA)
according to the manufacturer’s instructions. Each membrane was
stripped and reprobed with antibodies against actin to correct for
differences in protein loading. Quantitative data were expressed as
mean ± SD of the relative levels of the objective protein and
control β-actin of each group of cells from three independent
experiments.
Semi-quantitative RT-PCR assay
SW1990 cells were treated with the desired
concentration of oridonin in the absence or presence of inhibitors
for 48 h, and both adherent and floating cells were collected.
Subsequently, total cellular RNAs were isolated from the cells
using TRIzol reagent. The content of RNA was measured using a UV
spectrophotometer under 260 nm. cDNA was synthesized with 1 μg of
total RNA and oligo(dT) primer according to the manufacturer’s
instructions. PCR amplification conditions were: p38, 94°C for 60
sec, 61.8°C for 60 sec, 72°C for 60 sec, 35 cycles; p53, 94°C for
45 sec, 51.9°C for 1 min, 72°C for 90 sec, 35 cycles; p21, 94°C for
30 sec, 54°C for 30 sec, 72°C for 1 min, 30 cycles; caspase-9, 94°C
for 30 sec, 56°C for 30 sec, 72°C for 30 sec, 35 cycles; caspase-3,
94°C for 30 sec, 57°C for 30 sec, 72°C for 30 sec, 35 cycles;
GAPDH, 94°C for 30 sec, 54°C for 30 sec, 72°C for 20 sec, 25
cycles. GAPDH was used as an internal control. The primer pairs
used for the amplification are listed in Table I. Five microliters of the product
were added to the 1% agarose gel electrophoresis and images of the
results were captured.
 | Table IPrimer pairs used in the
semi-quantitative polymerase chain reaction. |
Table I
Primer pairs used in the
semi-quantitative polymerase chain reaction.
| Genes | Primer pairs
(5′→3′) | Product size
(bp) |
|---|
| p38 |
| Sense |
CGGAGTGGCATGAAGCTGTAG | 346 |
| Antisense |
CCCTAGGAAACCAACACAGCA | |
| p53 |
| Sense |
TCTGGGACAGCCAAGTCTGT | 435 |
| Antisense |
GGAGTCTTCCAGT-GTGATGA | |
| p21 |
| Sense |
AAACGGGAACCAGGACAC | 248 |
| Antisense |
AGCAGCGGAACAAGGAGT | |
| Caspase-9 |
| Sense |
GGTTCTGGAGGATTTGGTGA | 325 |
| Antisense |
GACAGCCGTGAGAGAGAATGA | |
| Caspase-3 |
| Sense |
AGCAAACCTCAGGGAAACATT | 309 |
| Antisense |
GTCTCAATGCCACAGTCCAGT | |
| GAPDH |
| Sense |
AACGGATTTGGTCGTATTGGG | 216 |
| Antisense |
TCGCTCCTGGAAGATGGTGAT | |
Statistical analysis
All results were confirmed in at least three
separate experiments. Statistical analysis was performed using SPSS
17.0. Significant differences were determined by ANOVA, followed by
the Student’s t-test for statistical analysis. The data were
expressed as mean ± SD. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
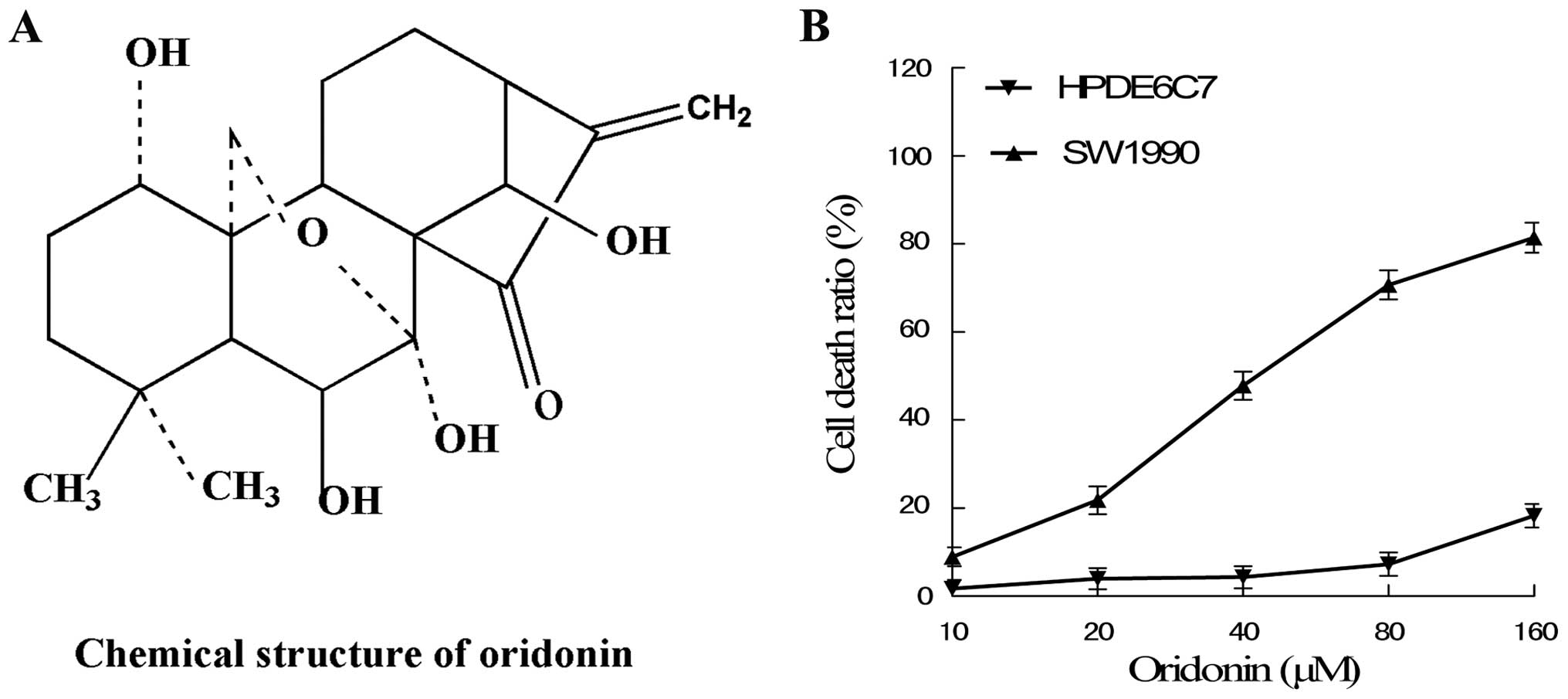
Cytotoxic effects of oridonin on SW1990
and HPDE6c7 cells
To detect the cytotoxic effects of oridonin on
SW1990 and HPDE6c7 cells, the cells were cultured with 10, 20, 40,
80 and 160 μM oridonin for 48 h. The results showed that oridonin
induced SW1990 cell death in a dose-dependent manner; the effects
at concentrations of 20–80 μM oridonin were apparent. In addition,
oridonin only exhibited a low cytotoxic effect on the human normal
pancreas cell line HPDE6c7 at below a concentration of 80 μM
oridonin (Fig. 1).
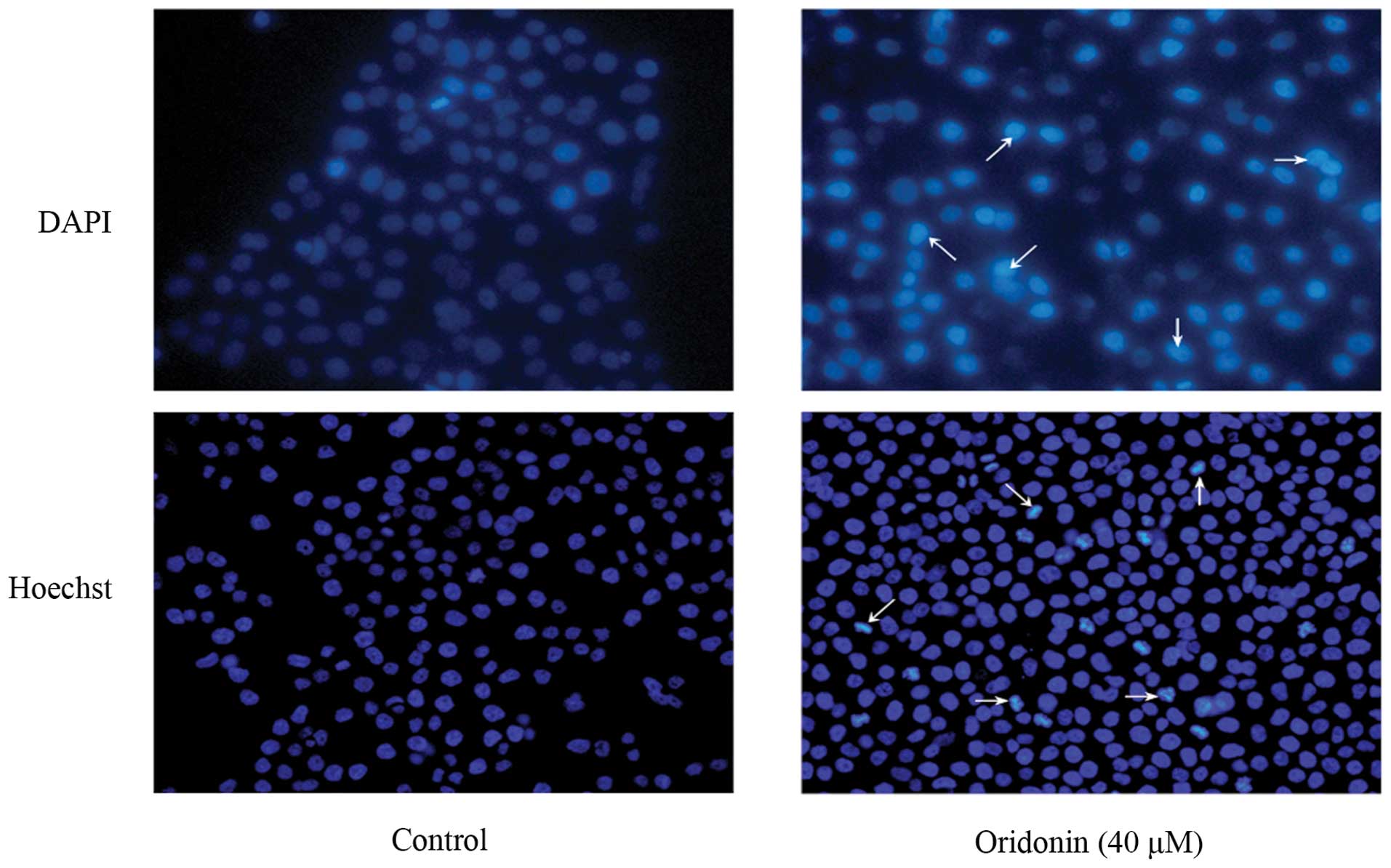
Oridonin induces apoptotic cell death in
SW1990 cells
To determine whether cell death induced by oridonin
in SW1990 cells was caused by apoptosis, SW1990 cells were treated
with 40 μM oridonin for 48 h, and the morphological changes were
examined with DAPI and Hoechst 33258 staining. As shown in Fig. 2, the nuclei of cells were round and
homogeneously stained in the control group; however, following
treatment with 40 μM oridonin, the cells displayed marked blebbing
of the nuclei and apoptotic bodies.
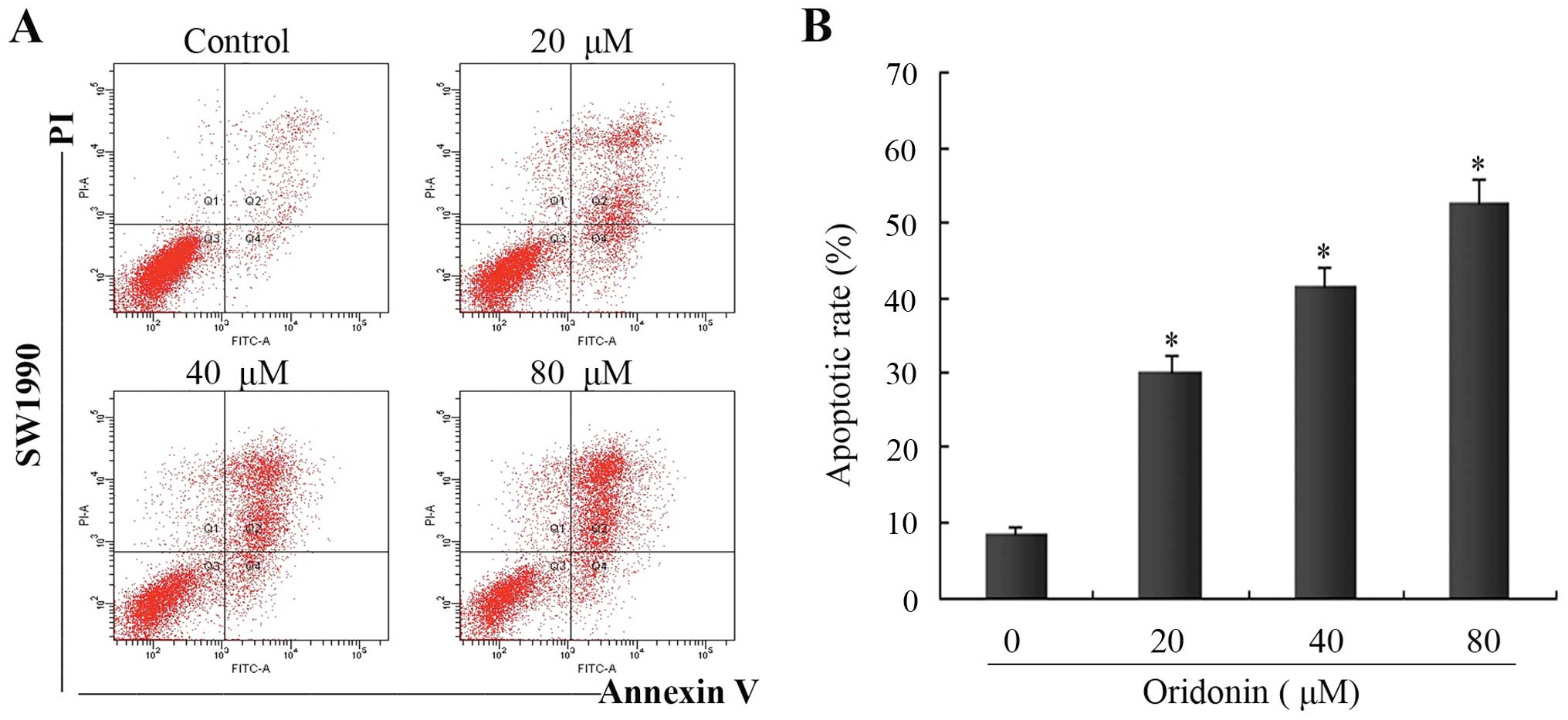
To further determine the features of SW1990 cell
death, flow cytometric analysis was performed using Annexin
V-FITC/PI-stained SW1990 cells. In the control group, the apoptotic
cell ratio was ~8.5±0.7%. In the presence of oridonin at 20, 40 and
80 μM, the numbers of apoptotic cells increased to 31.2±2.2,
41.4±2.9 and 50.5±3.4% at 48 h, respectively. There were
significant differences in the apoptotic ratio of cells between the
oridonin-treated groups and the control group (P<0.05) (Fig. 3). These results demonstrated that
oridonin treatment induced apoptosis in a dose-dependent manner;
one of the causes of SW1990 cell death induced by oridonin was
apoptosis.
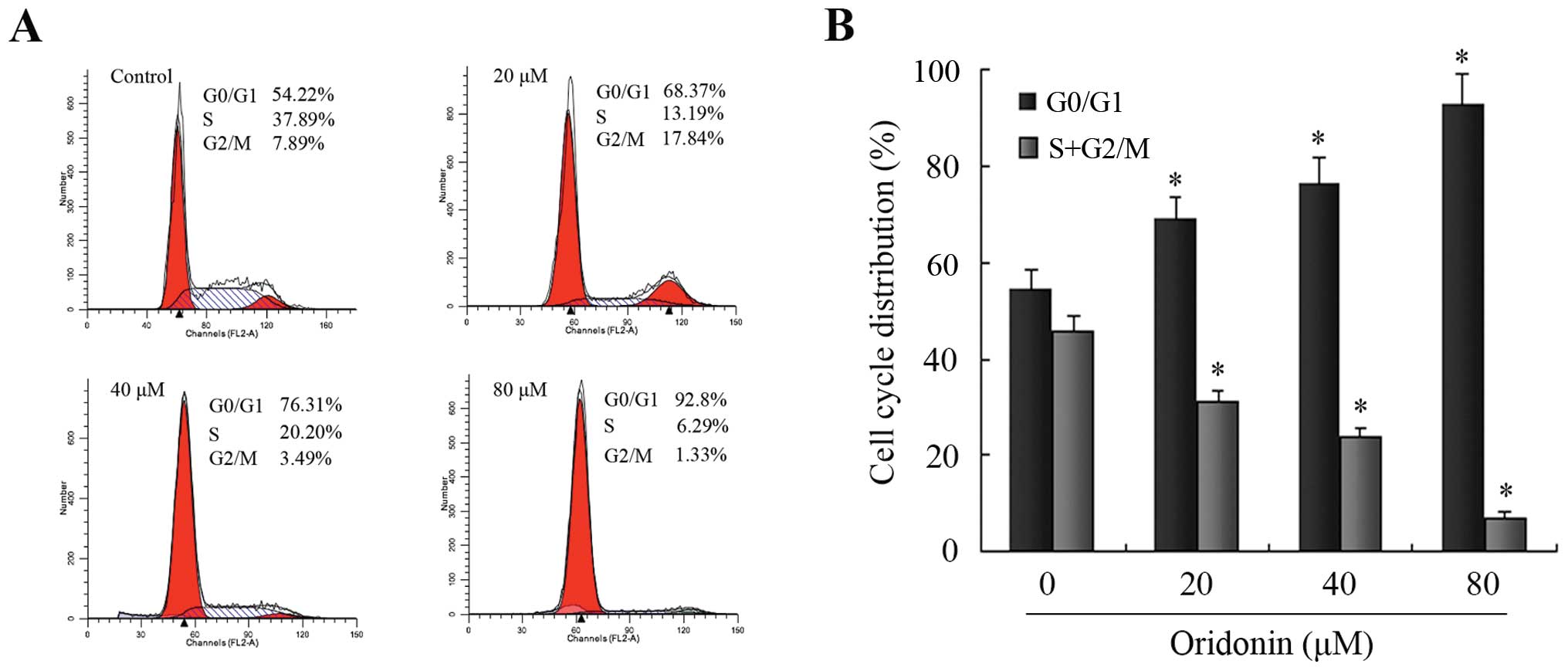
To further confirm the finding that oridonin induced
SW1990 apoptotic cell death, the DNA contents of SW1990 pancreatic
cancer cells treated with 0, 20, 40 and 80 μM oridonin for 48 h
were analyzed using a flow cytometer. As shown in Fig. 4, oridonin increased the ratio of
cells in the G0/G1 phase and decreased those in the S and G2/M
phase in a dose-dependent manner, and thus caused a significant
inhibition of cell cycle progression in SW1990 cells. There were
significant differences in the ratio of cells in the G0/G1 phase
and S plus the G2/M phases between the oridonin-treated groups and
the control group (P<0.05). These results suggested that
oridonin treatment caused SW1990 cell death by inducing apoptosis
associated with cell cycle arrest.
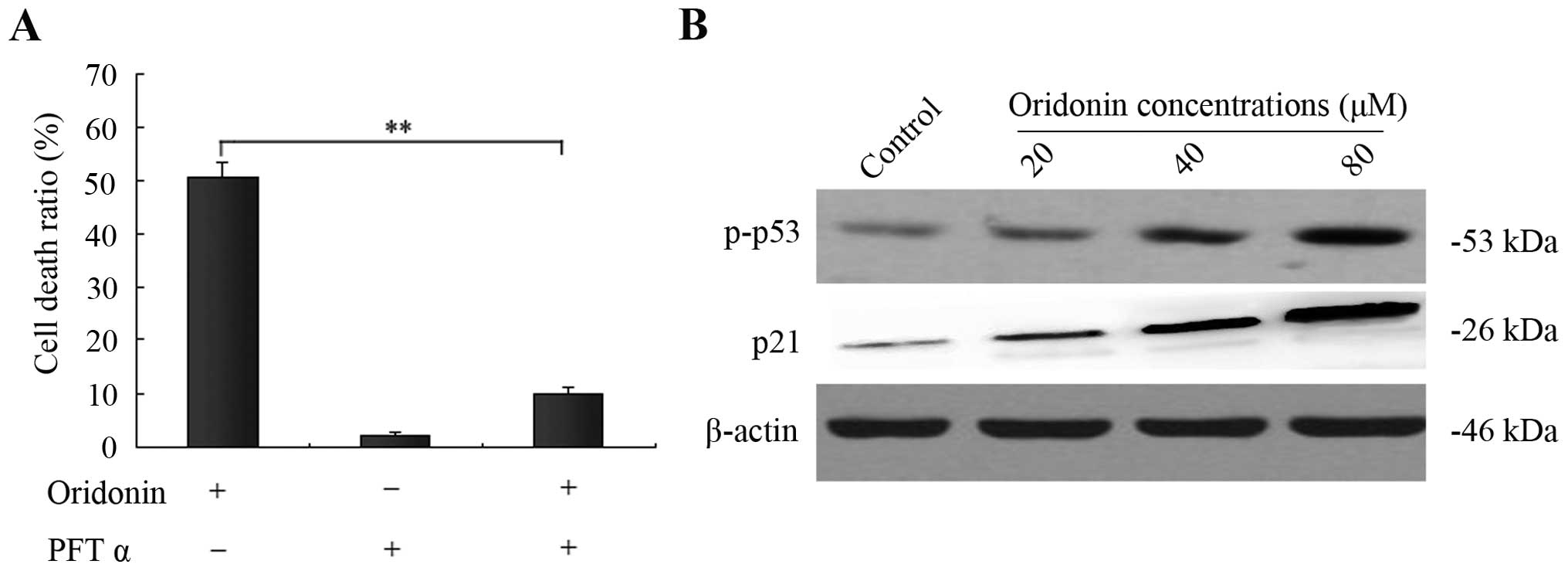
p53 and its downstream protein p21 are
involved in oridonin-induced SW1990 cell apoptosis
The tumor-suppressor gene product p53 has been
reported to mediate apoptosis in many experimental systems and is
capable of transcriptionally activating p21, which is responsible
for its tumor suppressive function (14). In the present study, the
p53-specific inhibitor PFT α was applied to evaluate the function
of p53 in oridonin-induced SW1990 cell apoptosis. After incubation
of SW1990 cells with 40 μM oridonin for 48 h, 15 μM PFT α
significantly reduced apoptosis from 50.5 to 9.8% (Fig. 5A). To further confirm this result,
western blot analysis was carried out to determine the p53 and
phospho-p53 expression. After treatment of SW1990 cells with
various concentrations of oridonin (0, 20, 40, 80 μM) for 48 h, the
expression of p53 did not change (data not shown), while the level
of phospho-p53 was markedly increased with increasing
concentrations of oridonin. Moreover, we further analyzed the
expression of p21, a downstream protein of p53, using western
blotting as above. Unexpectedly, expression of p21 was also
dramatically increased in a dose-dependent manner. These findings
revealed that p53 and its downstream protein p21 participated in
the oridonin-induced apoptosis (Fig.
5B).
Effects of inhibitors of ERK, p38, and
JNK on oridonin-treated SW1990 cells
To determine whether the MAPK family was involved in
the oridonin-induced SW1990 pancreatic cancer cell apoptosis, 10 μM
ERK inhibitor PD98059, 10 μM p38-MAPK inhibitor SB203580, and 10 μM
JNK inhibitor SP600125 were administered. SW1990 cells were
pretreated with 10 μM PD98059, 10 μM SB203580, and 10 μM SP600125
for 60 min, and then cultured with 40 μM oridonin for 48 h. The
results showed that the stimulatory effect of oridonin was
unaffected in the presence of PD98059 or SP600125. In contrast, 10
μM of the p38 inhibitor SB203580 partially inhibited the cell death
from 51.3 to 22.4% (Fig. 6A). On
the basis of these results, western blot analysis was carried out.
After treatment of SW1990 cells with various concentrations of
oridonin (0, 20, 40, 80 μM) for 48 h, the level of phospho-p38
increased with increasing concentrations of oridonin. However, in
the presence of 10 μM SB203580, SB203580 almost thoroughly reversed
the phosphorylation of p38 (Fig.
6B), indicating that the activation of p38 was also involved in
oridonin-induced SW1990 cell apoptosis.
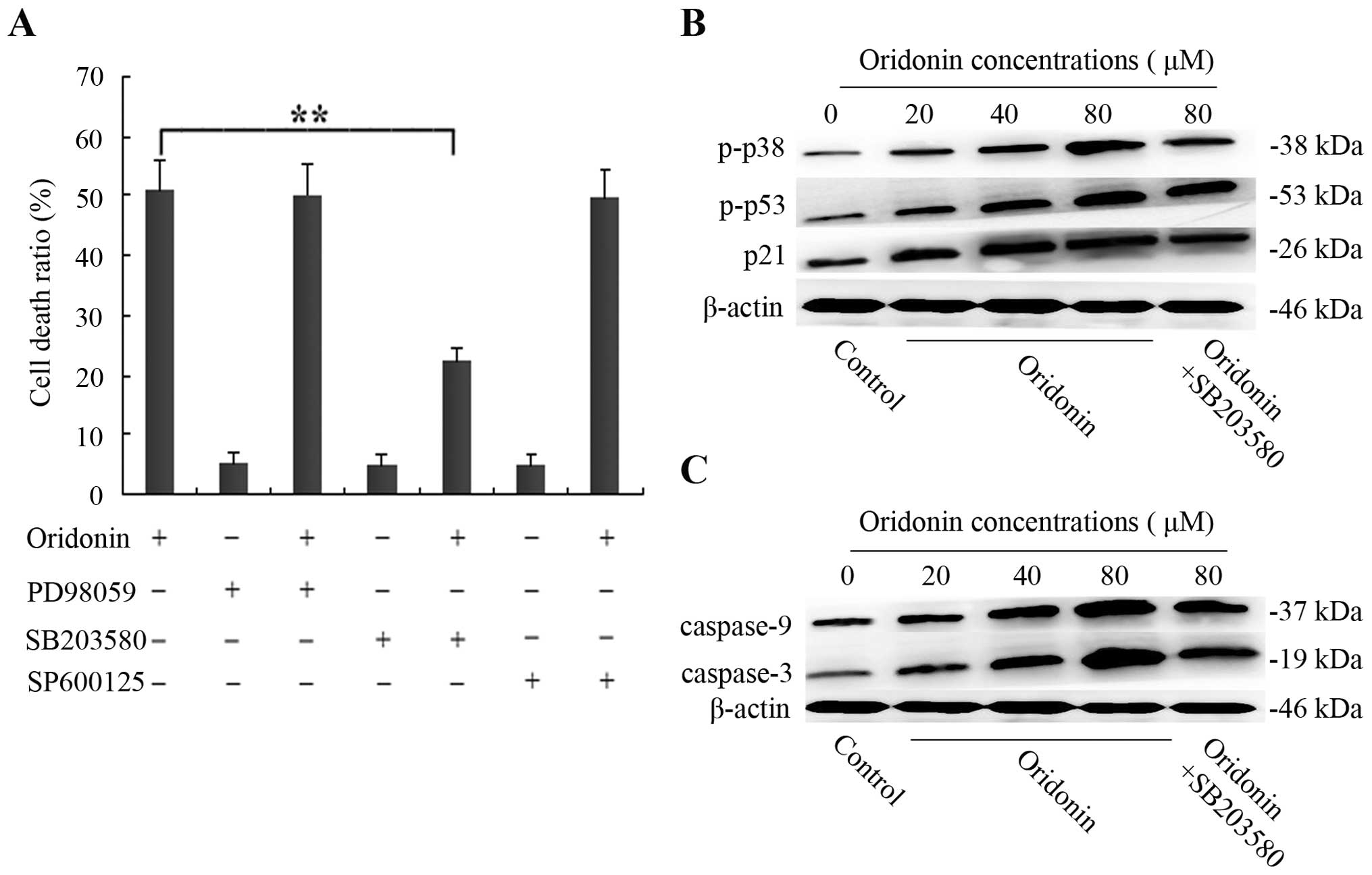 | Figure 6p38 is activated, which further
promotes the the activation of p53, p21, caspase-9 and caspase-3 in
SW1990 cells. (A) The SW1990 cells were pretreated with 10 μM
SB203580, 10 μM SP600125, or 10 μM PD98059 for 1 h and then
cultured with 40 μM oridonin for 48 h. Cell death ratio was
measured by CCK-8 assay. The results are representative of three
independent experiments. All data are presented as means ± SD.
**P<0.05 vs. the control. (B and C) The SW1990 cells
were treated with oridonin (20, 40, 80 μM) or vehicle for 48 h in
the absence or presence of 10 μM SB203580; and cell lysates were
separated by 12% SDS-PAGE electrophoresis, and the protein
expression of phospho-p38 and phospho-p53, p21, caspase-9 and
caspase-3 was detected by western blot analysis. The results are
representative of three independent experiments. All data are
presented as means ± SD. |
Activation of p38 kinase in
oridonin-treated SW1990 cells contributes to further activation of
p53 and p21
Previously, it has been reported that p38 kinase can
phosphorylate N-terminal serine residues of p53, thereby triggering
the proapoptotic transactivating function of p53, which in turn
leads to apoptosis (15). According
to this research, western blot analysis was performed to examine
the effects of SB203580 on the level of phospho-p53 and p21. As
shown in Fig. 6B, when SW1990 cells
were treated with various concentrations of oridonin (0, 20, 40, 80
μM) for 48 h, the enhanced phospho-p53 expression at 80 μM of
ordonin was significantly reduced by 10 μM SB203580.
Concomittantly, p21, the downstream protein of p53, diplayed a
similar trend. These observations indicated that oridonin-induced
SW1990 cell apoptosis did not require ERK and JNK activation;
however, it was dependent on p38-MAPK activity, which contributed
to further activation of p53 and its downstream protein p21.
Involvement of the caspase pathway in the
oridonin-induced SW1990 cell apoptosis
It is well known that caspases are required for
apoptosis. However, whether the caspase pathway was initiated by
p38 MAPK was unclear. We further examined the protein expression of
caspase-9, caspase-3 and the effects of SB203580 on the expression
of caspase-9 and -3. The expression of caspase-9 and caspase-3 in
SW1990 cells following the treatment with various concentrations of
oridonin (0, 20, 40, 80 μM) in the absence or presence of SB203580
for 48 h was examined by western blot analysis. The results showed
that the expression of caspase-9 and -3 was significantly increased
in a dose-dependent manner. However, in the presence of 10 μM
SB203580, the levels were unexpectedly decreased (Fig. 6C), indicating that oridonin induced
SW1990 apoptosis by p38 MAPK, which was dependent on the caspase
pathway.
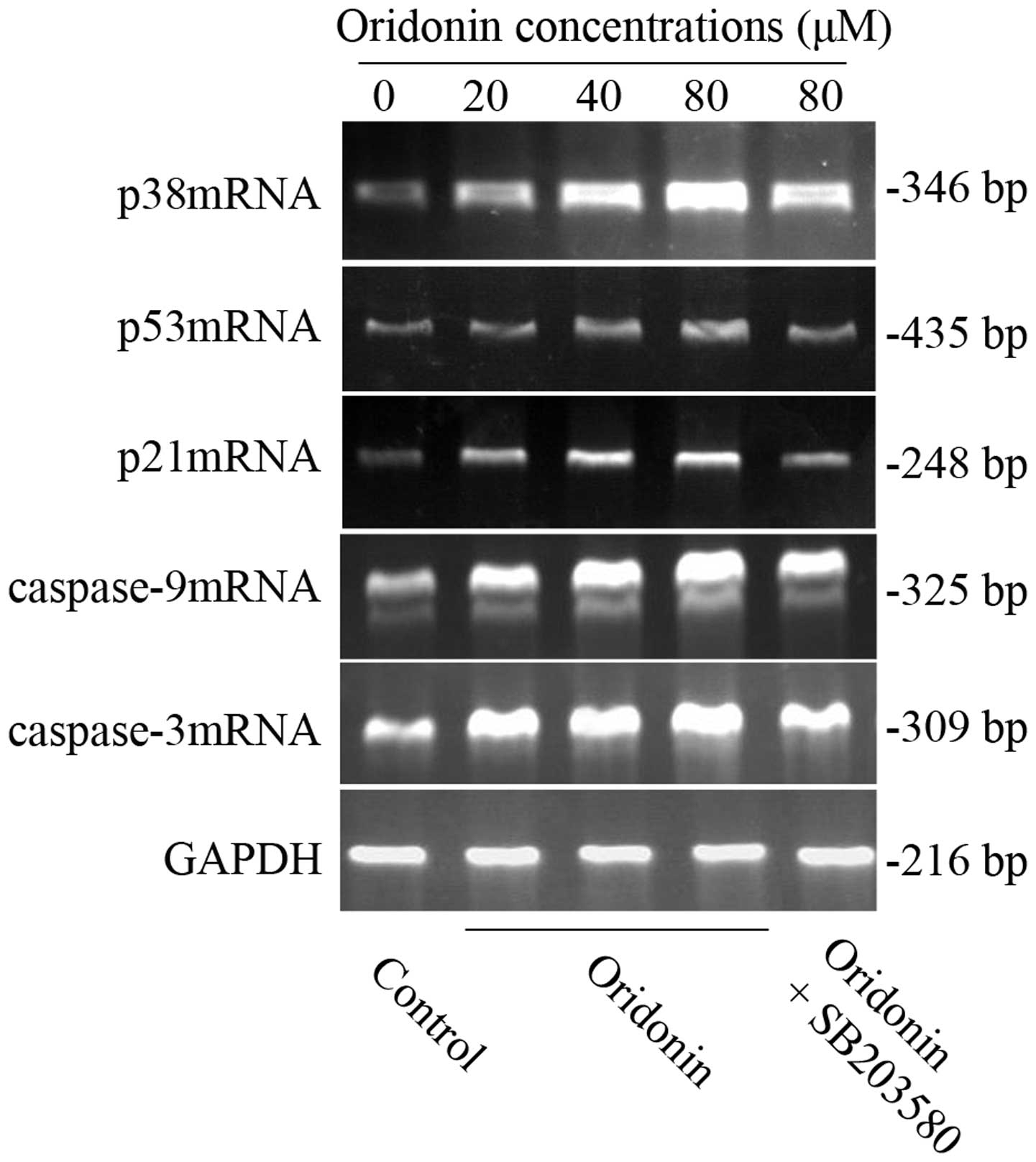
mRNA expression of p38, p53, p21,
caspase-9 and -3 in SW1990 cells
To further clarify whether the possible mechanism
was related to p38, p53, p21 caspase-9 and caspase-3 genes, RT-PCR
was carried out to detect the mRNA expression in the SW1990
pancreatic cancer cell line. After SW1990 cells were exposed to
various concentrations of oridonin (0, 20, 40, 80 μM) for 48 h, the
expression of p38, p53, p21, caspase-9 and caspase-3 mRNA all
increased in a dose-dependent manner. However, in the presence of
10 μM SB203580, their mRNA expression was similarly reduced
(Fig. 7) as well as the protein
expression. The results further confirmed our above conclusion.
Discussion
The aim of the present study was to determine
whether oridonin has potential in the treatment of pancreatic
cancer, one of the most lethal cancers. In the SW1990 pancreatic
cancer cell line, we found that oridonin was able to induce cell
death in a dose-dependent manner, and this in vitro effect
was through cell cycle arrest in the G0/G1 phase and induction of
apoptosis. More importantly, we found that p38, not ERK or JNK, was
activated, and the expression levels of p53, p21 and caspase-9 and
-3 were upregulated, which were reduced in the presence of SB203580
in oridonin-induced SW1990 cell apoptosis. The activation of p38
MAPK was involved in the induction of apoptosis in SW1990
pancreatic cancer cells and this may be one of the mechanisms.
These results provide strong evidence to support the notion that
oridonin has strong activity against pancreatic cancer. To the best
of our knowledge, this is the first in vitro study
concerning the chemotherapeutic effects of oridonin against SW1990
pancreatic cancer cells.
The p53 pathway is targeted for inactivation in most
human cancers either directly or indirectly, highlighting its
critical function as a tumor-suppressor gene (16). The p53 tumor suppressor is essential
for maintaining genomic stability in mammals. p53 function is
usually switched off; although when cells are subjected to stress
signals such as hypoxia, radiation, or chemotherapeutic drugs, p53
is activated, and its ubiquitin-dependent degradation is blocked
leading to an accumulation of active p53 transcription factor
(17). Upon activation, p53
mediates a growth-suppressive effect on cells by blocking the cell
cycle or it can lead cells to undergo programmed cell death
primarily by binding to particular DNA sequences and activating
transcription of specific genes (18). In addition, the tumor suppressor p53
is implicated in induction of growth arrest. Following DNA damage,
p53 increases the transcription of p21, which plays a pivotal role
in controlling cell-cycle progression by binding directly to
CDK/cyclin complexes (19). The
expression of cyclin-dependent kinase inhibitor p21 has been
implicated in chemotherapy-induced cell cycle arrest in numerous
human cancers (20,21). In our study, we confirmed that
oridonin is capable of activating p53 and p21 in pancreatic cancer
cells. CCK-8, flow cytometric analysis and western blot analysis
assay showed that the activation of p53 was accompanied by the
upregulation of p21, which was able to arrest the cell cycle in the
G0/G1 phase, thus transmitting apoptotic signals in
oridonin-treated SW1990 cells, as verified by the evidence that the
ratio of cell death was effectively suppressed by PFT α. This is in
agreement with previous findings that the p53 inhibitor pifithrin
was able to significantly reduce oridonin-induced apoptosis in
HepG2 cells (7).
Mitogen-activated protein kinases (MAPKs) are
serine/threonine kinases that mediate intracellular signaling
associated with a variety of cellular activities including cell
proliferation, differentiation, survival, death and transformation
(22,23). The three main members that integrate
the MAPK family in mammalian cells are stress-activated protein
kinase c-Jun NH2-terminal kinase (JNK), stress-activated protein
kinase 2 (SAPK2, p38), and the extracellular signal-regulated
protein kinases (ERK1/2, p44/p42). The p38 signaling pathways are
activated by proinflammatory (TNF-α, IL-6 or IL-1) or
anti-inflammatory (EGF, TGF-β) cytokines, but also in response to
cellular stresses such as genotoxic, osmotic, hypoxic or oxidative
stress (24). Activation of p38
kinase has also been implicated in anticancer drug-induced
apoptosis (25). Of note, in our
study, pretreatment of SB203580 was effective in preventing
oridonin-induced apoptosis, indicating that p38 was involved in
this process. This is consistent with previous studies that the
p38-MAPK pathway was involved in cell cycle regulation and/or
apoptosis (26).
MAPKs have been shown to be responsive to different
stress stimuli. Upon activation, p38 phosphorylates and regulates
various transcription factors (including ATF-2, NF-κB, Elk-1, Max,
Mac, p53 or Stat1) (27,28) and other cell cycle and apoptotic
mediators (e.g., p21, Cdc25A, Bcl-2) (29), but, of particular note, it has been
demonstrated to phosphorylate the tumor suppressor p53, which can
initiate the p53 response, leading to cell cycle arrest and
apoptosis (30,31). Previous studies have documented that
activation of the p38-MAPK pathway may lead to p53-induced
apoptosis (32). In the present
study, we found that in response to oridonin treatment increased
expression of p-p38 was accompanied by the upregulation of p-p53
and p21, suggesting that the activation of p38 further contributed
to the activation of p53 and p21. This was further verified by the
evidence that high levels of phospho-p53 and p21 were effectively
inhibited by SB203580. This is consistent with a previous study
which found that a p38 inhibitor was able to partially inhibit cell
death and the activation of p53 in oridonin-treated HepG2 cells
(7).
Caspases are a family of cysteine proteases, which
play key roles in promoting the degradative changes associated with
apoptosis and are divided into two classes based on the lengths of
their N-terminal prodomains, including upstream caspases such as
caspase-8 and -10 and downstream caspases such as caspase-3, -6 and
-9 (33). In general, caspase
activation is believed to be involved in apoptosis. In the present
study, western blot analysis and RT-PCR showed that caspase-9 and
active-caspase-3 expression was significantly upregulated in
oridonin-treated SW1990 cells. However, the high levels of
caspase-9 and -3 were also correspondingly suppressed by SB203580.
These results indicated that p38 was responsible for causing the
activation of caspase-9 and -3, which executed the oridonin-induced
SW1990 apoptosis.
Collectively, the present study suggests that the
MAPK-p38 pathway was selectively activated in SW1990 pancreatic
cancer cells following treatment with oridonin, and the induction
of apoptosis was dependent on p53 and caspase activation. However,
certain molecular links remain to be clarified, such as whether p53
is linked to caspase, and how p38 signaling acts on the caspase
pathway. The specific mechanisms involved require further study.
Based on these results, further clinical studies are necessary to
confirm our findings in patients with pancreatic cancer.
Acknowledgements
We are grateful for the financial support from the
Administration of Traditional Chinese Medicine of Zhejiang, China
(grant no. 2013ZQ026) and The National Natural Science Foundation
of China (grant no. 63412018).
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics. CA Cancer J Clin. 59:225–249. 2009.
|
|
2
|
Löhr M: Is it possible to survive
pancreatic cancer? Nat Clin Pract Gastroenterol Hepatol. 3:236–237.
2006.PubMed/NCBI
|
|
3
|
Omura N and Goggins M: Epigenetics and
epigenetic alterations in pancreatic cancer. Int J Clin Exp Pathol.
2:310–326. 2009.PubMed/NCBI
|
|
4
|
Tan W, Lu J, Huang M, et al: Anti-cancer
natural products isolated from Chinese medicinal herbs. Chin Med.
6:272011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao FH, Hu XH, Li W, et al: Oridonin
induces apoptosis and senescence in colorectal cancer cells by
increasing histone hyperacetylation and regulation of p16, p21, p27
and c-myc. BMC Cancer. 10:6102010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheng Y, Qiu F, Ye YC, Tashiro S, Onodera
S and Ikejima T: Oridonin induces G2/M arrest and apoptosis via
activating ERK-p53 apoptotic pathway and inhibiting PTK-RAS-RAF-JNK
survival pathway in murine fibrosarcoma L929 cells. Arch Biochem
Biophys. 490:70–75. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang J, Wu L, Tashiro S, Onodera S and
Ikejima T: Reactive oxygen species mediate oridonin-induced HepG2
apoptosis through p53, MAPK, and mitochondrial signaling pathways.
J Pharmacol Sci. 107:370–379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jin S, Shen JN, Wang J, Huang G and Zhou
JG: Oridonin induced apoptosis through AKT and MAPKS signaling
pathways in human osteosarcoma cells. Cancer Biol Ther. 6:261–268.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li D, Wu LJ, Tashiro S, Onodera S and
Ikejima T: Oridonin induced A431 cell apoptosis partially through
blockage of the RAS/RAF/ERK signal pathway. J Pharmacol Sci.
103:56–66. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu HZ, Yang YB, Xu XD, et al: Oridonin
induces apoptosis via PI3K/Akt pathway in cervical carcinoma HeLa
cell line. Acta Pharmacol Sin. 28:1819–1826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu YQ, Mu ZQ, You S, et al: Fas/FasL
signaling allows extracelluar-signal regulated kinase to regulate
cytochrome c release in oridonin-induced apoptotic U937 cells. Biol
Pharm Bull. 29:1873–1879. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hsieh TC, Wijeratne EK, Liang JY,
Gunatilaka AL and Wu JM: Differential control of growth, cell cycle
progression, and expression of NF-kappaB in human breast cancer
cells MCF-7, MCF-10A, and MDA-MB-231 by ponicidin and oridonin,
diterpenoids from the Chinese herb Rabdosia rubescens.
Biochem Biophys Res Commun. 337:224–231. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ikezoe T, Chen SS, Tong XJ, et al:
Oridonin induces growth inhibition and apoptosis of a variety of
human cancer cells. Int J Oncol. 23:1187–1193. 2003.PubMed/NCBI
|
|
14
|
Bunz F, Dutriaux A, Lengauer C, et al:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Perfettini JL, Castedo M, Nardacci R, et
al: Essential role of p53 phosphorylation by p38 MAPK in apoptosis
induction by the HIV-1 envelope. J Exp Med. 201:279–289. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuribayashi K and El-Deiry WS: Regulation
of programmed cell death by the p53 pathway. Adv Exp Med Biol.
615:201–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fuster JJ, Sanz-González SM, Moll UM and
Andrés V: Classic and novel roles of p53: prospects for anticancer
therapy. Trends Mol Med. 13:192–199. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Burns TF, Fei P, Scata KA, et al:
Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic
catastrophe in paclitaxel (taxol)-exposed cells. Mol Cell Biol.
23:5556–5571. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar
|
|
20
|
Shankar S, Singh G and Srivastava RK:
Chemoprevention by resveratrol: molecular mechanisms and
therapeutic potential. Front Biosci. 12:4839–4854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee JT, Lehmann BD, Terrian DM, et al:
Targeting prostate cancer based on signal transduction and cell
cycle pathways. Cell Cycle. 7:1745–1762. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McCubrey JA, LaHair MM and Franklin RA:
Reactive oxygen species-induced activation of the MAP kinase
signaling pathways. Antioxid Redox Signal. 8:1775–1789. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kholodenko BN and Birtwistle MR:
Four-dimensional dynamics of MAPK information processing systems.
Wiley Interdiscip Rev Syst Biol Med. 1:28–44. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rodríguez-Berriguete G, Fraile B,
Martínez-Onsurbe P, et al: MAP kinases and prostate cancer. J
Signal Transduct. 2012:1691702012.PubMed/NCBI
|
|
25
|
Schaeffer HJ and Weber MJ:
Mitogen-activated protein kinases: specific messages from
ubiquitous messengers. Mol Cell Biol. 19:2435–2444. 1999.PubMed/NCBI
|
|
26
|
Zarubin T and Han J: Activation and
signaling of the p38 MAP kinase pathway. Cell Res. 15:11–18. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Whyte J, Bergin O, Bianchi A, McNally S
and Martin F: Key signalling nodes in mammary gland development and
cancer. Mitogen-activated protein kinase signalling in experimental
models of breast cancer progression and in mammary gland
development. Breast Cancer Res. 11:2092009. View Article : Google Scholar
|
|
28
|
Royuela M, Rodríguez-Berriguete G, Fraile
B and Paniagua R: TNF-α/IL-1/NF-κB transduction pathway in human
cancer prostate. Histol Histopathol. 23:1279–1290. 2008.
|
|
29
|
Thornton TM and Rincon M: Non-classical
p38 map kinase functions: cell cycle checkpoints and survival. Int
J Biol Sci. 5:44–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Harris SL and Levine AJ: The p53 pathway:
positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu GS: The functional interactions between
the p53 and MAPK signaling pathways. Cancer Biol Ther. 3:156–161.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bulavin DV and Fornace AJ Jr: p38 MAP
kinase’s emerging role as a tumor suppressor. Adv Cancer Res.
92:95–118. 2004.
|
|
33
|
Degterev A, Boyce M and Yuan J: A decade
of caspases. Oncogene. 22:8543–8567. 2003. View Article : Google Scholar : PubMed/NCBI
|