Introduction
Colorectal cancer (CRC) is one of the leading causes
of cancer-related morbidity and mortality worldwide. Although
surgery is the most effective treatment for advanced CRC,
recurrence frequently occurs within a few years after surgery. In
addition to surgical treatment, chemotherapy has been widely used
to improve the survival rate, but the existing chemotherapy
regimens such as oxaliplatin or irinotecan plus 5-fluorouracil have
not achieved satisfactory results (1,2). It is
well known that several signaling pathways such as the epidermal
growth factor receptor (EGFR) pathway are involved in the
carcinogenesis and progression of CRC (3,4).
Developing novel chemotherapeutic strategies targeting these
signaling pathways is critical for reducing the carcinogenic
progression of CRC.
EGFR is a transmembrane tyrosine kinase receptor
that is thought to control cell growth, differentiation and
survival (5). Two main
intracellular pathways activated by the EGFR are the
mitogen-activated protein kinase (MAPK) pathway and the
phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT)
pathway, which are involved in cell proliferation, migration,
differentiation and apoptosis (3,6). The
EGFR and its downstream signaling pathways are frequently
overexpressed in CRC, and are associated with a high risk of
metastasis and poor prognosis of CRC (3,7).
Antitumor agents targeting the EGFR such as gefitinib have been
used for the treatment of CRC (8).
Gefitinib (Iressa, ZD-1839), an EGFR-tyrosine kinase inhibitor,
binds at the ATP site on the EGFR, and thus blocks its downstream
signaling pathway that is involved in the proliferation and
survival of cancer cells (9,10).
Gefitinib has been used for the treatment of non-small cell lung
cancer, and improves the survival rate of patients with lung cancer
with acceptable toxicity (11–13).
Gefitinib has also been reported to reduce tumor regression in
patients with metastatic CRC (14).
However, a randomized phase II study demonstrated that gefitinib is
inactive as a single agent in patients with previously treated CRC
(15). In addition, adding
gefitinib into the chemotherapy regime using capecitabine (16), raltitrexed (17) or 5-fluorouracil, folinic acid and
irinotecan (18) does not improve
the efficacy in patients with CRC. These studies suggest that CRC
cells may exhibit resistance to gefitinib.
The adenomatous polyposis coli (APC) gene
encodes a 310-kDa tumor-suppressor protein that plays a role in
cell growth, differentiation, migration, adhesion and apoptosis
(19). Mutations in the APC
gene have been found in more than 80% of sporadic CRCs and are
responsible for hereditary forms of CRC (19,20).
The best-known function of the APC protein is as a scaffolding
protein in a protein complex, including GSK-3β, axin and APC, that
regulates the cellular level of β-catenin in the Wnt signaling
pathway (21). Mutations in the APC
genes lead to aberrant activation of β-catenin, which causes
transcriptional activation of several target genes of the Wnt
signaling pathway such as cyclin D1 and c-Myc, thus, promoting cell
proliferation and differentiation (19,20).
In addition, the Wnt/β-catenin and EGFR signaling pathways can
crosstalk in cancer (22). However,
it remains unclear whether changes in APC can regulate EGFR
signaling in CRC cells and alter their sensitivity to
gefitinib.
In the present study, we aimed to investigate the
effect of APC on the sensitivity of CRC cells to gefitinib, using
human HCT-116 (wild-type APC) and HT-29 (mutant APC) CRC cells. We
found that HT-29 cells exhibited enhanced sensitivity to gefitinib
when compared to HCT-116 cells. Knockdown of APC expression
increased the sensitivity of HCT-116 cells to gefitinib, whereas
overexpression of APC decreased the sensitivity of HT-29 cells to
gefitinib. Our data suggest that inhibiting APC may represent a
potential novel therapeutic method for increasing gefitinib
sensitivity in the treatment of CRC.
Materials and methods
Cell culture
The human colorectal cancer cells, HCT-116
containing wild-type APC, HT-29/APC (APC inducible) containing
mutant APC, and HT-29/βGal (β-galactosidase inducible), were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). Cells were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin, and 100 mg/ml streptomycin in a humidified atmosphere
in a 37°C, 5% CO2 incubator.
RNAi and cell transfection
The APC-specific siRNA sequence was
5′-GATCCATACACATTCAAACACTTATTCAAGAG ATAAGTGTTTGAATGTGTATGGA-3′, and
was cloned into the pSilencer 4.1-CMV neo Vector. Scramble siRNA
was used as a negative control. HCT-116 cells were transfected with
the pSilencer 4.1-CMV neo Vector containing either APC-specific
shRNA or the scramble control, using Lipofectamine (Invitrogen).
Forty-eight hours after transfection, cells were used for the
following experiments. To test the effect of overexpression of APC
on the sensitivity of HT-29 cells to gefitinib, HT-29/APC and
HT-29/βGal cells were cultured in the presence of 0, 50 or 100 μM
ZnCl2 for 24 h. Cells were then used for the following
experiments.
Cell viability assay
MTT assay was used to measure cell viability
following exposure to gefitinib. Cells were seeded in 96-well
plates at a density of 5×103 cells/well, and allowed to
grow in RPMI-1640 medium for 24 h. Gefitinib was added at final
concentrations of 0–100 μM. After 0, 24, 48 and 72 h, cells were
incubated with 5 mg/ml MTT for 4 h, and subsequently solubilized in
DMSO (100 μl/well). The absorbance at 570 nm was then measured
using an ELISA reader. Experiments were repeated at least three
times.
RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from the HCT-116 and HT-29
cells using TRIzol reagent (Gibco-Invitrogen, Carlsbad, CA, USA).
RNA was reverse transcribed into complementary DNA using a reverse
transcription kit (Takara, Dalian, China) according to the
manufacturer’s protocol. PCR was performed in a mixture containing
2 μl cDNA, 2 mM MgCl2, 0.03 U/l Taq DNA
polymerase, 0.4 mmol/l dNTP, and 0.5 μM primers at a final volume
of 25 μl. Primers used for amplification of APC were
5′-CTGCAGCTTCATATGATC-3′ (sense) and 5′-AGAGTCTTTGTCATTGCAT-3′
(antisense). β-actin was used as an internal control. The reaction
condition was as follow: 94°C for 2 min; 35 cycles of 94°C for 30
sec, 50°C for 30 sec, and 72°C for 1 min; and 72°C for 10 min. The
mRNA expression of APC was normalized to the expression of β-actin.
The relative expression of APC was calculated using the
2−ΔΔCT method.
Western blot analysis
Cells were homogenized on ice in lysis buffer. The
lysates were centrifuged at 12,000 rpm for 30 min at 4°C. Protein
concentrations were determined using the BCA method. Proteins were
separated by electrophoresis in 10% SDS-PAGE, and transferred onto
polyvinylidene fluoride membranes by electroblotting. Membranes
were incubated with primary antibodies against APC, EGFR, ERK,
p-ERK, AKT and p-AKT (dilution 1:1,000) at 4°C overnight. β-actin
was used as a loading control. Membranes were then incubated with
horseradish peroxidase-linked goat anti-rabbit secondary antibodies
(dilution 1:2,500) at room temperature for 2 h. All antibodies were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Bands were visualized using a chemiluminescence detection
system.
Flow cytometry
Flow cytometric analysis was performed on a
FACSCalibur (Becton-Dickinson). For determination of the cell
cycle, 500 μl of cell culture was incubated with 30 μg/ml propidium
iodide (PI) for 30 min at 37°C before analysis. For detection of
apoptotic cells, cells were harvested, washed twice with
phosphate-buffered saline (PBS), and then incubated for 30 min at
37°C with a solution of fluorescence isothiocyanate
(FITC)-conjugated Annexin V (2.5 μg/ml) and PI (5 μg/ml) (Bio-Sea,
China) before analysis for apoptosis.
Wound healing assay
Cells were seeded at a density of 2.5×105
cells/ml in 6-well plates. Cells were cultured until reaching full
confluency. A cell monolayer was carefully scratched across the
diameter of the wells using a sterile 1-ml pipette tip. The cells
were further cultured for 48 h, and the wound closure was examined
under a microscope. Experiments were repeated three times.
Transwell assay
Cell invasion assays were performed using Transwell
membrane filter inserts (Corning Costar Corp., Corning, NY USA).
The upper surface of the Transwell membrane was coated with
Matrigel™ Basement Membrane Matrix (BD Biosciences, San Jose, CA,
USA) for 4 h. The membrane was placed in the upper compartment of
24-well culture plates. Cells in serum-free medium were harvested
and seeded at a density of 2.5×105 cells/ml onto
Transwell inserts. The lower compartment was filled with cell
culture medium containing 20% FBS. Cells were allowed to migrate
for 24 h at 37°C. The Matrix was erased and the inserts were washed
with PBS for 3 times. The chamber was then fixed in 95% methanol
for 20 min. Cells on the upper surface of the membrane were removed
by wiping with a cotton swap. Membranes were stained in 0.1%
hexamethyl pararosaniline for another 20 min. The membrane was then
mounted onto a glass slide. Cell invasion was determined by
counting the number of invasive cells. Ten random fields per
membrane (×200) were counted for each assay, and three separated
assays were performed.
Statistical analyses
Statistical analyses were performed using SPSS 13.0.
All values are presented as the mean and standard deviation. The
Student’s t-test was used to compare differences between two
groups. A probability value <0.05 was considered to indicate a
statistically significant result.
Results
HCT-116 and HT-29 cells exhibit
differential sensitivity to gefitinib
We investigated the effect of APC on the sensitivity
of human HCT-116 (wild-type APC) and HT-29 (mutant APC) CRC cells
to gefitinib. Cells were treated with various concentrations of
gefitinib (0–100 μM) for 0–72 h, and cell viability was examined
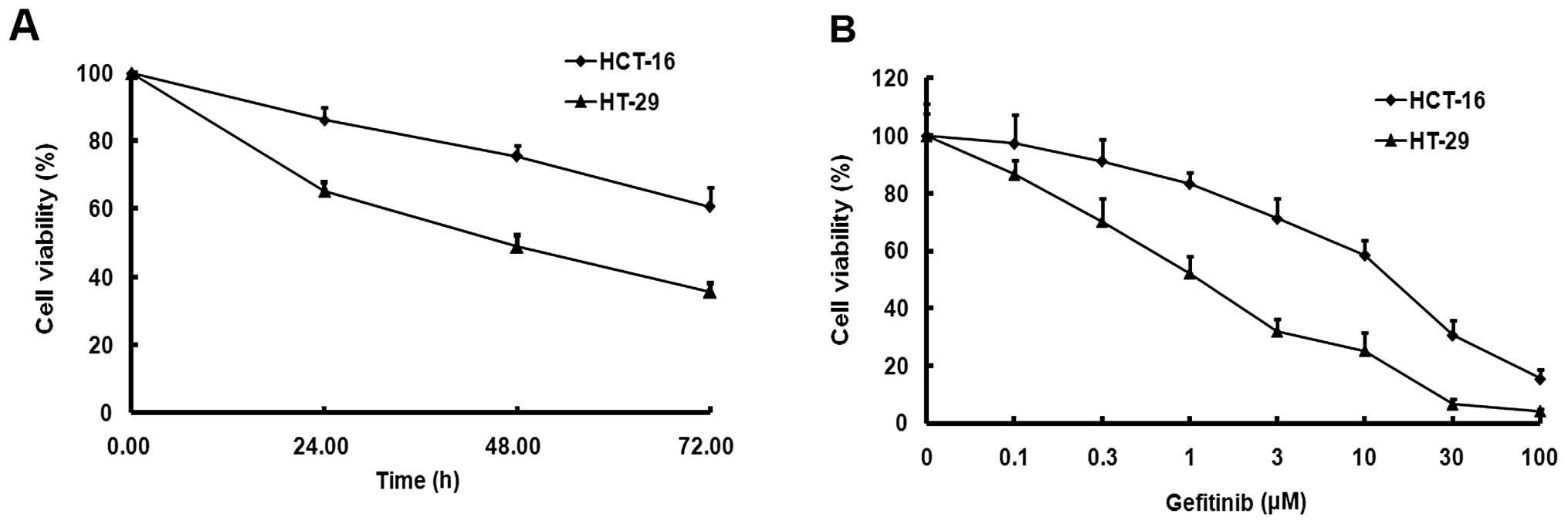
using MTT assay. Gefitinib inhibited the cell viability of the
HCT-116 and HT-29 cells in a time-dependent manner (Fig. 1A). At 24, 48 and 72 h after
gefitinib treatment, the survival rate of HCT-116 cells was
significantly higher than that of HT-29 cells (P<0.05). The
survival rates of both cells decreased in a dose-dependent manner.
The IC50 value for gefitinib in HCT-116 cells was
15.64±1.48 μM, which was ~15-fold higher than that of the HT-29
cells (1.19±0.04 μM) (Fig. 1B).
Gefitinib induces increased apoptosis in
HT-29 cells when compared with the HCT-116 cells
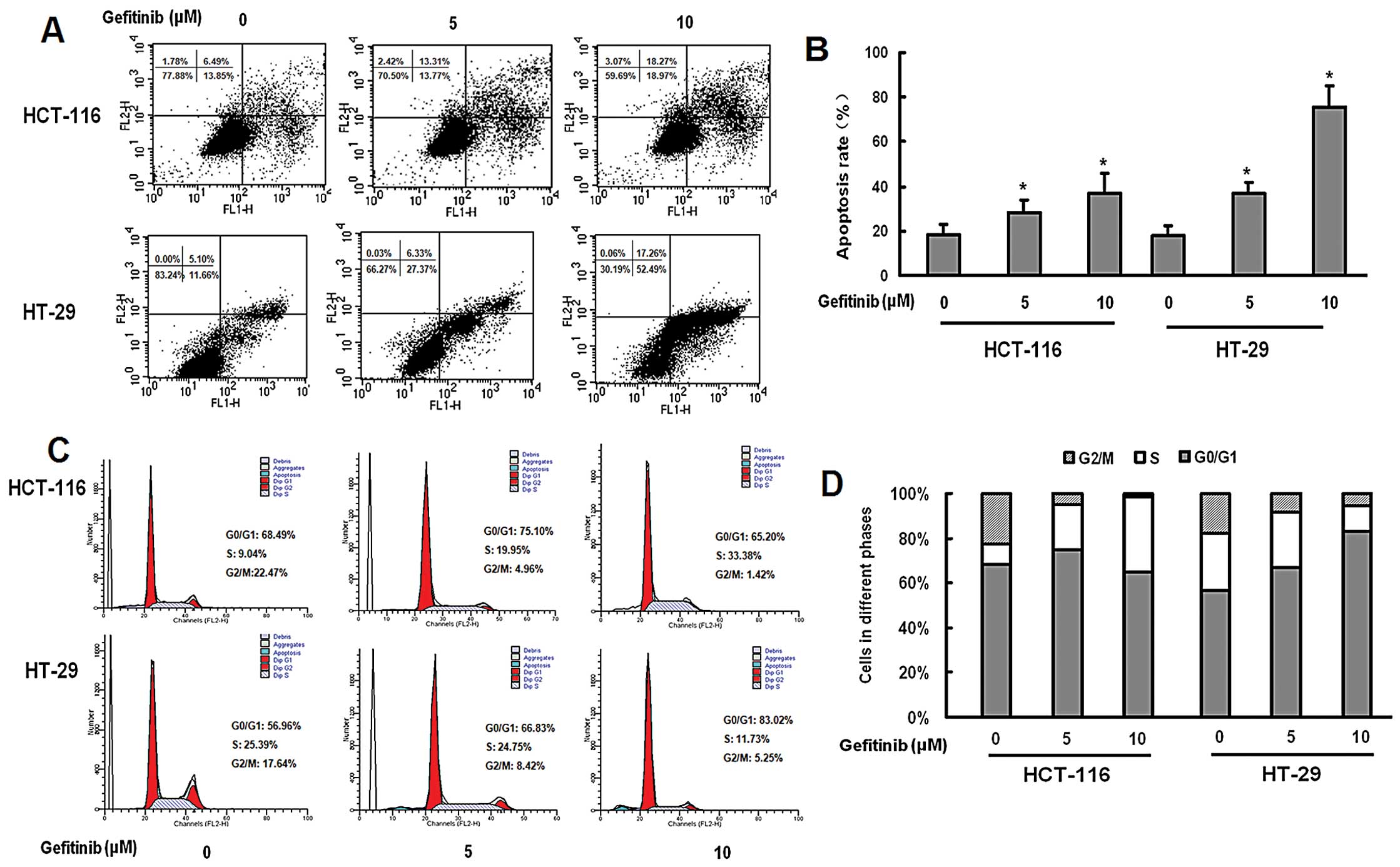
We further examined the effect of gefitinib on the
apoptosis of HCT-16 and H-29 cells, using flow cytometry. Gefitinib
treatment increased apoptosis of both cell lines in a
dose-dependent manner (Fig. 2A).
Gefitinib (10 μM) significantly induced increased apoptosis in
HT-29 cells when compared with that in HCT-116 cells (P<0.05;
Fig. 2A). Exposure of HCT-116 cells
to gefitinib resulted in an increase in the percentage of
G0/G1-phase cells in a dose-dependent manner. Gefitinib-treated
HT-29 cells were arrested in the S phase of the cell cycle in a
dose-dependent manner (Fig.
2B).
Gefitinib-induced inhibition of cell
migration is greater in HT-29 cells than that in HCT-116 cells
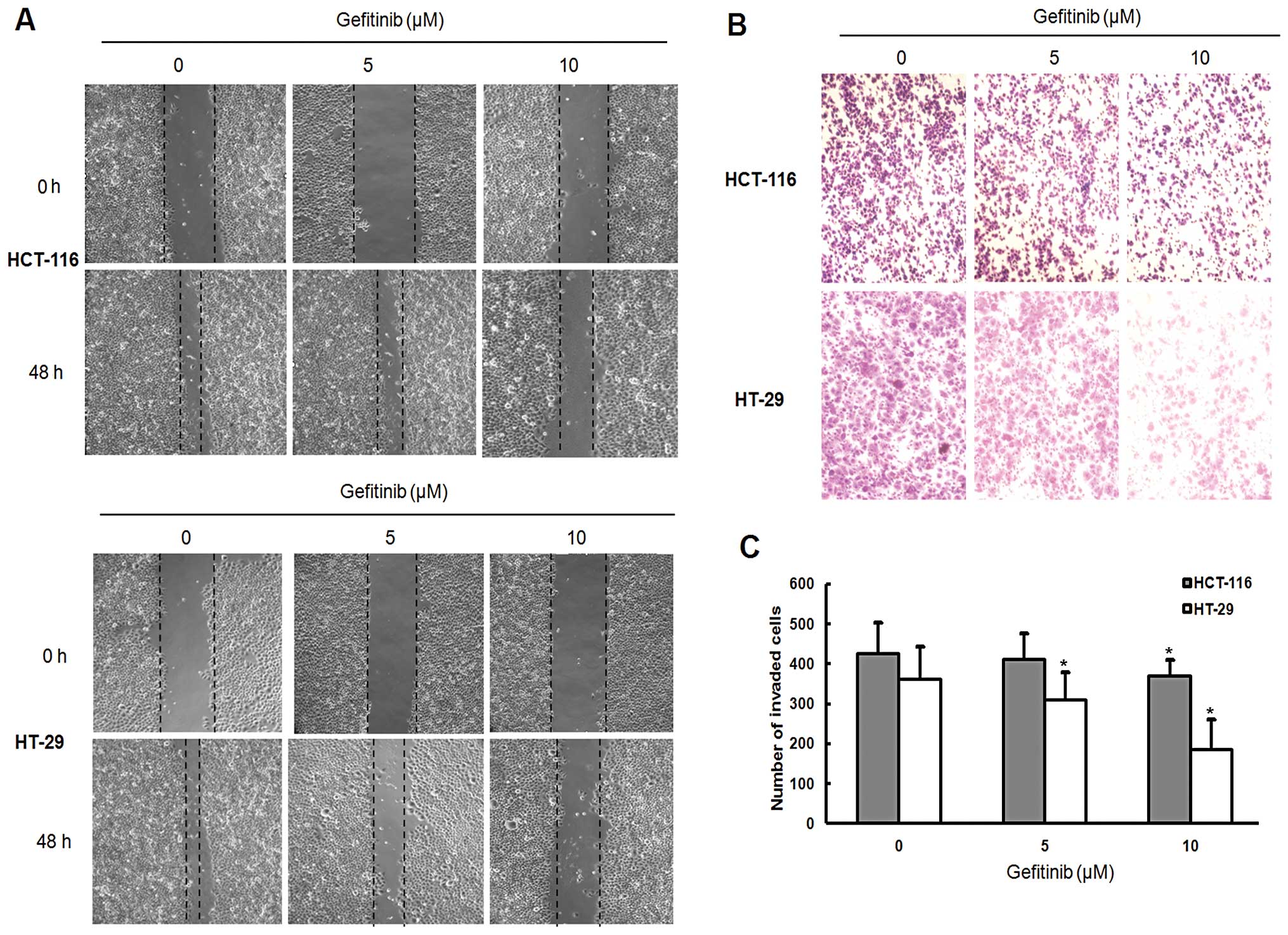
We investigated the effect of gefitinib on the cell
mobility of HCT-116 and HT-29 cells using a monolayer wound-healing
assay. Gefitinib inhibited the cell migration of both HCT-116 and
HT-29 cells in a dose-dependent manner. Following gefitinib
treatment for 48 h, HCT-116 cells migrated more slowly than HT-29
cells (Fig. 3A). Gefitinib-induced
inhibition of cell migration was further confirmed by Transwell
migration assay (Fig. 3B). After
gefitinib treatment for 48 h, the number of HCT-116 cells that
migrated into the lower chamber was significantly higher when
compared with that of HT-29 cells (P<0.05; Fig. 3B).
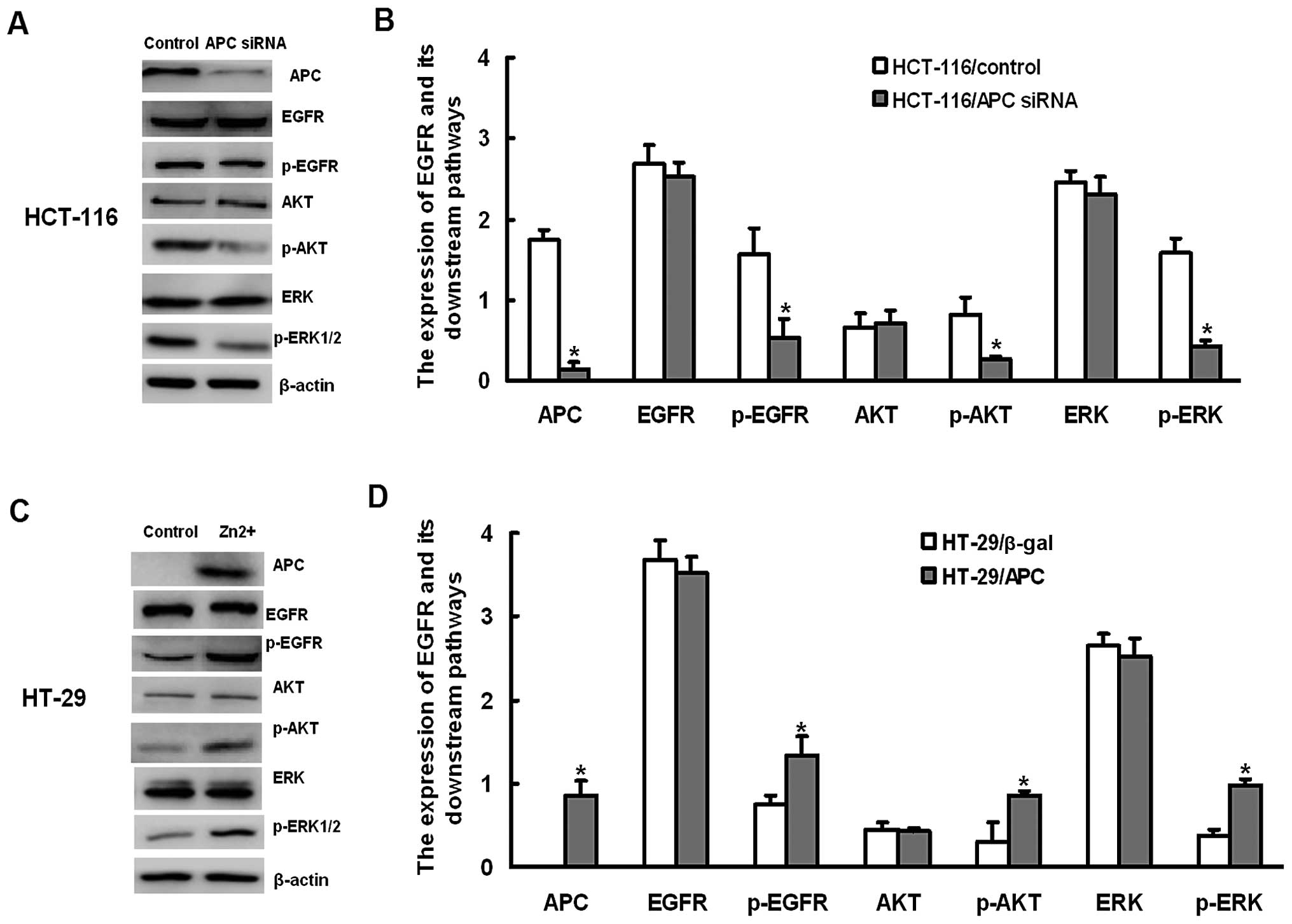
Knockdown of APC increases the
sensitivity of HCT-116 cells to gefitinib
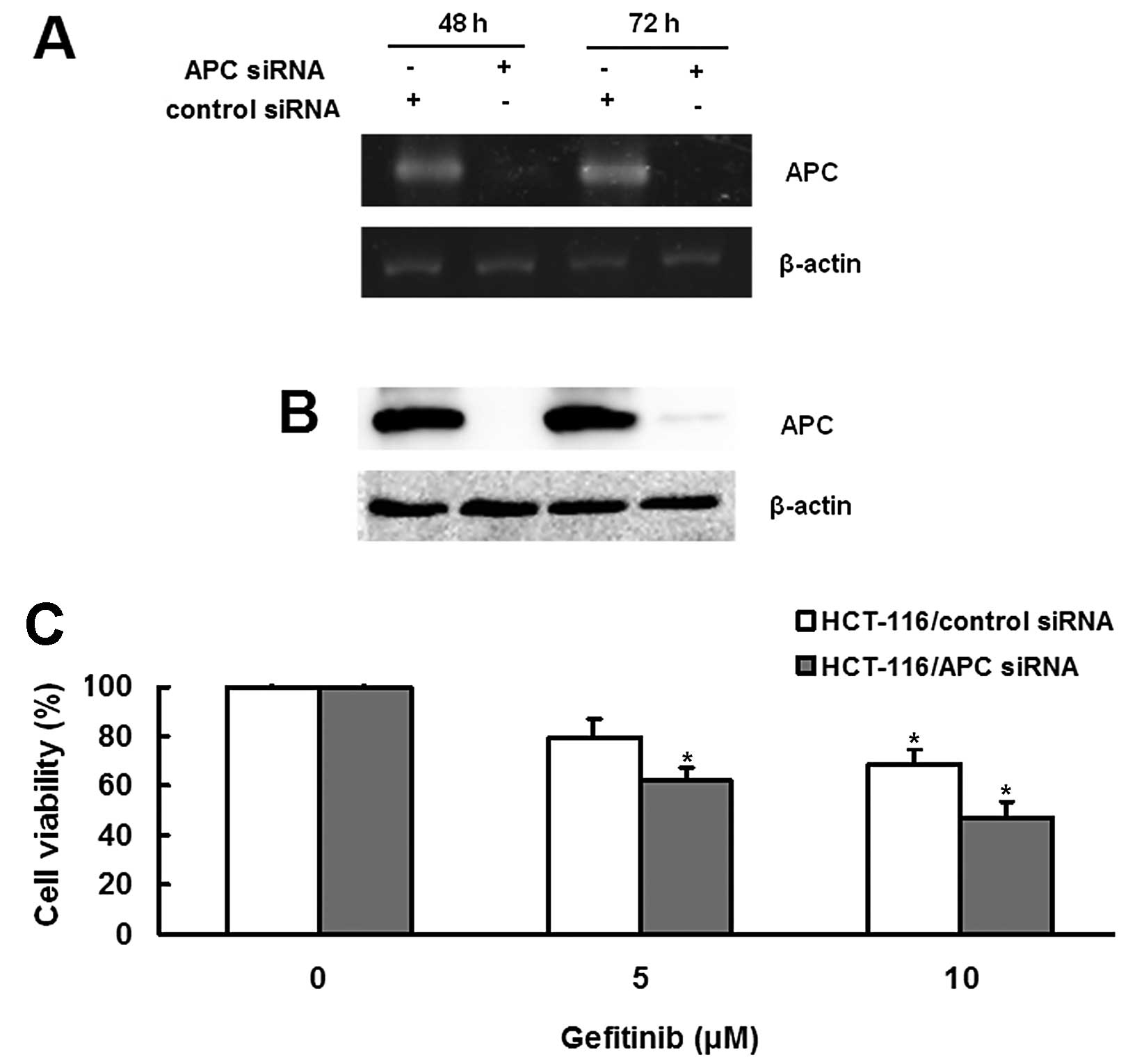
We further tested whether APC contributes to the
sensitivity of HCT-116 cells to gefitinib using APC siRNA to knock
down endogenously expressed APC. RT-PCR results showed that APC
siRNA decreased the expression of APC in HCT-116 cells (Fig. 4A). The APC siRNA-induced inhibition
of APC expression was further confirmed by western blot analysis
(Fig. 4B). We then examined the
sensitivity of HCT-116 cells to gefitinib after reducing APC
expression. Following gefitinib treatment, the survival rate of
HCT-116 cells treated with APC siRNA was significantly reduced when
compared with cells treated with scramble siRNA (P<0.05;
Fig. 4C), suggesting that knockdown
of APC increased the sensitivity of HCT-116 cells to gefitinib.
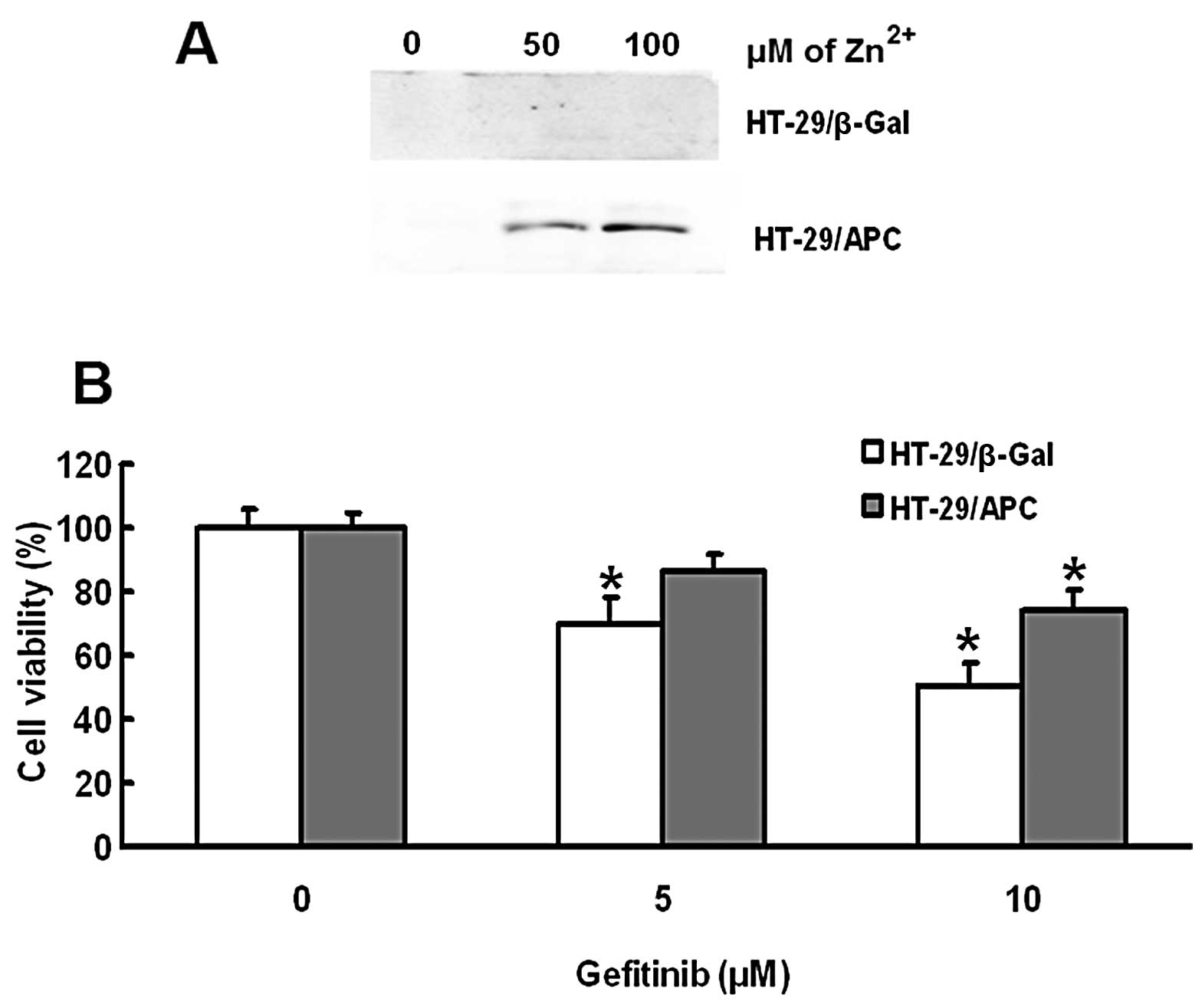
Enhanced expression of APC decreases the
sensitivity of HT-29 cells to gefitinib
We further investigated whether enhanced expression
of APC in HT-29 cells improves their sensitivity to gefitinib.
Western blot results showed that ZnCl2 treatment
upregulated the expression of APC in the HT-29/APC cells in a
concentration-dependent manner (Fig.
5A). Gefitinib treatment dose-dependently decreased the
survival rate of HT-29/βGal cells after exposure to 100 μm
ZnCl2. The gefitinib-induced decrease in the survival
rate was significantly reduced in HT-29/APC cells compared with
that in HT-29/βGal cells (P<0.05; Fig. 5B), suggesting that upregulation of
APC inhibited the sensitivity of HT-29 cells to gefitinib.
APC regulates the EGFR and its downstream
pathways
We then explored the potential role of APC on the
EGFR signaling pathway. Knockdown of APC significantly
downregulated the expression of pEGFR, p-AKT and pERK1/2 in HCT-116
cells, whereas overexpression of APC significantly upregulated the
expression of pEGFR, p-AKT and pERK1/2 in HT-29 cells (Fig. 6).
Discussion
EGFR kinase inhibitors have been clinically used for
the treatment of lung cancer with improved survival (23–25).
However, several clinical studies have shown that EGFR kinase
inhibitors have not achieved satisfactory outcomes in CRC patients
(15–18). The mechanisms underlying CRC
resistance to EGFR kinase inhibitors remain unknown. Mutations in
APC genes have been associated with CRC (19,20).
However, it remains to be determined whether APC is involved in the
sensitivity of CRC cells to EGFT kinase inhibitors. In the present
study, we investigated the role of APC in the sensitivity of human
CRC cells to gefitinib, using HCT-116 cells containing wild-type
APC and HT-29 cells containing mutant APC. We found that gefitinib
inhibited the viability, promoted apoptosis, and reduced the
migration of HCT-116 and HT-29 cells. HT-29 cells exhibited more
sensitivity to gefitinib than HCT-116 cells. Furthermore, knockdown
of APC expression improved the sensitivity of HCT-16 cells to
gefitinib, whereas overexpression of APC decreased the sensitivity
of HT-29 cells to gefitinib. The present study suggests that APC is
critical for determining the sensitivity of CRC cells to
gefitinib.
APC is known to play an important role in the
pathogenesis of CRC (20).
Mutations in APC genes have been found in ~80% of patients
with sporadic CRC (19,20). Abnormal production of mutant APC
proteins leads to accumulation of β-catenin, which promotes
transcriptional activation of many proliferation genes (19,20).
The role of APC in CRC is largely attributed to its inhibition of
Wnt/β-catenin signaling, which has been found to be associated with
resistance to chemotherapy in cancers (26–28).
It has been reported that knockdown of the expression of β-catenin
increases the sensitivity of lung cancer cells to gefitinib
(29). If β-catenin-mediated
resistance to gefitinib occurs in CRC, we would expect that the
knockdown of the expression of APC, which leads to upregulation of
β-catenin, may result in CRC resistance to gefitinib. However, in
the present study, we found that the knockdown of APC expression
increased the sensitivity of HCT-16 cells to gefitinib. Our results
suggest that APC may mediate gefitinib resistance in CRC, at least
in part, via a mechanism different from lung cancer. It has been
reported that EGFR mutations are associated with the sensitivity of
non-small cell lung cancer to gefitinib (23–25).
The incidence of EGFR mutations in lung cancer varies in different
regions, and has been reported to be 15% in Korea (30), 28% in Greece (31) and 45% in Japan (32). However, the incidence of EGFR
mutations in CRC is ~0.34–12% depending on different regions
(33,34), which is much lower than that in lung
cancer. The low incidence of EGFR mutations in CRC suggests that
CRC may exhibit a different mechanism underlying gefitinib
resistance compared with lung cancer. Our findings that APC
mediated gefitinib resistance suggest that APC may represent a
novel mechanism underlying gefitinib resistance.
Gefitinib resistance presents a great challenge for
its clinical application for the treatment of cancer. The
mechanisms underlying gefitinib resistance in cancer cells remain
poorly understood. Altered EGFR and its downstream signaling
pathways are likely to contribute to gefitinib resistance. It has
been reported that inhibiting sialylation of EGFR increases the
sensitivity of CRC cells to gefitinib (35). In addition, mutations in cancer
cells with reduced phosphorylation levels of EGFR exhibit enhanced
sensitivity to gefitinib (29,36).
In agreement with these reports, we found that the phosphorylation
level of EGFR was increased in CRC cells with low expression of
APC, accompanied with more sensitivity to gefitinib. Furthermore,
we found that downregulation of APC increased the activity of the
EGFR and its downstream AKT and ERK1/2 signaling pathways.
Conversely, overexpression of APC reduced the activity of the EGRF
and its downstream pathways. However, the mechanisms underlying
APC-mediated regulation of EGFR signaling remain unclear. It has
been reported that overexpression of β-catenin confers lung cancer
cells resistance to gefitinib (29). Our present finding that the
knockdown of APC expression, which should upregulate the expression
of β-catenin, increased the sensitivity of HCT-116 cells to
gefitinib suggests that APC may not regulate the EGFR signaling via
the Wnt/β-catenin. APC has also been reported to regulate
cytoskeletal proteins such as F-action (19), which can bind to the EGFR (37), suggesting that APC may regulate EGFR
via F-action. Further studies are required to demonstrate the
signaling pathway that is involved in APC-mediated regulation of
EGFR signaling.
In summary, we found that APC plays an important
role in the sensitivity of CRC cells to gefitinib. Our finding that
the inhibition of APC increased the sensitivity of CRC cells to
gefitinib suggests that APC may represent a potential therapeutic
target for the treatment of CRC. Further animal studies and
clinical trials are required to verify the efficacy of the
inhibition of APC in the treatment of CRC.
Acknowledgements
The present study was supported by grants from the
Scientific and Technological Projects of Liaoning Province Science
and the Technology Department (no. 2009225011-2), and it was also
supported by the Specialized Research Fund for the Doctoral Program
of Higher Education (no. 20102104110004).
References
|
1
|
de Castro-Carpeno J, Belda-Iniesta C,
Casado Saenz E, Hernandez Agudo E, Feliu Batlle J and Gonzalez
Baron M: EGFR and colon cancer: a clinical view. Clin Transl Oncol.
10:6–13. 2008.PubMed/NCBI
|
|
2
|
Fischer von Weikersthal L, Schalhorn A,
Stauch M, et al: Phase III trial of irinotecan plus infusional
5-fluorouracil/folinic acid versus irinotecan plus oxaliplatin as
first-line treatment of advanced colorectal cancer. Eur J Cancer.
47:206–214. 2011.PubMed/NCBI
|
|
3
|
Krasinskas AM: EGFR signaling in
colorectal carcinoma. Patholog Res Int. 2011:9329322011.PubMed/NCBI
|
|
4
|
Fodde R and Brabletz T: Wnt/β-catenin
signaling in cancer stemness and malignant behavior. Curr Opin Cell
Biol. 19:150–158. 2007.
|
|
5
|
Wieduwilt MJ and Moasser MM: The epidermal
growth factor receptor family: biology driving targeted
therapeutics. Cell Mol Life Sci. 65:1566–1584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Citri A and Yarden Y: EGF-ERBB signalling:
towards the systems level. Nat Rev Mol Cell Biol. 7:505–516. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yarom N and Jonker DJ: The role of the
epidermal growth factor receptor in the mechanism and treatment of
colorectal cancer. Discov Med. 11:95–105. 2011.PubMed/NCBI
|
|
8
|
Albanell J and Gascon P: Small molecules
with EGFR-TK inhibitor activity. Curr Drug Targets. 6:259–274.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wakeling AE, Guy SP, Woodburn JR, et al:
ZD1839 (Iressa): an orally active inhibitor of epidermal growth
factor signaling with potential for cancer therapy. Cancer Res.
62:5749–5754. 2002.PubMed/NCBI
|
|
10
|
Ono M and Kuwano M: Molecular mechanisms
of epidermal growth factor receptor (EGFR) activation and response
to gefitinib and other EGFR-targeting drugs. Clin Cancer Res.
12:7242–7251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou C, Wu YL, Chen G, et al: Erlotinib
versus chemotherapy as first-line treatment for patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase
3 study. Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maemondo M, Inoue A, Kobayashi K, et al:
Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi Y, Zhang L, Liu X, et al: Icotinib
versus gefitinib in previously treated advanced non-small-cell lung
cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority
trial. Lancet Oncol. 14:953–961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mackenzie MJ, Hirte HW, Glenwood G, et al:
A phase II trial of ZD1839 (Iressa) 750 mg per day, an oral
epidermal growth factor receptor-tyrosine kinase inhibitor, in
patients with metastatic colorectal cancer. Invest New Drugs.
23:165–170. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rothenberg ML, LaFleur B, Levy DE, et al:
Randomized phase II trial of the clinical and biological effects of
two dose levels of gefitinib in patients with recurrent colorectal
adenocarcinoma. J Clin Oncol. 23:9265–9274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Trarbach T, Reinacher-Schick A,
Hegewisch-Becker S, et al: Gefitinib in combination with
capecitabine as second-line therapy in patients with advanced
colorectal cancer (aCRC): a phase I/II study of the
Arbeitsgemeinschaft Internistische Onkologie (AIO). Onkologie.
33:89–93. 2010. View Article : Google Scholar
|
|
17
|
Vieitez JM, Valladares M, Pelaez I, et al:
A randomized phase II study of raltitrexed and gefitinib versus
raltitrexed alone as second line chemotherapy in patients with
colorectal cancer. (1839IL/0143). Invest New Drugs. 29:1038–1044.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Santoro A, Comandone A, Rimassa L, et al:
A phase II randomized multicenter trial of gefitinib plus FOLFIRI
and FOLFIRI alone in patients with metastatic colorectal cancer.
Ann Oncol. 19:1888–1893. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nathke IS: The adenomatous polyposis coli
protein: the Achilles heel of the gut epithelium. Annu Rev Cell Dev
Biol. 20:337–366. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fodde R: The APC gene in colorectal
cancer. Eur J Cancer. 38:867–871. 2002. View Article : Google Scholar
|
|
21
|
Barker N: The canonical Wnt/β-catenin
signalling pathway. Methods Mol Biol. 468:5–15. 2008.
|
|
22
|
Hu T and Li C: Convergence between
Wnt-β-catenin and EGFR signaling in cancer. Mol Cancer.
9:2362010.
|
|
23
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paez JG, Janne PA, Lee JC, et al: EGFR
mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cappuzzo F, Magrini E, Ceresoli GL, et al:
Akt phosphorylation and gefitinib efficacy in patients with
advanced non-small-cell lung cancer. J Natl Cancer Inst.
96:1133–1141. 2004. View Article : Google Scholar
|
|
26
|
Yuan G, Regel I, Lian F, et al: WNT6 is a
novel target gene of caveolin-1 promoting chemoresistance to
epirubicin in human gastric cancer cells. Oncogene. 32:375–387.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang X and Guo B: Adenomatous polyposis
coli determines sensitivity to histone deacetylase
inhibitor-induced apoptosis in colon cancer cells. Cancer Res.
66:9245–9251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Narayan S, Jaiswal AS and Balusu R: Tumor
suppressor APC blocks DNA polymerase β-dependent strand
displacement synthesis during long patch but not short patch base
excision repair and increases sensitivity to methylmethane
sulfonate. J Biol Chem. 280:6942–6949. 2005.
|
|
29
|
Fang X, Gu P, Zhou C, et al: β-Catenin
overexpression is associated with gefitinib resistance in non-small
cell lung cancer cells. Pulm Pharmacol Ther. May 23–2013.(Epub
ahead of print).
|
|
30
|
Park SH, Ha SY, Lee JI, et al: Epidermal
growth factor receptor mutations and the clinical outcome in male
smokers with squamous cell carcinoma of lung. J Korean Med Sci.
24:448–452. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kalikaki A, Koutsopoulos A, Trypaki M, et
al: Comparison of EGFR and K-RAS gene status between
primary tumours and corresponding metastases in NSCLC. Br J Cancer.
99:923–929. 2008.
|
|
32
|
Sasaki H, Shimizu S, Endo K, et al: EGFR
and erbB2 mutation status in Japanese lung cancer patients. Int J
Cancer. 118:180–184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barber TD, Vogelstein B, Kinzler KW and
Velculescu VE: Somatic mutations of EGFR in colorectal
cancers and glioblastomas. N Engl J Med. 351:28832004.PubMed/NCBI
|
|
34
|
Nagahara H, Mimori K, Ohta M, et al:
Somatic mutations of epidermal growth factor receptor in colorectal
carcinoma. Clin Cancer Res. 11:1368–1371. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park JJ, Yi JY, Jin YB, et al: Sialylation
of epidermal growth factor receptor regulates receptor activity and
chemosensitivity to gefitinib in colon cancer cells. Biochem
Pharmacol. 83:849–857. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Milligan SA, Burke P, Coleman DT, et al:
The green tea polyphenol EGCG potentiates the antiproliferative
activity of c-Met and epidermal growth factor receptor inhibitors
in non-small cell lung cancer cells. Clin Cancer Res. 15:4885–4894.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
den Hartigh JC, van Bergen en Henegouwen
PM, Verkleij AJ and Boonstra J: The EGF receptor is an
actin-binding protein. J Cell Biol. 119:349–355. 1992.PubMed/NCBI
|