Introduction
Quaking (QKI) is a KH-type RNA-binding protein
encoded by the quaking (qk) locus (1). There are three isoforms of QKI, namely
QKI-5, QKI-6 and QKI-7 (2,3). QKI-5 is the only nuclear isoform and
shuttles between the nucleus and the cytoplasm, whereas QKI-6 and
QKI-7 are localized in the cytoplasm (4,5).
As an RNA-binding protein, QKI
post-transcriptionally regulates the mRNA stability, translation
efficiency or RNA transportation of target genes via specifically
binding to the cis-elements within the 3′-untranslated
region (3′UTR) (6–10). Among these targets, MAG and MBP,
which are essential for postnatal myelination are well
characterized. Overexpression of QKI-5 caused MBP mRNA retention in
the nucleus, resulting in a reduction in its protein level and
defects in myelination development (10,11).
In adult mammals, qki mRNAs can be detected
in tissues, such as the heart, skeletal, lung, testes and immune
cells in addition to the central nervous system (CNS) (9,12–15).
In a search for putative RNA targets recognized by QKI, a bipartite
consensus sequence ACUAAY-N(1–20)-UAAY
was defined as a QKI response element (QRE), and 1,430 putative
mRNA targets were identified by bioinformatic analysis (16). Thirteen percent were found to be
associated with cell proliferation and cell cycle regulation. Among
them, forkhead box O1 (FOXO1) appeared to be an appealing candidate
due to its potent roles in regulating the cell cycle, apoptosis,
stem cells and the aging process (17–23).
Low and even undetectable FOXO1 expression has been observed in
prostate, breast and colon cancers, suggesting its tumor-suppressor
role (24–29). Nevertheless, the molecular mechanism
for the aberrant expression of FOXO1 in those cancers remains to be
elucidated.
In the present study, we provide initial evidence
showing that QKI-5 repressed FOXO1 expression via decreasing its
mRNA stability. ATRA and 5-FU increased FOXO1 expression at the
post-transcriptional level through inhibition of QKI. Based on the
findings, we suggest that targeting QKI may provide a new insight
into novel therapies for the treatment of breast cancers.
Materials and methods
Cell culture and reagents
Human breast carcinoma cell lines, MCF-7, T47-D,
MDA-MB-231 and SKBR-3, were cultured in Dulbecco’s modified Eagle’s
medium (low glucose, Gibco, UK) supplemented with 10% fetal bovine
serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM
L-glutamine at 37°C in a humidified atmosphere of 5%
CO2. ATRA (Sigma, St. Louis, MO, USA) was dissolved in
ethanol at the concentration of 10−3 mol/l as a stock.
5-FU (Sigma) was dissolved in DMSO at the concentration of 5 mg/ml
as a stock and maintained at −20°C. Transcription inhibitor
actinomycin D (Sigma) was dissolved in DMSO at the concentration of
5 mg/ml.
Plasmids, siRNAs and
oligonucleotides
The FOXO1 3′UTR sequence containing three QKI
response elements (QREs) corresponding to base pairs 3275–3279,
3337–3341 and 5298–5302 (wild-type, denoted pGL3-F) or its 3′UTR
sequence with deleted QREs (mutant, denoted pGL3-FM) were amplified
from MCF-7 cDNA and cloned into our previously recombined pGL3
vector with EcoRI, EcoRV, PstI inserted
downstream of the XbaI site. pcDNA3.1(+)-flag-QKI-5 was
kindly provided by Professor Feng Yue (Emory University, Atlanta,
GA). Stealth™ siRNAs targeting QKI were synthesized by Invitrogen
(Invitrogen Life Technologies, Carlsbad, CA, USA) and were
dissolved in DEPC-treated H2O at a concentration of 20
pmol/μl as a stock.
Adenovirus
Recombinant adenoviruses expressing flag-tagged
QKI-5 were generated using the AdEasy system (MP Biomedicals, Inc.,
Solon, OH, USA) according to the manufacturer’s instructions. MCF-7
cells were infected with the adenovirus-expressing vectors for 2 h
and subsequently cultured for 24 h before being harvested.
RT-PCR
Following the indicated treatments, cells were
harvested for isolation of RNA using TRIzol reagent (Invitrogen)
according to the manufacturer’s instructions. First-strand cDNA
synthesis was performed using random primers catalyzed by murine
leukemia virus (M-MLV) reverse transcriptase. PCR conditions were
as follows: 95°C for 5 min followed by 25 cycles of 95°C for 20
sec, 55°C for 20 sec, and 72°C for 30 sec. The primers for
amplification were: QKI-5, sense 5′-atacagaccgctgtcatgc-3′ and
antisense 5′-tcacagtaactgcctctgtc-3′; FOXO1, sense
5′-atcaccaaggccatcgagag-3′ and antisense
5′-tggccagactggagagatgc-3′; β-actin, sense
5′-gaaaatctggcaccacacct-3′ and antisense
5′-ggccggactcgtcatactc-3′.
Western blotting
Following the indicated treatments, cells were
harvested and lysed in RIPA buffer containing 50 mM Tris-HCl, pH
7.5, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate and
50 mM NaF. One tablet of protease inhibitor mixture (Complete Mini,
Roche Applied Science, Burgess Hill, UK) was added just prior to
use. Then 100 μg of the protein lysates was separated by 12%
SDS-PAGE and transferred onto nitrocellulose membranes. After
blocking in a 5% non-fat dried milk solution in washing buffer
containing 10 mmol/l Tris (pH 7.5), 50 mmol/l NaCl and 0.02%
Tween-20 (TBST), membranes were incubated overnight at 4°C with
rabbit polyclonal anti-QKI (1:1,500; produced by our laboratory),
anti-FOXO1 (1:2,500; Abcam, Cambridge, UK) and mouse polyclonal
anti-α-tubulin (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA,
USA) antibodies. After being washed three times with TBST, the
membranes were incubated for 1 h with horseradish
peroxidase-coupled secondary antibodies (1:1,000 dilution; Santa
Cruz Biotechnology) at room temperature. Signals were detected with
the ECL kit (Amersham Pharmacia Biotech, Amersham, UK). The scanned
images were quantified using Kodak Digital Science one-dimensional
software (Eastman Kodak Co., New Haven, CT, USA).
Reporter gene assay
To detect the interaction between FOXO1 3′UTR and
QKI-5, HEK-293 cells were seeded in 24-well plates and transfected
with 400 ng of pGL3-F or pGL3-FM in combination with increased
doses of pcDNA3.1-QKI-5, respectively. Cells were cotransfected
with 50 ng of pBIND (a plasmid constitutively expressing
Renilla luciferase) to normalize for the transfection
efficiency. After 24 h of transfection, cells were lysed using
passive lysis buffer and analyzed for firefly and Renilla
luciferase activities using the Dual-Luciferase Reagent Assay kit
(Promega, Madison, WI, USA) according to the manufacturer’s
instructions.
Real-time PCR assay for FOXO1 mRNA
decay
To analyze the stability of FOXO1 mRNA,
transcription inhibitor actinomycin D (2 μg/ml) was added to the
MCF-7 cells with the indicated treatment to stop further
transcription. At the times indicated, total RNA was harvested
using TRIzol reagent (Invitrogen). First-strand cDNA synthesis was
performed using random primers catalyzed by murine leukemia virus
(M-MLV) reverse transcriptase. Real-time PCR was performed with the
SYBR® Premix Ex Taq™ (Takara Biotechnology, Dalian,
China) in 25 μl reactions using ABI PRISM® 7500
Real-Time PCR System (Applied Biosystems, Grand Island, NY, USA).
The primers for amplification were: FOXO1, sense
5′-cacacagtgtcaagacaacgaca-3′ and antisense
5′-ttctctcagttcctgctgtcagac-3′. The reactions were amplified for 40
cycles of 95°C for 5 sec and 60°C for 34 sec with predenaturation
at 95°C for 30 sec. FOXO1 mRNA relative abundance was determined by
normalizing to β-actin using the ΔCT method, where
CT is the threshold cycle. The relative amount of FOXO1
mRNA without actinomycin D treatment was set to 100%.
RNA immunoprecipitation
MCF-7 cells were seeded in 100-mm dishes and
transfected with recombinant adenoviruses expressing flag-tagged
QKI-5 for 24 h. After protein-RNA crosslinking with 1%
formaldehyde, cells were lysed in a buffer containing 10 mM HEPES
(pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.1%
NP-40, 50 mM NaF, 10 mM Na3VO4, 10 mM sodium
pyrophosphate, 50 mM disodium glycerol phosphate, 10 nM okadaic
acid, 0.2% VRC, 100 U/ml RNasin and 1/25 v/v Complete EDTA-free
protease inhibitor cocktail. The lysed cells were centrifuged at
12,000 × g for 10 min at 4°C. The supernatants were pre-cleaned for
60 min at 4°C by using 5 μl of Protein-A Sepharose beads (BD
Biosciences, San Jose, CA, USA). After centrifugation, the
supernatant was incubated with 30 μg of unrelated antibody (IgG,
Sigma) or anti-flag at 4°C for 60 min. After incubation, 5 μl of
protein-A Sepharose beads was added to all tubes and incubated for
a further 60 min at 4°C. The tubes were centrifuged at 12,000 × g
for 5 min, and the precipitated beads were washed with lysis buffer
three times. The RNA in the immunoprecipitated complex and the RNA
in the previously saved input fraction were released by reversing
the cross-linking at 65°C for 2 h with 200 mM NaCl and 20 μg of
proteinase K. The RNA was extracted as described above. Specific
primers used to detect the presence of FOXO1 mRNAs were synthesized
as follows: sense 5′-ttgttacatagtcagcttg-3′ and antisense
5′-tcactttcctgcccaaccag-3′. PCR conditions were: 95°C for 5 min
followed by 25 cycles of 95°C for 15 sec, 55°C for 20 sec, and 72°C
for 1 min. All experiments described in this study were performed
in triplicate.
Statistical analysis
Data are expressed as mean ± SD from three
independent experiments. Statistical analysis was performed using
Student’s t test to assess the differences between the experimental
groups. p<0.05 was considered to indicate a statistically
significant result.
Results
FOXO1 as a potential target of QKI
Due to limited information concerning the function
of QKI-5 outside the CNS system, seeking for and verifying
potential target genes may be an efficient way to research the
function of QKI-5. We selected multiple potential targets of QKI,
which are closely related to cell cycle regulation, from the 1430
target genes identified by Galarneau and Richard (16). Among them, FOXO1 was found to be the
most significant.
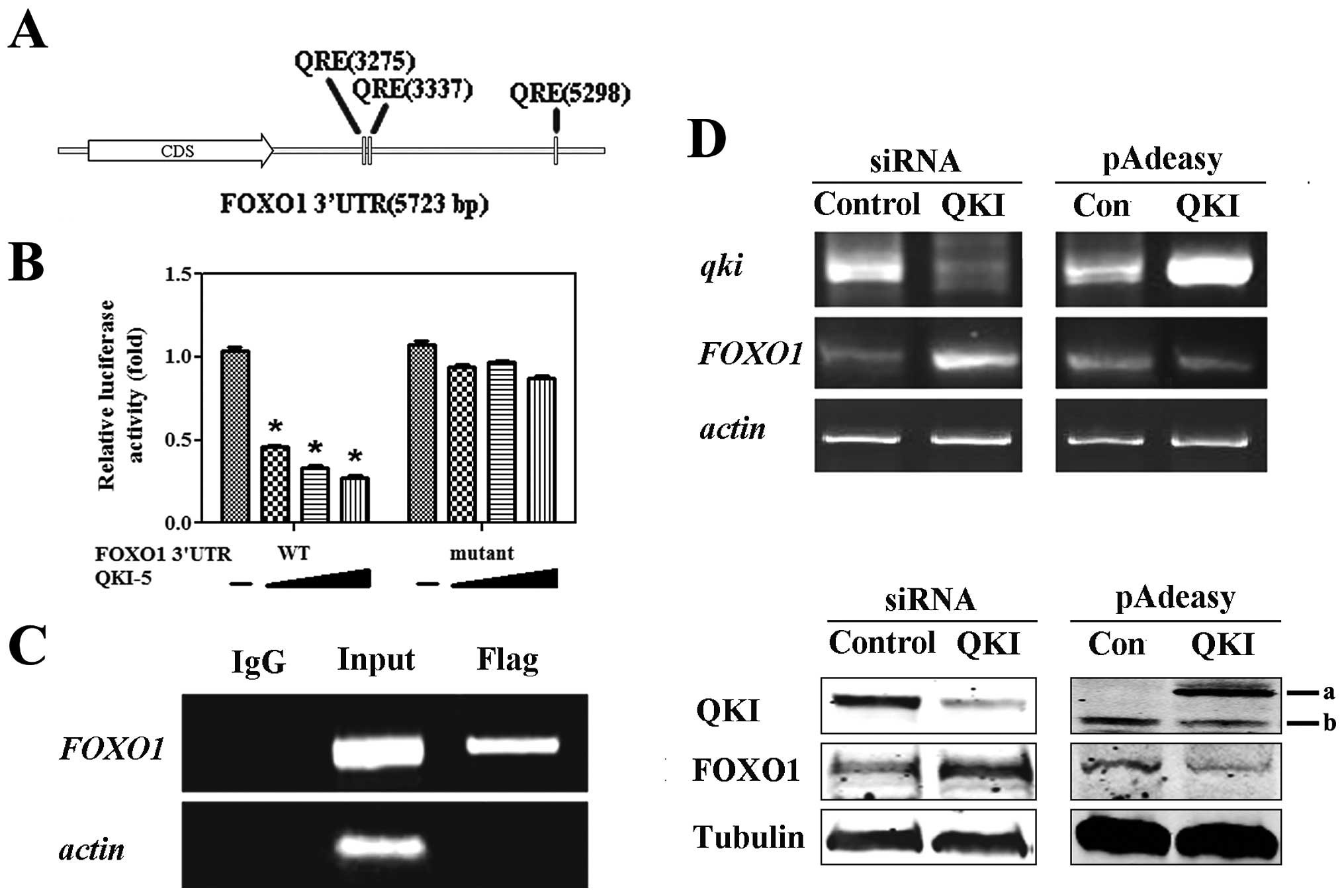
Bioinformatics analysis revealed that FOXO1 3′UTR
contained three conserved QREs (Fig.
1A). To test whether FOXO1 is a target of QKI, two constructs
with FOXO1 3′UTR downstream of firefly luciferase in the pGL3
vector were included. pGL3-F harbored the wild-type QRE while
mutant FOXO1 3′UTR (pGL3-FM) had none of the three QREs. As shown
in Fig. 1B, the luciferase
activities of pGL3-F were gradually reduced by QKI in a
dose-dependent manner, whereas that of GL3-FM was rarely changed by
QKI. On the basis of the above observations, we hypothesized that
QKI may mediate a negative regulation of FOXO1.
In order to detect the in vivo interaction
between QKI and FOXO1 3′UTR, an RNA co-immunoprecipitation
experiment was performed. MCF-7 cells were transfected with
pcDNA3.1(+)-flag-QKI for 24 h, and total cellular extracts were
then incubated with either an anti-flag or a nonspecific antibody
(IgG). By RT-PCR analysis, only the anti-Flag immunoprecipitate
contained FOXO1 RNA whereas the one with non-specific IgG did not
(Fig. 1C).
To verify the negative regulation of FOXO1 by QKI,
small interfering RNA-mediated specific knockdown of QKI increased
the mRNA and protein levels of FOXO1, whereas overexpression of QKI
inhibited FOXO1 expression (Fig.
1D).
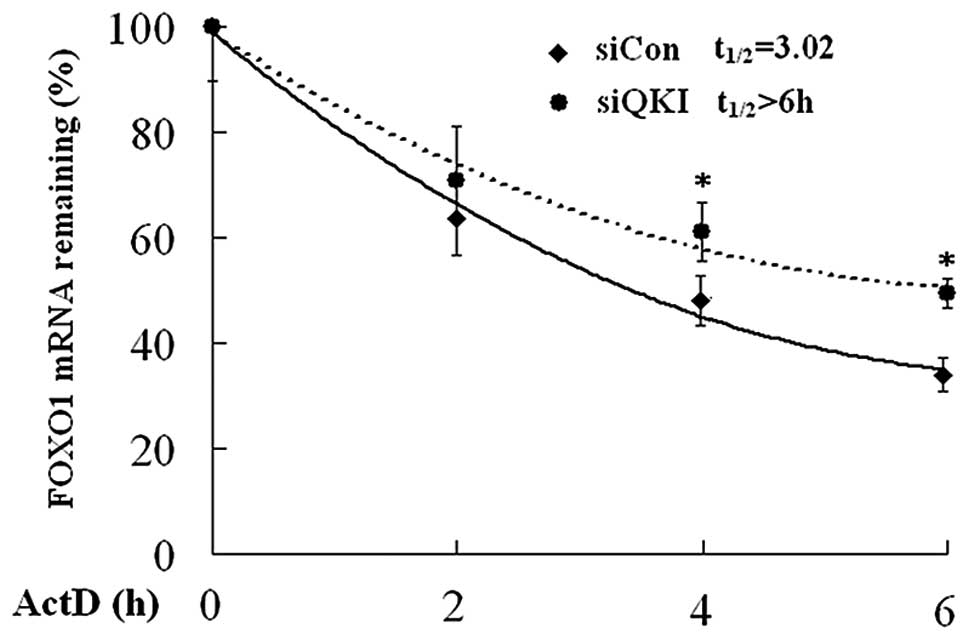
QKI decreases the mRNA stability of
FOXO1
As QKI reduced FOXO1 mRNA level to a great extent,
it is reasonable to deduce that QKI, as an RNA binding protein, may
regulate the mRNA stability of FOXO1. To test this, actinomycin D,
a transcription inhibitor, was used to block the transcription
initiation in cells transfected with negative control (siCon) and
QKI siRNA (siQKI). Cells were harvested at different time points,
and real-time PCR was employed to determine the decaying rate of
FOXO1 mRNA. As shown in Fig. 2, the
half-life of FOXO1 mRNA was 3.02 h in siCon cells, whereas it was
prolonged to 6 h in siQKI cells. These data suggest that QKI
destabilizes FOXO1 mRNA.
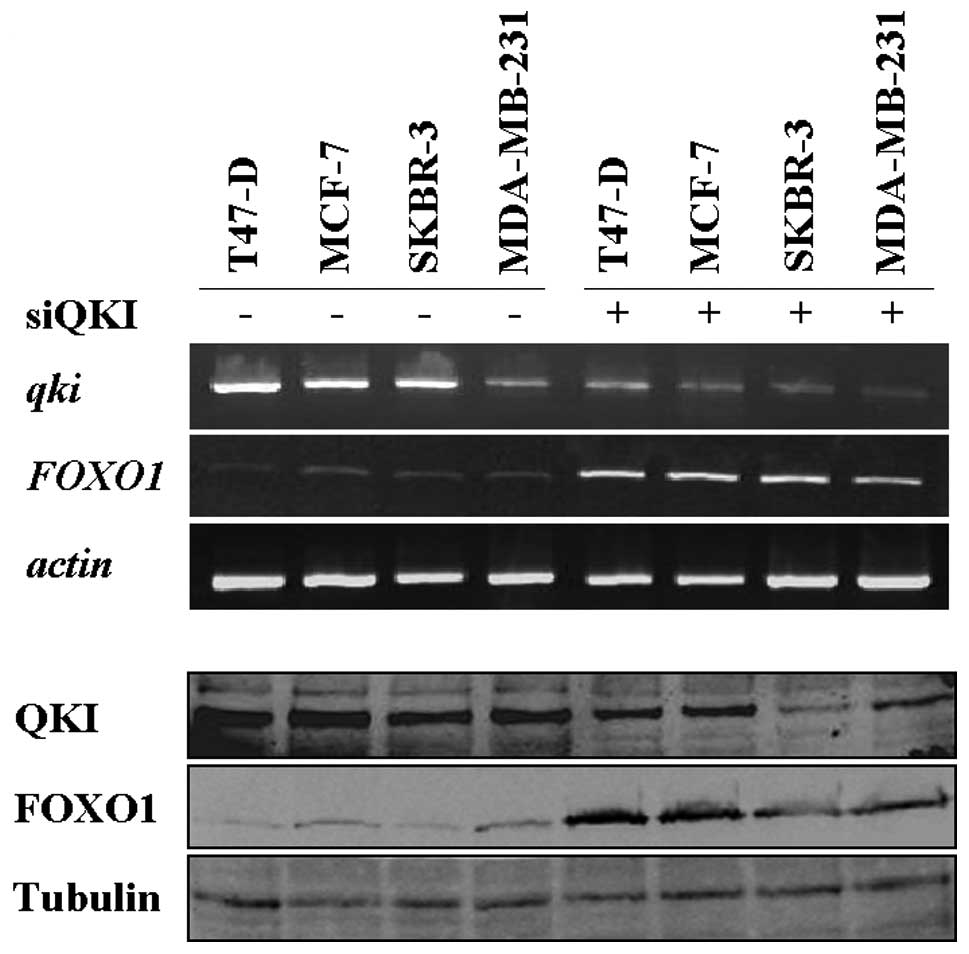
Inverse expression of QKI and FOXO1 in
breast cancer cell lines
Strong evidence indicates that FOXO1 expression is
extremely low and even undetectable in several types of cancers,
including breast, prostate and endometrial cancers (24–29).
To determine whether RNA binding protein QKI is responsible for the
aberrant expression of FOXO1 in breast cancers, QKI and FOXO1
expression was detected in a panel of breast cancer cell lines.
Consistent with previous findings, the FOXO1 expression level in
the four cell lines was extremely low, with trace expression in
MDA-MB-231, T47-D and SKBR-3 cells (Fig. 3). In sharp contrast, the expression
of QKI was extremely high in these cells (Fig. 3). Furthermore, knockdown of QKI
resulted in a significant increase in endogenous FOXO1 expression
in these cell lines (Fig. 3). Our
data suggest that negative regulation of FOXO1 by QKI may serve as
one of the molecular mechanisms contributing to the low expression
of FOXO1 in breast cancer cells.
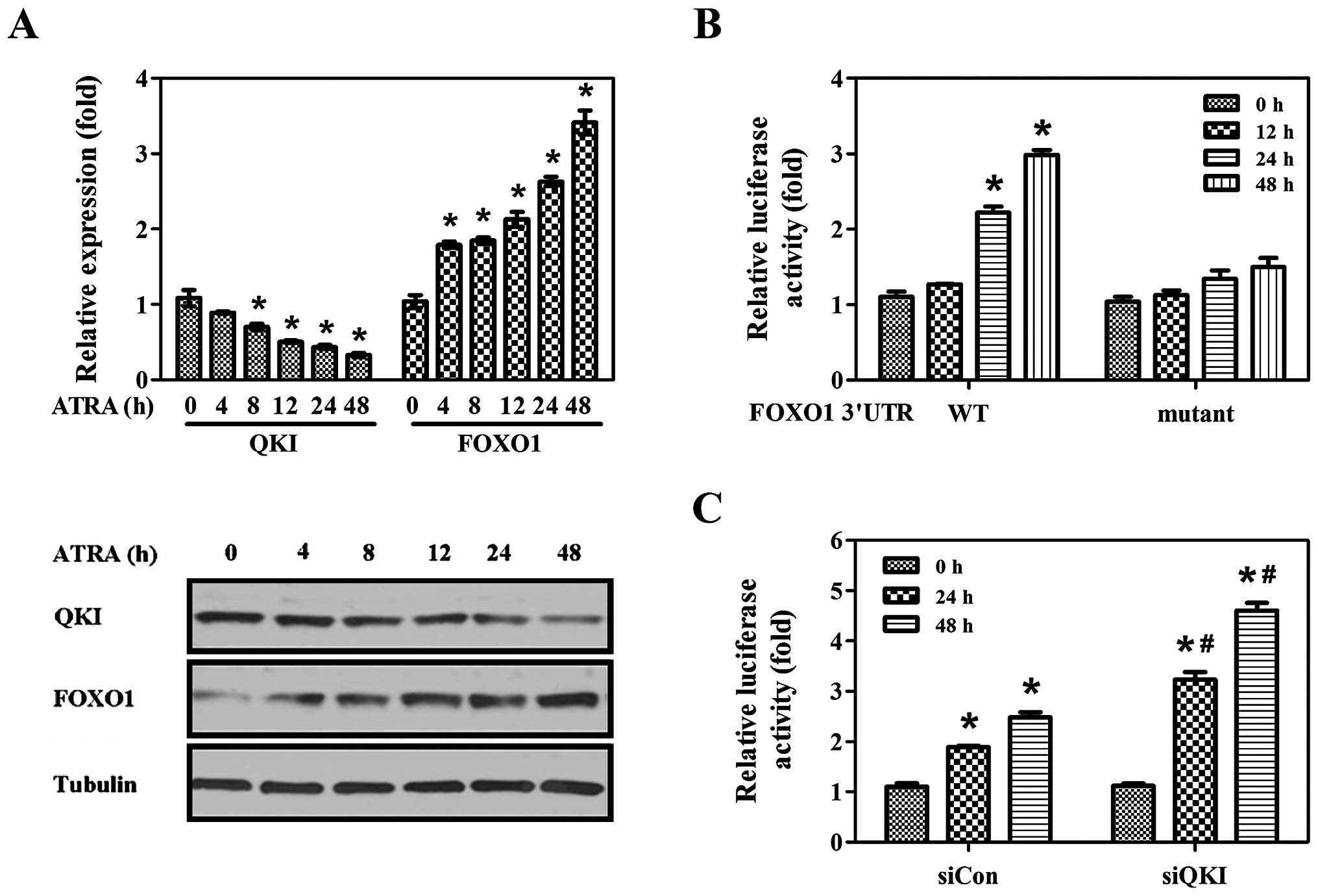
Upregulation of FOXO1 by ATRA or 5-FU is
dependent on QKI
ATRA is capable of inhibiting cell proliferation,
inducing cell cycle arrest and cell differentiation (30–34).
Dysregulation of FOXO1 expression has been observed to decrease the
sensitivity of various carcinoma cells to differentiation and
apoptosis inducers (24,26,35–37).
To evaluate whether QKI-mediated post-transcriptional repression of
FOXO1 indeed plays a role in cancer cells, we detected the
expression pattern of QKI and FOXO1 following ATRA treatment. QKI
expression showed a dramatic decrease upon ATRA induction, whereas
FOXO1 expression was gradually increased, exhibiting a
time-dependent trend (Fig. 4A,
upper and low panel). To ascertain whether enhanced FOXO1
expression by ATRA is dependent on QKI, pGL3-F and pGL3-FM plasmids
were transfected, and the luciferase activities were determined
following ATRA treatment. As expected, upon ATRA induction, the
luciferase activities of FOXO1 3′UTR were gradually induced almost
50% (Fig. 4B). However, the
luciferase activities of pGL3-FM were rarely changed (Fig. 4B). Furthermore, ATRA induced a
marked increase in luciferase activities of FOXO1 3′UTR in siQKI
cells compared to the siCon group (Fig.
4C). The above data indicate that QKI-mediated
post-transcriptional regulation is indeed responsible for enhanced
FOXO1 expression by ATRA induction.
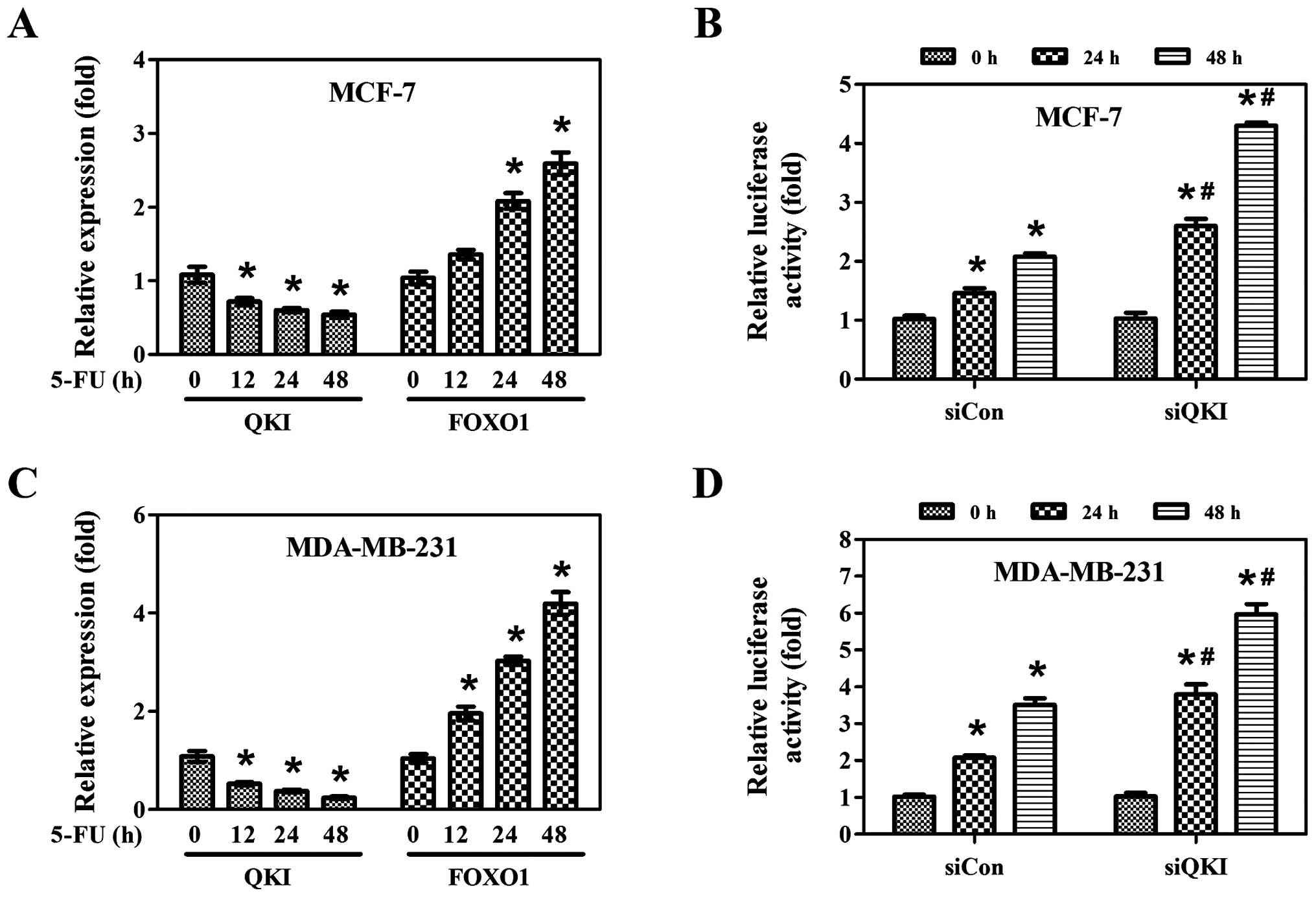
To ascertain whether the post-transcriptional
regulation of FOXO1 by QKI is not ATRA-specific, MCF-7 and
MDA-MB-231 cells were treated with 5-FU. As shown in Fig. 5A and C, 5-FU induced FOXO1
expression whereas it inhibited QKI expression in both cell lines.
Moreover, knockdown of QKI dramatically increased the luciferase
activities of FOXO1 3′UTR upon 5-FU treatment (Fig. 5B and D), indicating that 5-FU
enhances FOXO1 expression at the post-transcriptional level through
inhibition of QKI expression.
Discussion
Previous studies have shown that QKI is an
RNA-binding protein known to be important for myelination in the
central nervous system (CNS) and peripheral nervous system, through
regulating mRNA stability, translation, splicing and
cytoplasmic/nuclear localization of target genes (1,4,6,8,10,13,38).
In addition, QKI was previously found to bind p53 3′UTR and inhibit
its translation (39). Due to the
limited information concerning QKI-5 function outside the CNS
system, seeking for and verifying potential target genes may be an
efficient way to elucidate the function of QKI-5.
FOXO1 was an appealing candidate among thousands of
targets due to its diverse roles in controlling cell cycle,
apoptosis, stem cells and the aging process (17–23,27).
Previous studies found that FOXO1 function was closely related with
its phosphorylation and acetylation modification which determined
its transcriptional activity (40–44).
Recent studies found that microRNAs, such as miR-96 and miR-370,
downregulated FOXO1 expression (28,29).
Our current data demonstrated that QKI, an RNA-binding protein,
repressed the expression of FOXO1 via decreasing its mRNA stability
(Figs. 1 and 2). Our study, for the first time,
elucidated the detailed molecular mechanism by which FOXO1 mRNA was
regulated at the post-transcriptional level.
FOXO1 is a critical tumor suppressor, and its
expression is often dysregulated in cancers, such as breast,
bladder, prostate and endometrial cancers (24–29).
Our study observed that QKI and FOXO1 were inversely expressed in
breast cancer cells, and the low expression level of FOXO1 mRNA
could be enhanced by knockdown of QKI (Figs. 1D and 3), elucidating a novel mechanism of how
FOXO1 is aberrantly expressed in breast cancer. As a critical
transcription factor, FOXO1 orchestratedly regulates genes involved
in cell proliferation, apoptotic response, cell cycle checkpoints
and cellular metabolism (17,21,23).
We hypothesized that a low level of FOXO1 expression may make these
cancers resistant to apoptosis- or differentiation-inducing agents.
Of note, ATRA and 5-FU significantly induced an increase in FOXO1
expression through inhibition of QKI (Figs. 4 and 5). Our further study will disclose whether
QKI-mediated post-transcription repression of FOXO1 is involved in
ATRA or 5-FU-mediated effects and knockdown of QKI indeed
sensitizes breast cancer cells to those agents.
In contrast to our previous research in which QKI
functioned as a tumor suppressor in colonic epithelial cells and
gastric cancer (45,46), the present study suggests that QKI
may function as an oncogene in breast cancer. The reason for this
discrepancy may be due to the cell-context dependent role of QKI.
As we know, QKI is an RNA-binding protein belonging to the STAR
family, and its function is dependent on the accessibility of its
targets and the cellular environment. Thus, it is reasonable to
suggest that QKI functions differently in different cells.
In summary, our study provides initial evidence that
dysregulation of FOXO1 by QKI via post-transcriptional inhibition
may be one of the factors contributing to the oncogenesis and
progression of breast carcinoma. Thus, targeting QKI may serve as a
novel strategy to sensitize breast cancers to chemotherapy and/or
radiotherapy.
Acknowledgements
This study was supported by the National Science
Fund of China (NSF: 31000520; NSF: 30470387; NSF: 30570396; NSF:
C030200303) and Program for Changjiang Scholars and Innovative
Research Team in the University (IRT0459).
References
|
1
|
Sidman RL, Dickie MM and Appel SH: Mutant
mice (Quaking and Jimpy) with deficient myelination in the central
nervous system. Science. 144:309–311. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ebersole T, Rho O and Artzt K: The
proximal end of mouse chromosome 17: new molecular markers identify
a deletion associated with quakingviable. Genetics. 131:183–190.
1992.PubMed/NCBI
|
|
3
|
Kondo T, Furuta T, Mitsunaga K, et al:
Genomic organization and expression analysis of the mouse qkI
locus. Mamm Genome. 10:662–669. 1999.PubMed/NCBI
|
|
4
|
Pilotte J, Larocque D and Richard S:
Nuclear translocation controlled by alternatively spliced isoforms
inactivates the QUAKING apoptotic inducer. Genes Dev. 15:845–858.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McInnes LA and Lauriat TL: RNA metabolism
and dysmyelination in schizophrenia. Neurosci Biobehav Rev.
30:551–561. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Larocque D, Pilotte J, Chen T, et al:
Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron.
36:815–829. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang Y and Feng Y: Distinct molecular
mechanisms lead to diminished myelin basic protein and 2′,3′-cyclic
nucleotide 3′-phosphodiesterase in qk(v) dysmyelination. J
Neurochem. 77:165–172. 2001.PubMed/NCBI
|
|
8
|
Zhao L, Mandler MD, Yi H and Feng Y:
Quaking I controls a unique cytoplasmic pathway that regulates
alternative splicing of myelin-associated glycoprotein. Proc Natl
Acad Sci USA. 107:19061–19066. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van der Veer EP, de Bruin RG, Kraaijeveld
AO, et al: The RNA-binding protein quaking is a critical regulator
of vascular smooth muscle cell phenotype. Circ Res. 113:1065–1075.
2013.PubMed/NCBI
|
|
10
|
Li Z, Zhang Y, Li D and Feng Y:
Destabilization and mislocalization of myelin basic protein mRNAs
in quaking dysmyelination lacking the QKI RNA-binding proteins. J
Neurosci. 20:4944–4953. 2000.PubMed/NCBI
|
|
11
|
Rosenbluth J and Bobrowski-Khoury N:
Structural bases for central nervous system malfunction in the
quaking mouse: dysmyelination in a potential model of
schizophrenia. J Neurosci Res. 91:374–381. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chubb C: Oligotriche and quaking gene
mutations. Phenotypic effects on mouse spermatogenesis and
testicular steroidogenesis. J Androl. 13:312–317. 1992.PubMed/NCBI
|
|
13
|
Hall MP, Nagel RJ, Fagg WS, et al: Quaking
and PTB control overlapping splicing regulatory networks during
muscle cell differentiation. RNA. 19:627–638. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo W, Shi X, Liu A, et al: RNA binding
protein QKI inhibits the ischemia/reperfusion-induced apoptosis in
neonatal cardiomyocytes. Cell Physiol Biochem. 28:593–602. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fu H, Yang G, Wei M, et al: The
RNA-binding protein QKI5 is a direct target of C/EBPalpha and
delays macrophage differentiation. Mol Biol Cell. 23:1628–1635.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Galarneau A and Richard S: Target RNA
motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol
Biol. 12:691–698. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Diep CH, Charles NJ, Gilks CB, Kalloger
SE, Argenta PA and Lange CA: Progesterone receptors induce
FOXO1-dependent senescence in ovarian cancer cells. Cell Cycle.
12:1433–1449. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Coffer PJ and Burgering BM: Forkhead-box
transcription factors and their role in the immune system. Nat Rev
Immunol. 4:889–899. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Paik JH, Kollipara R, Chu G, et al: FoxOs
are lineage-restricted redundant tumor suppressors and regulate
endothelial cell homeostasis. Cell. 128:309–323. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pohl BS, Schön C, Rössner A and Knöchel W:
The FoxO-subclass in Xenopus laevis development. Gene Expr
Patterns. 5:187–192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee SY, Lee GR, Woo DH, et al: Depletion
of Aurora A leads to upregulation of FoxO1 to induce cell cycle
arrest in hepatocellular carcinoma cells. Cell Cycle. 12:67–75.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Yalcin S, Lee DF, et al: FOXO1 is
an essential regulator of pluripotency in human embryonic stem
cells. Nat Cell Biol. 13:1092–1099. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guttilla IK and White BA: Coordinate
regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast
cancer cells. J Biol Chem. 284:23204–23216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Yang L, Song L, et al: Astrocyte
elevated gene-1 is a proliferation promoter in breast cancer via
suppressing transcriptional factor FOXO1. Oncogene. 28:3188–3196.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Silva J, Cavazos DA, Donzis E, Friedrichs
WE, Marciniak R and deGraffenried LA: Akt-induced tamoxifen
resistance is associated with altered FKHR regulation. Cancer
Invest. 25:569–573. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arden KC: Multiple roles of FOXO
transcription factors in mammalian cells point to multiple roles in
cancer. Exp Gerontol. 41:709–717. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu Z, Sun H, Zeng W, He J and Mao X:
Upregulation of MircoRNA-370 induces proliferation in human
prostate cancer cells by downregulating the transcription factor
FOXO1. PLoS One. 7:e458252012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo Y, Liu H, Zhang H, Shang C and Song Y:
miR-96 regulates FOXO1-mediated cell apoptosis in bladder cancer.
Oncol Lett. 4:561–565. 2012.PubMed/NCBI
|
|
30
|
Lotan R: Retinoids in cancer
chemoprevention. FASEB J. 10:1031–1039. 1996.PubMed/NCBI
|
|
31
|
Yang QJ, Zhou LY, Mu YQ, et al:
All-trans retinoic acid inhibits tumor growth of human
osteosarcoma by activating Smad signaling-induced osteogenic
differentiation. Int J Oncol. 41:153–160. 2012.
|
|
32
|
Chen S, Fang Y, Ma L, Liu S and Li X:
Realgar-induced apoptosis and differentiation in all-trans
retinoic acid (ATRA)-sensitive NB4 and ATRA-resistant MR2 cells.
Int J Oncol. 40:1089–1096. 2012.PubMed/NCBI
|
|
33
|
Lainey E, Wolfromm A, Sukkurwala AQ, et
al: EGFR inhibitors exacerbate differentiation and cell cycle
arrest induced by retinoic acid and vitamin D 3 in acute myeloid
leukemia cells. Cell Cycle. 12:2978–2991. 2013. View Article : Google Scholar
|
|
34
|
Bengtsson AM, Jonsson G, Magnusson C,
Salim T, Axelsson C and Sjolander A: The cysteinyl leukotriene 2
receptor contributes to all-trans retinoic acid-induced
differentiation of colon cancer cells. BMC Cancer. 13:3362013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schuur ER, Loktev AV, Sharma M, Sun Z,
Roth RA and Weigel RJ: Ligand-dependent interaction of estrogen
receptor-alpha with members of the forkhead transcription factor
family. J Biol Chem. 276:33554–33560. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han CY, Cho KB, Choi HS, Han HK and Kang
KW: Role of FoxO1 activation in MDR1 expression in
adriamycin-resistant breast cancer cells. Carcinogenesis.
29:1837–1844. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tzivion G and Hay N: PI3K-AKT-FoxO axis in
cancer and aging. Biochim Biophys Acta. 1813:19252011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saccomanno L, Loushin C, Jan E, Punkay E,
Artzt K and Goodwin EB: The STAR protein QKI-6 is a translational
repressor. Proc Natl Acad Sci USA. 96:12605–12610. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schumacher B, Hanazawa M, Lee MH, et al:
Translational repression of C. elegans p53 by GLD-1
regulates DNA damage-induced apoptosis. Cell. 120:357–368.
2005.PubMed/NCBI
|
|
40
|
Kim DH, Kim JM, Lee EK, et al: Modulation
of FoxO1 phosphorylation/acetylation by baicalin during aging. J
Nutr Biochem. 23:1277–1284. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Birkenkamp KU and Coffer PJ: Regulation of
cell survival and proliferation by the FOXO (Forkhead box, class O)
subfamily of Forkhead transcription factors. Biochem Soc Trans.
31:292–297. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Finlay D, Patel S, Dickson LM, et al:
Glycogen synthase kinase-3 regulates IGFBP-1 gene transcription
through the thymine-rich insulin response element. BMC Mol Biol.
5:152004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang W, Yan C, Zhang J, et al: SIRT1
inhibits TNF-alpha-induced apoptosis of vascular adventitial
fibroblasts partly through the deacetylation of FoxO1. Apoptosis.
18:689–701. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu P, Kao TP and Huang H: CDK1 promotes
cell proliferation and survival via phosphorylation and inhibition
of FOXO1 transcription factor. Oncogene. 27:4733–4744. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang G, Fu H, Zhang J, et al: RNA-binding
protein quaking, a critical regulator of colon epithelial
differentiation and a suppressor of colon cancer. Gastroenterology.
138:231–240.e1–5. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bian Y, Wang L, Lu H, et al:
Downregulation of tumor suppressor QKI in gastric cancer and its
implication in cancer prognosis. Biochem Biophys Res Commun.
422:187–193. 2012. View Article : Google Scholar : PubMed/NCBI
|