Introduction
Human papillomavirus (HPV) infection is a major
cause of cervical cancer, one of the most common cancers in women
worldwide (1,2). HPV DNA integration into the host
genome results in constitutive expression of the viral oncoproteins
E6 and E7, which deregulate cell cycle control through interactions
with the tumor-suppressor protein p53 and pRb-type oncoproteins
(3). The HPV E6 protein induces the
degradation of p53, thereby inhibiting p53-dependent signaling and
contributing to tumorigenesis (4).
The HPV E7 protein binds to the retinoblastoma family of proteins
(pRb, p107 and p130) and prevents G1 arrest in response to a
variety of antiproliferative signals (5).
pRb is important for progression of the cell cycle
from the G1 to the S phase, which is regulated by the
cyclin-dependent kinases (CDKs) cyclin D-CDK4/6 (6) and cyclin E-CDK2 (7). Cyclin and CDK inhibitors, such as
p16INK4A and p21CIP1, regulate CDK activity.
p21CIP1 activation by p53 inhibits the formation of
cyclin E-CDK2 complexes and triggers cell cycle arrest in the G1
phase (8). Therefore, many tumors
exhibit increased cyclin D1 or CDK4 levels, and a loss of
p16INK4A, Rb and p53. A previous study demonstrated that
the cell cycle regulator proteins cyclin D1 and cyclin E1 are
overproduced in cervical cancer cells (9). When TC-1 cells, which are immortalized
mouse lung epithelial cells, were transduced with HPV E6 and E7,
p53 and Rb expression in the TC-1 cells was reduced as a result of
degradation mediated by interaction with HPV16 E6 and E7 (10). Combining bortezomib treatment with
DNA vaccines was found to result in improved immunity against TC-1
cells in mice compared to monotherapy, and treatment with
bortezomib increased TC-1 tumor cell apoptosis (11). However, the present study did not
identify the mechanism of apoptosis since cyclin-dependent kinase
inhibitors (CKIs), such as p21, are transcriptionally activated
primarily by p53 during the G1 phase.
Bortezomib reversibly inhibits the 26S proteasome,
preventing the degradation of pro-apoptotic proteins related to the
regulation of cell proliferation and survival. Many previous
studies have shown that bortezomib decreases NF-κB and STAT3
activation as well as activation of their downstream target genes,
ICAM-1, MCP-1 and cyclin D1 (12,13).
For these reasons, many clinical trials are in progress to test the
effect of bortezomib alone or in combination with chemo/radiation
to treat various NF-κB-dependent solid tumor models.
Cyclooxygenase-2 (COX-2) is known to play an important role in
carcinogenesis and cancer progression (14). Several studies have shown that COX-2
is overexpressed in cervical intraepithelial neoplasia, but not in
normal cervical tissue (15,16).
The HPV16 E6 and E7 oncoproteins contribute to carcinogenesis by
increasing COX-2 transcription by activating the EGFR-Ras-MAP
kinase pathway (17). COX-2 and
Ki-67 expression levels were found to be downregulated and
neoangiogenesis was inhibited in cervical cancer patients treated
with celecoxib (400 mg) twice daily (18). Moreover, celecoxib induced apoptosis
in various cancer cell lines via a COX-2-independent mechanism,
which may involve inhibition of NF-κB and STAT3 phosphorylation
(19). From these results, we
predicted that celecoxib, a specific inhibitor of COX-2, may have
an important role in cancer treatment in various malignancies when
combined with bortezomib.
We, therefore, investigated whether bortezomib
treatment acts synergistically with celecoxib to increase
therapeutic efficacy against TC-1 cells, in an HPV16 E6- and
E7-expressing cervical cancer model. We assessed various
combinations of bortezomib and celecoxib in TC-1 cells to determine
the most effective cancer therapy with drugs in vitro and
in vivo.
Materials and methods
Cells, reagents and animals
Bortezomib (provided by Janssen Korea) was prepared
as a 7.8 mM stock in PBS and stored at −20°C. Celecoxib (Toronto
Research Chemicals Inc., Toronto, Ontario, Canada) was prepared as
a 200 mM stock in DMSO and stored at −20°C. Antibodies to STAT3,
p21, cyclin D1, CHOP and β-actin, and horseradish peroxidase
(HRP)-conjugated goat anti-rabbit/mouse IgG were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to CDK2,
CDK4, survivin, p38, p-p38, Bid, Bcl-2, Bax and caspase-3, -8 and
-9 were purchased from Cell Signaling Technology (Beverly, MA,
USA). An antibody to COX-2 was purchased from Cayman Chemical Co.
(Ann Arbor, MI, USA). An antibody to BiP was purchased from Abcam
(Cambridge, UK). SB203580 was purchased from Calbiochem (Darmstadt,
Germany). C57BL/6 mice were acquired from the Chung-Ang Laboratory
Animal Service (Seoul, Korea). All procedures were performed
according to the approved protocols and in accordance with the
recommendations of the Ethics Committee of the College of Medicine,
Inje University and Chung-Ang University for the proper use and
care of laboratory animals.
Cell culture conditions and cell
viability assay
The TC-1 cells were cultured in RPMI-1640 medium
(HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum
(FBS; HyClone), 1% penicillin-streptomycin, 1% glutamine, 1%
MEM-non essential amino acids, and 1% sodium pyruvate (Life
Technologies, Grand Island, NY, USA) in a humidified incubator
supplied with 5% CO2 at 37°C (10). TC-1 cells were seeded into 96-well
plates at a density of 5×103 cells/well. The cells were
pre-treated with bortezomib for 6 h and then treated with celecoxib
for an additional 18 h. For comparison, TC-1 cells were treated
either with bortezomib or celecoxib alone, or co-treated for 24 h.
Finally, TC-1 cells pre-exposed to celecoxib for 6 h were then
treated with bortezomib for 18 h. Cell viability was determined by
an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide] assay using the Vybrant MTT cell assay kit (Molecular
Probes, Eugene, OR, USA). The analysis was performed according to
the manufacturer’s instructions. The absorbance at 560 nm was
measured using a microplate reader (Molecular Probes).
Western blot analysis for
apoptosis-related proteins and cell cycle regulators
TC-1 cells were seeded into 24-well plates at a
density of 3×104 cells/well. The TC-1 cells were then
treated with bortezomib and celecoxib for the indicated times.
After the cells were pelleted and resuspended in lysis buffer, 40
μg of total cell protein per sample was subjected to 8–15% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The
proteins were then transferred to an Immobilon polyvinylidene
difluoride membrane (Millipore, Bedford, MA, USA) for
immunoblotting. The primary antibodies used were: anti-Bip,
anti-CDK2, anti-CDK4, anti-CHOP, anti-CyclinD1, anti-cleaved
caspase-3, anti-caspase-8, anti-caspase-9, anti-p38, anti-p53,
anti-p21, anti-Stat3 (1:1,000); and anti-Cox-2, anti-Bid, anti-Bax,
anti-Bcl-2 and anti-β-actin (1:2,000). The primary antibodies were
detected using horseradish peroxidase-conjugated goat anti-rabbit
(1:2,000) or goat anti-mouse (1:10,000) secondary antibodies.
Western blot analysis was performed using standard techniques, and
the bands were visualized with the ECL system (Amersham Pharmacia
Biotech, UK).
Flow cytometric analysis of apoptosis and
the cell cycle
TC-1 cells were treated with bortezomib and
celecoxib singly, in combination, and sequentially for 12 h. The
cells were then analyzed using an Annexin V-FITC detection kit (BD
Pharmingen, San Diego, CA, USA) according to the manufacturer’s
instructions. The TC-1 cells were harvested and fixed in cold 70%
ethanol at 4°C overnight. The next day, the cells were stained with
a 50 μg/ml propidium iodide (PI) solution (Sigma, St. Louis, MO,
USA) and 500 μl 10 μg/ml RNase A (Sigma) for 30 min at 37°C in the
dark. The cells were analyzed immediately using a FACSCalibur flow
cytometer (Becton-Dickinson, San Diego, CA, USA).
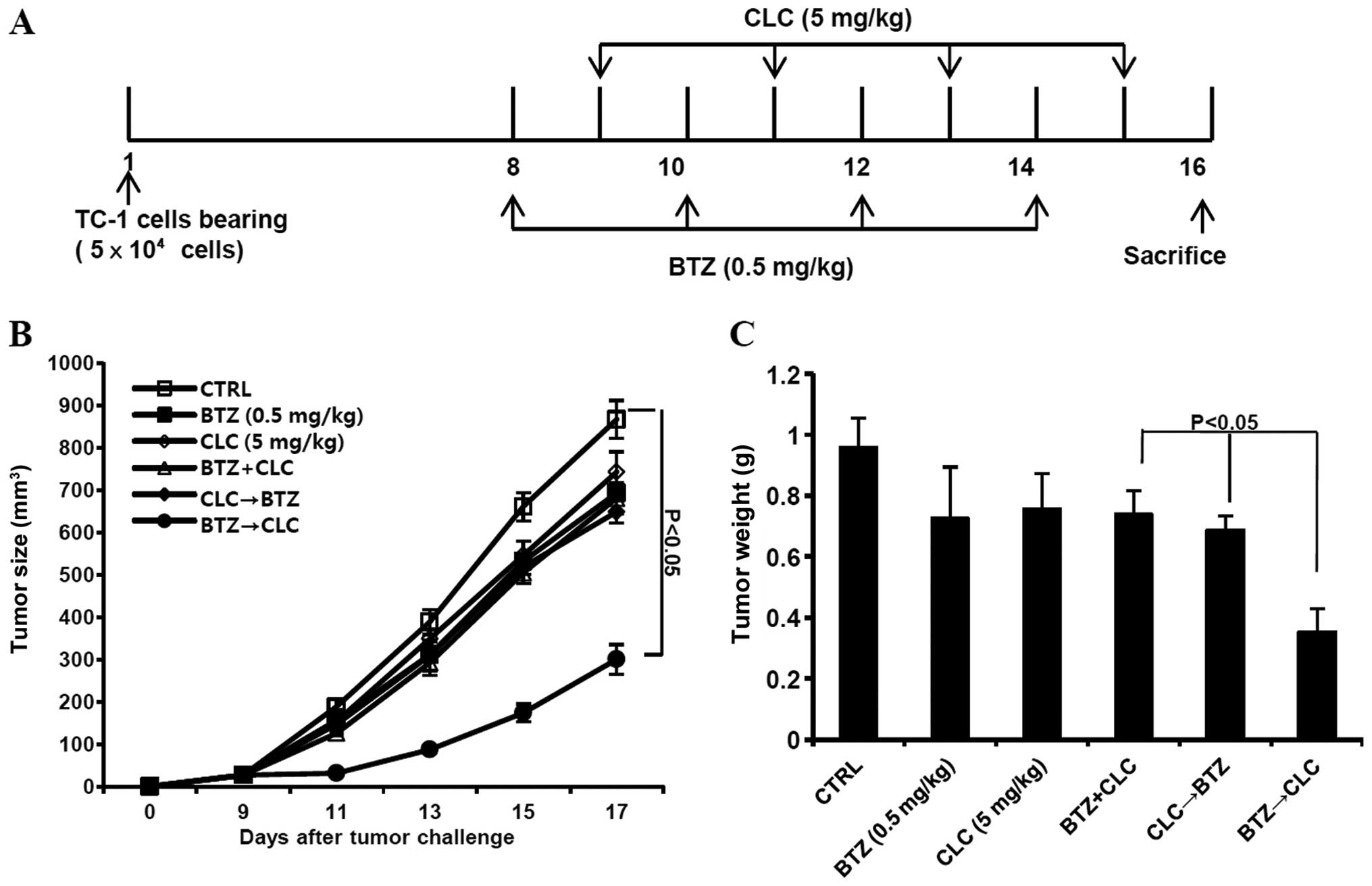
In vivo tumor treatment using bortezomib
and celecoxib
C57BL/6 mice (5–6 per group) were challenged with
5×104 TC-1 tumor cells by subcutaneous (s.c.) injection
for the in vivo tumor treatment experiment. By day 8, the
average tumor size was ~90–100 mm3. The mice were then
divided into the following groups: control (no treatment), single
treatment, co-treatment, and sequential treatment with bortezomib
(0.5 mg/kg) and celecoxib (5 mg/kg). Drug treatment was
administered via intraperitoneal (i.p.) injection. The sequential
treatment group was administered bortezomib (day 8, 10, 12 and 14)
followed by celecoxib (day 9, 11, 12 and 14) or celecoxib (day 8,
10, 12 and 14) followed by bortezomib (day 9, 11, 12 and 14) daily.
The mice in the co-treatment groups were injected with the
appropriate drug four times every other day (day 8, 10, 12 and 14).
The tumor size was measured at intervals of approximately two days.
At the end of the treatment, the mice were sacrificed, and the
tumors were collected and weighed.
Statistics
The mean value for cell viability or mean
fluorescence intensity for the apoptosis assay with standard error
of the mean (SEM) from representative experiments is shown in the
figures. Data were confirmed by at least three independent
experiments. All data shown are representative of at least two
different experiments. The data were compared by analysis of
variance (one-way ANOVA). Statistical significance was defined by a
P-value of <0.05.
Results
Sequential treatment with bortezomib and
celecoxib increases apoptosis via an intrinsic pathway in TC-1
cells
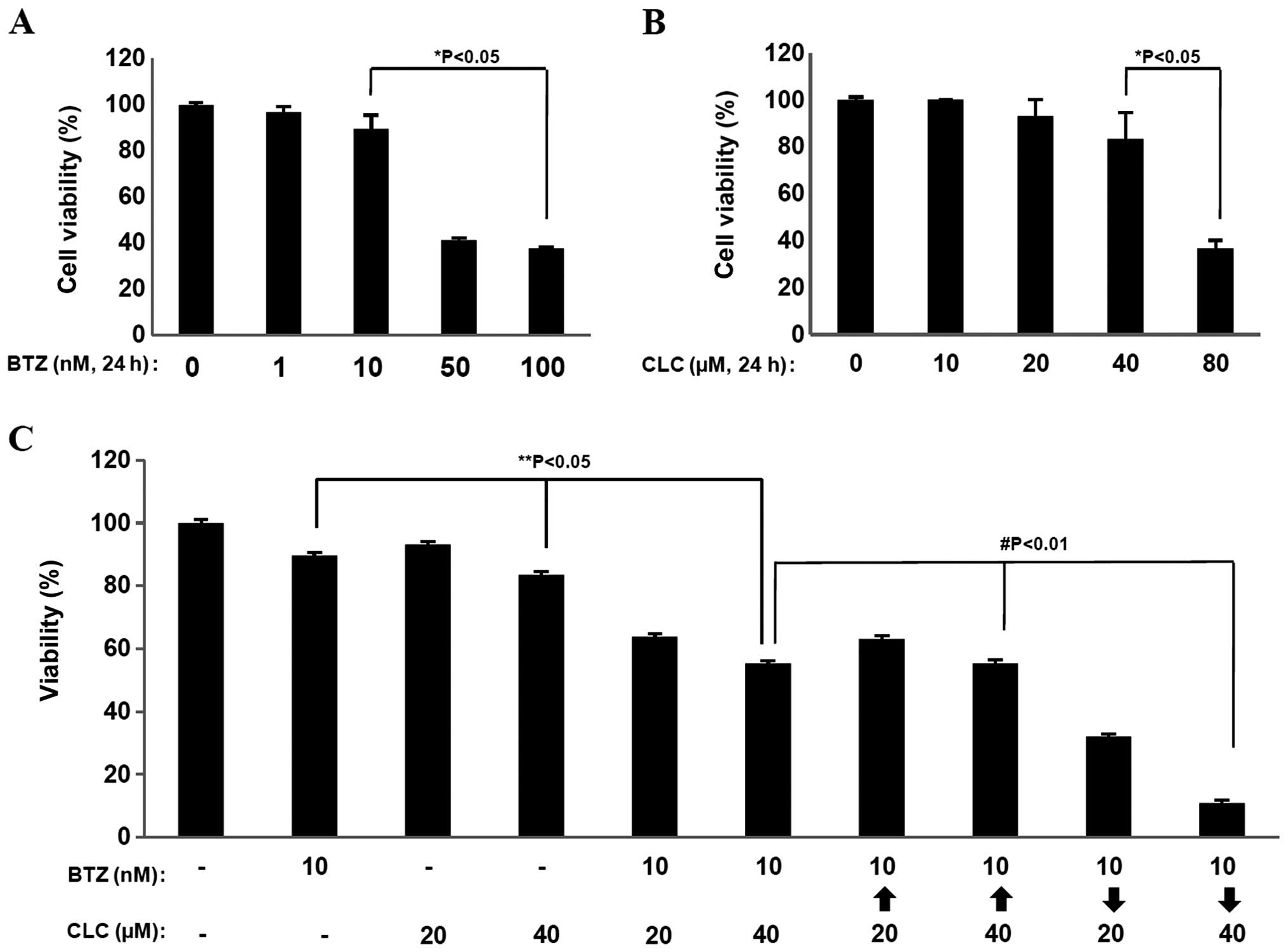
Treatment with bortezomib (BTZ) or celecoxib (CLC)
significantly reduced cell viability at relative high doses (50–100
nM of bortezomib and 80 μM of celecoxib) based on MTT assays
(P<0.05) (Fig. 1A and B).
However, co-treatment with relatively low doses of BTZ (10 nM) and
CLC (20 and 40 μM) induced significant levels of cell death in the
MTT assays compared to single treatment (P<0.05, individual drug
treatment vs. co-treatment) (Fig.
1C). Notably, the effects of combination treatment with BTZ (10
nM) and CLC (40 μM) on cell viability showed strong differences
depending on the treatment order (P<0.01, sequential drug
treatment vs. co-treatment). When TC-1 cells were treated with
bortezomib and then celecoxib (BTZ→CLC), cell viability was
significantly reduced compared to the group treated simultaneously
with BTZ and CLC or the group treated with celecoxib and then
bortezomib (CLC→BTZ) (P<0.01, co-treatment or CLC→BTZ treatment
vs. BTZ→CLC treatment) (Fig. 1C).
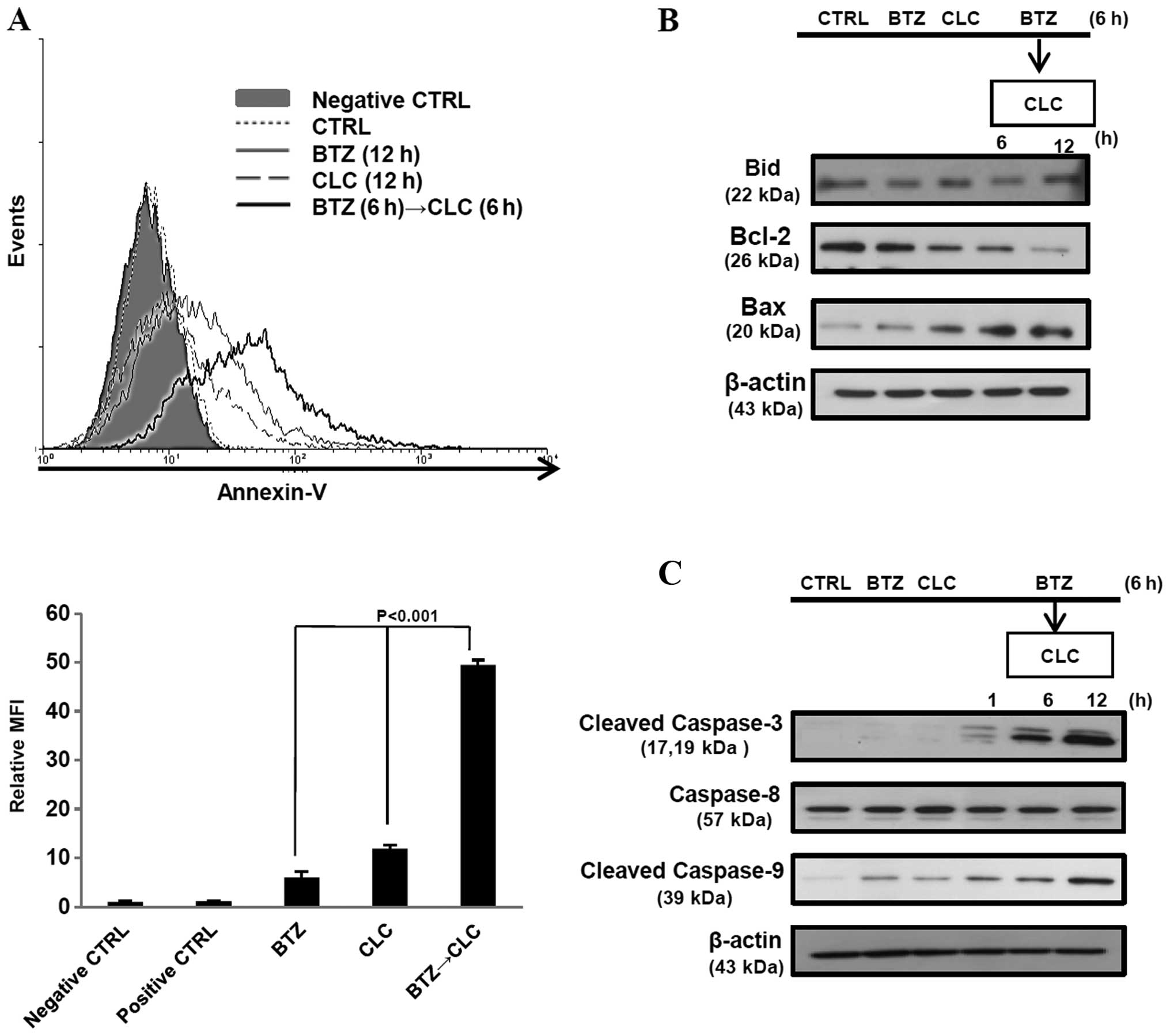
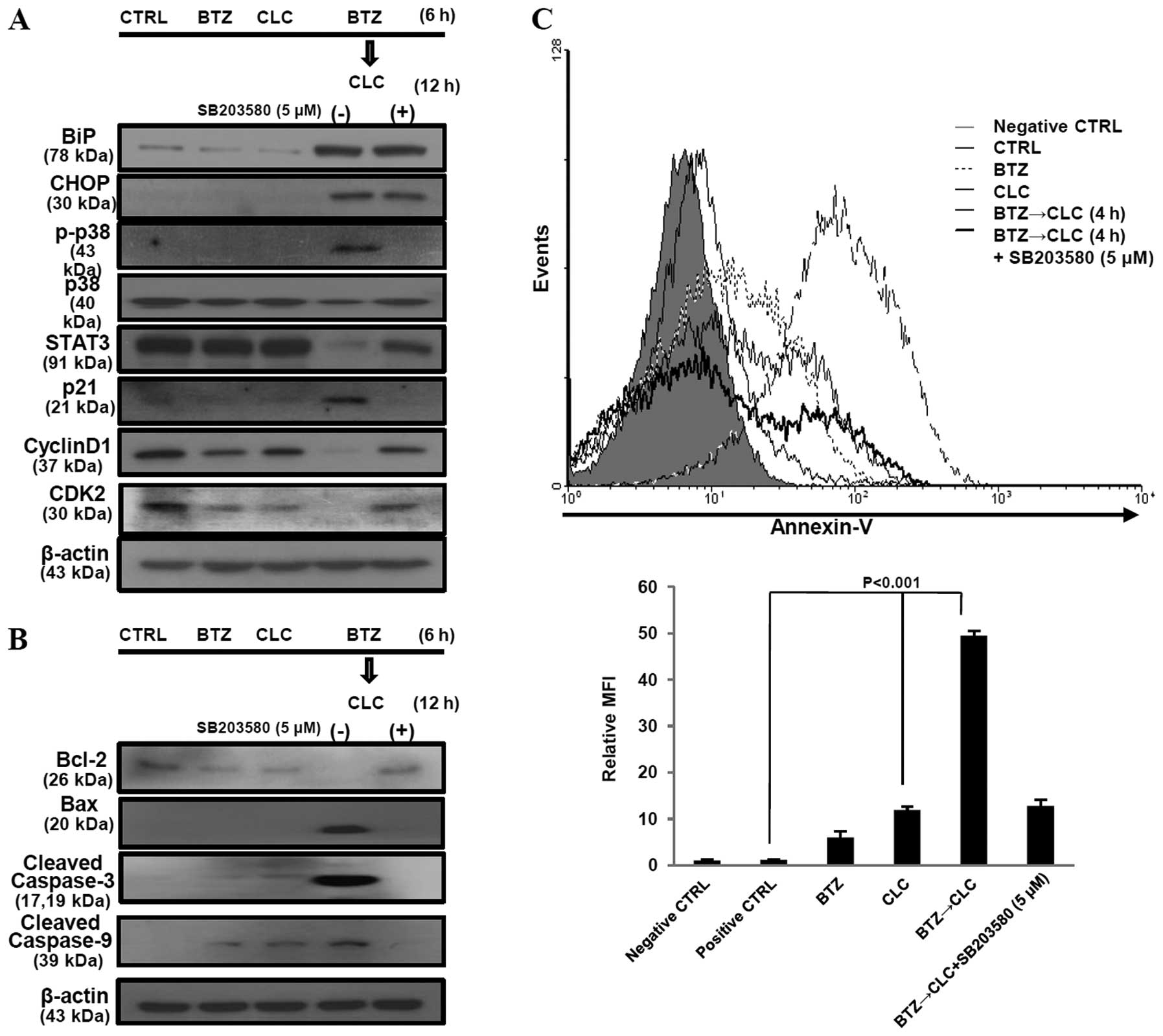
The number of Annexin V-positive TC-1 cells increased after
sequential treatment with bortezomib and celecoxib (P<0.001, BTZ
or CLC treatment vs. BTZ→CLC treatment) (Fig. 2A). Furthermore, production of
pro-apoptotic protein Bax increased, whereas production of the
anti-apoptotic protein Bcl-2 decreased after pre-treatment with
bortezomib followed by treatment with celecoxib. Bid and caspase-8
levels, which are related to the extrinsic apoptotic pathway, did
not change after bortezomib and celecoxib treatment, while levels
of cleaved caspase-3 and -9 increased in the TC-1 cells that were
sequentially treated with bortezomib and celecoxib (Fig. 2B and C). These results suggest that
sequential treatment with relatively low doses of bortezomib and
celecoxib may effectively induce TC-1 cell death through intrinsic
apoptosis.
The apoptosis of TC-1 cells sequentially
treated with bortezomib and celecoxib is mediated by STAT3, cyclin
D1, CDK2 and ER stress
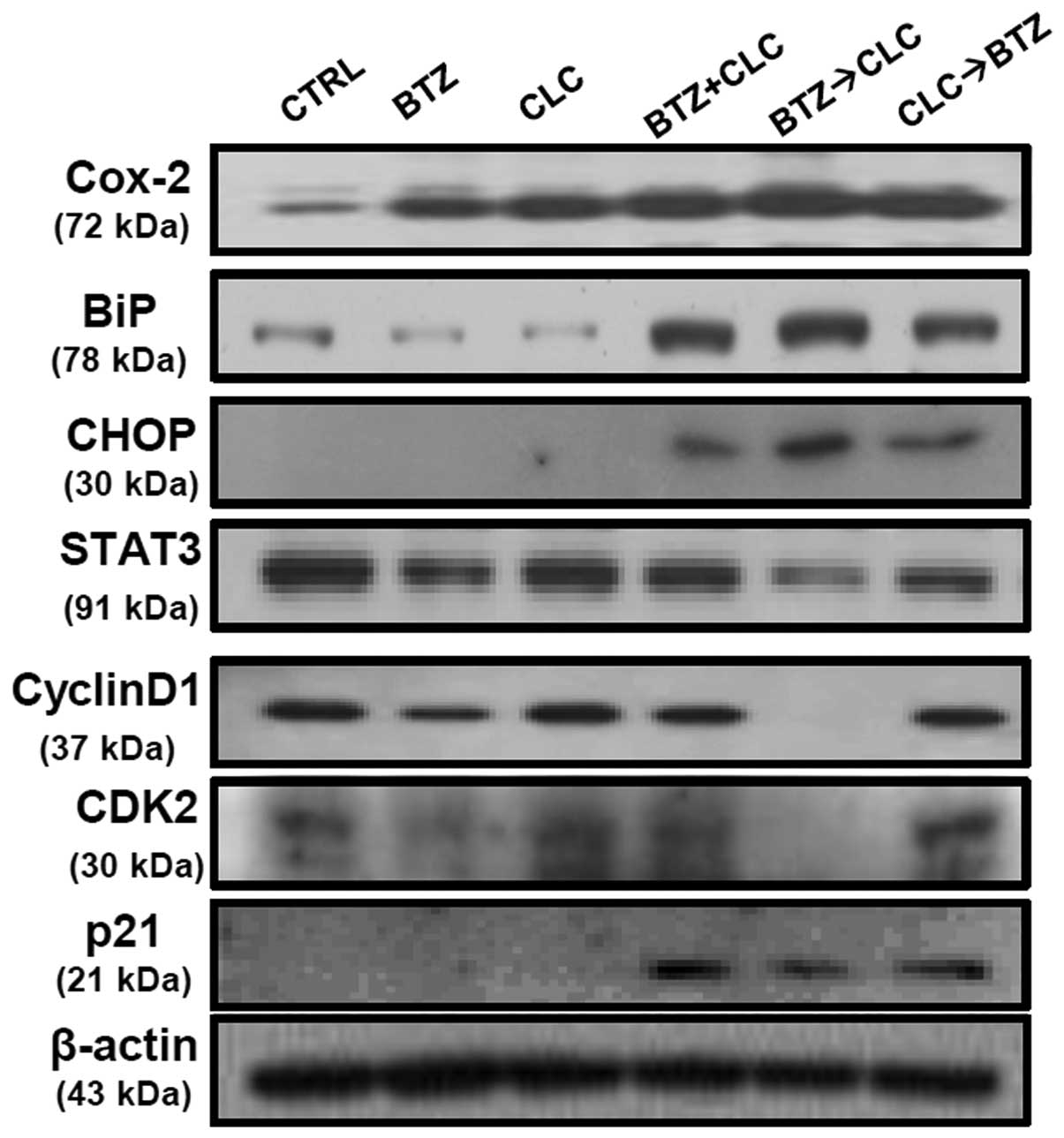
Co-treatment with bortezomib and celecoxib has been
reported to induce apoptosis via production of the ER
stress-related proteins, glucose-regulated protein 78 (GRP78/BiP)
and CCAAT/enhancer binding protein homologous transcription factor
(CHOP/GADD153), but not through inhibition of COX-2 production
(20). Our results are consistent
with these earlier studies, including the presence of increased
COX-2 levels after combined treatment with bortezomib and celecoxib
(Fig. 3). However, the significant
production of BiP and CHOP is not sufficient to explain the high
levels of apoptosis in TC-1 cells since TC-1 cells co-treated with
bortezomib and celecoxib or sequential celecoxib and bortezomib
treatment also exhibited elevated BiP and CHOP protein levels
(Fig. 3). STAT3 and cyclin D1
expression was significantly decreased in the TC-1 cells
sequentially treated with bortezomib and celecoxib compared to the
levels in the TC-1 cells co-treated with bortezomib and celecoxib
or treated with a single drug. p21 production was not significantly
upregulated in the TC-1 cells after single treatment with
bortezomib or celecoxib. Co-treatment with bortezomib and celecoxib
or sequential celecoxib and bortezomib treatment also enhanced p21
production in the p53-degraded TC-1 cells, while the levels of the
cell cycle regulator proteins cyclin D1 and CDK2 did not change at
all (Fig. 3). Notably, we observed
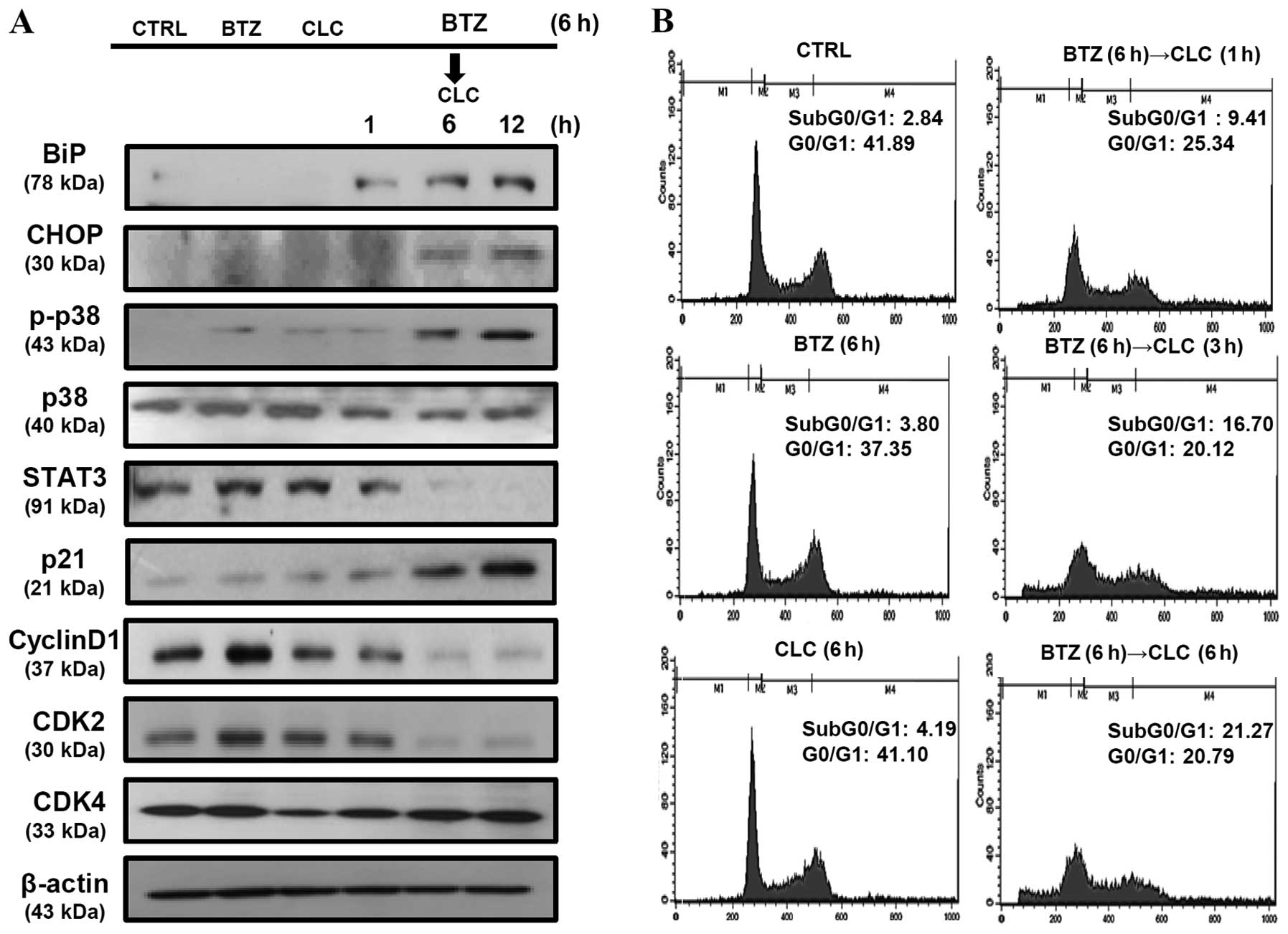
that in the TC-1 cells sequentially treated with bortezomib and
celecoxib, p21 production increased in a time-dependent manner and
was associated with the downregulation of STAT3, cyclin D1, and
CDK2, but not CDK4. BiP and CHOP levels were also significantly
increased in the TC-1 cells sequentially treated with bortezomib
and celecoxib in a time-dependent manner (Fig. 4A). In addition, we found that
apoptosis in TC-1 cells sequentially treated with bortezomib and
celecoxib was associated with cell cycle arrest in the G0/G1 phase,
and the apoptosis rate increased in a time-dependent manner
(Fig. 4B). These results suggest
that the apoptosis of TC-1 cells sequentially treated with
bortezomib and celecoxib may be related to cell cycle regulation as
well as ER stress.
Sequential treatment with bortezomib and
celecoxib enhances TC-1 cell apoptosis via activation of p38 MAPK
and inhibition of the cell cycle
p-p38 MAPK activation has been reported to mediate
cell cycle arrest via an increase in p21 and p27 production and a
decrease in the production of G1-specific cyclins (cyclin A, D1 and
D3) and CDK4 and CDK6. Moreover, activation of p-p38 MAPK is
associated with caspase activation as well as ER stress-induced
apoptosis (21). We found that
sequential treatment with bortezomib and celecoxib increased the
expression of BiP, CHOP, p21 and phosphorylated p38 MAPK, but not
unphosphorylated p38 MAPK, in a time-dependent manner in TC-1 cells
(Fig. 4A). Next, we investigated
the effects of activated p-p38 MAPK on BiP, CHOP, p21, STAT3,
cyclin D1 and CDK2 using SB203580, an inhibitor of p-p38, in the
TC-1 cells sequentially treated with bortezomib and celecoxib. When
p-p38 was inhibited in these cells, production of STAT3, cyclin D1,
CDK2 and anti-apoptotic protein Bcl-2 was restored. In contrast,
p21, Bax, cleaved caspase-3 and -9 were downregulated after
treatment with SB203580 in these cells. However, SB203580 did not
have an effect on BiP and CHOP production in the TC-1 cells
sequentially treated with bortezomib and celecoxib (Fig. 5A and B). The rate of apoptosis was
also significantly decreased when p-p38 MAPK was inhibited by
treatment with SB203580 (P<0.001, BTZ→CLC treatment vs. BTZ→CLC
treatment after exposure to SB203580) (Fig. 5C). These results suggest that
sequential treatment with bortezomib and celecoxib may induce
apoptosis in TC-1 cells through a p-p38-mediated increase in p21
expression and cell cycle arrest, rather than through ER
stress.
Sequential treatment with bortezomib and
celecoxib retards tumor growth in TC-1 cell-inoculated mice
Finally, we determined whether sequential treatment
with bortezomib and celecoxib inhibits the growth of TC-1 cells
in vivo. C57BL/6 mice were challenged with an s.c. injection
of TC-1 cells (5×104/mouse) 1 week prior to treatment
with bortezomib or celecoxib or sequential treatment (Fig. 6A). We observed that the growth of
TC-1 tumors sequentially treated with bortezomib and celecoxib was
retarded compared to tumors co-treated with bortezomib and
celecoxib or sequentially treated with celecoxib and bortezomib
(P<0.05, co-treatment or CLC→BTZ treatment vs. BTZ→CLC
treatment) (Fig. 6B and C).
Therefore, our data suggest that sequential treatment with
bortezomib and celecoxib has greater therapeutic anti-tumor effects
than each single treatment alone or any other bortezomib and
celecoxib combination.
Discussion
The proteasome inhibitor bortezomib is currently
used to treat various malignancies (11,22),
and the selective COX-2 inhibitor celecoxib has been used widely
for chemoprevention (23). In the
present study, we demonstrated that pre-treatment with bortezomib
followed by treatment with celecoxib enhanced apoptotic cell death
through p38 MAPK-dependent dysregulation of cell cycle-related
proteins including cyclin D1, CDK2 and p21, as well as ER stress in
the HPV E6 and E7 protein-expressing TC-1 cervical cancer model.
The growth of tumors sequentially treated with bortezomib and
celecoxib was retarded compared to tumors exposed to a single
treatment with bortezomib or celecoxib in mice challenged with TC-1
cells.
Recent studies have shown that induction of
apoptotic death by bortezomib is triggered via a c-Jun NH2-terminal
kinase (JNK)-dependent mechanism and the accumulation of unfolded
or misfolded proteins, which results in ER stress (24,25).
Bortezomib is thought to induce cell death by stimulating the
expression of CHOP and other pro-apoptotic components (26). Celecoxib has shown potent anticancer
activity in various animal tumor models. However, the pro-apoptotic
effects of celecoxib are not entirely understood, as normal or
upregulated levels of COX-2 have been detected in various cancers
after treatment (27–29). We also observed that TC-1 cells were
overall slightly less sensitive to bortezomib or celecoxib at
relatively low doses, and detected increased COX-2 levels after
co-treatment or sequential treatment with bortezomib and celecoxib.
Notably, sequential treatment with bortezomib (10 nM) and celecoxib
(40 μM) effectively induced apoptosis, compared to co-treatment or
sequential treatment with celecoxib and bortezomib.
The intrinsic apoptotic pathway involves
dysregulation of pro- and anti-apoptotic mitochondrial proteins,
and the extrinsic apoptotic pathway is triggered by the binding of
tumor necrosis factor family death ligands to their appropriate
death receptors (DRs) on the cell surface, followed by caspase-8
cleavage (30). Caspase-8 and Bid,
a pro-apoptotic BH3 family member, are then cleaved by the death
ligands. Expression of STAT3 is correlated with the production of
high levels of anti-apoptotic gene products in cancer (31). Similarly, our results showed that
sequential treatment with bortezomib and celecoxib increased the
production of intrinsic apoptotic-related molecules, such as Bax
and the cleaved form of caspase-9, while it decreased levels of
Bcl-2 and did not affect the levels of Bid and caspase-8.
STAT3 plays an important role in tumor progression
by promoting cell growth and angiogenesis as well as immune evasion
and inflammation. Constitutive activation of STAT3 had been
reported in many cancer cell types, including cervical cancer cells
(32). Activation of STAT3 has an
effect on the expression of cell cycle regulator genes as well as
the expression of Bcl-2 and Bax. STAT3 production also affects
tumor growth through control of transient G1 to S phase molecules,
particularly cyclin D1, which is a G0/G1 cell cycle-related protein
(33). It has been reported that
cyclin D1-dependent repression of STAT3 may induce apoptosis
(34). Transition through G1 into S
phase requires activation of cyclin D through the binding of CDK4
(4). In the next phase, cyclin E is
activated by CDK2 binding (5), and
cell cycle progression is regulated by CKIs such as p21. p21 in
particular, when activated by p53, binds to the cyclin-CDK complex
and subsequently blocks progression of the cell cycle at the G0/G1
checkpoint (35). Many studies have
demonstrated that STAT3 overproduction in tumor cells is related to
tumor survival (30), and decreased
cyclin D1 and CDK2 levels are known to induce apoptosis in various
cancer cells (36,37). A previous study has shown that
withaferin A, the active component of the medicinal plant
Withania somnifera, significantly downregulates the
expression of HPV E6 and E7 proteins in CaSki cells and restores
the p53 pathway. Withaferin A also decreases the levels of STAT3
(38). Our results showed that
STAT3, cyclin D1, and CDK2 levels were decreased in TC-1 cells
sequentially treated with bortezomib and celecoxib. Moreover,
increased levels of p21 led to cell cycle arrest, despite the lack
of a concurrent upregulation in p53 expression. These results
indicate that apoptosis in TC-1 cells sequentially treated with
bortezomib and celecoxib may be mediated by decreased production of
STAT3, cyclin D1 and CDK2 in the G1 phase even in p53-deficient
cancer cells.
We detected an increase in p21 protein expression
with all combined treatment regimens, which correlates with
previous research indicating that p21 is positively regulated by
CHOP under conditions of ER stress (39). The ER maintains a balance between
protein synthesis and degradation (40). BiP is a major component of the
pro-survival pathway in the unfolded protein response (UPR) and
plays an important role in the ER in protein folding and assembly
(41,42). BiP dissociates from the three ER
transmembrane receptors (PERK, ATF-6 and IRE-1α), leading to their
activation and triggering the UPR. The activated PERK pathway
induces downstream CHOP expression, triggering apoptosis (43). CHOP is usually produced at very low
levels in the cytosol, but is produced at high levels in response
to ER stress, leading to the induction of CHOP and its accumulation
in the nucleus (39). p38 MAPK has
been reported to promote the ER stress response (44) and cell cycle arrest in the G1 phase
(45). In the present study, we
observed an increased expression of CHOP and phosphorylated p38
(p-p38) after co-treatment with bortezomib and celecoxib in TC-1
cells. Notably, our results also showed that the apoptosis of TC-1
cells sequentially treated with bortezomib and celecoxib was more
closely related to cell cycle arrest and p-p38 levels than to
increased CHOP expression. SB203580, an inhibitor of p-p38, affects
the expression of G0/G1 cell cycle molecules, cyclin D1 and CDK2,
but has no effect on CHOP. These results suggest that sequential
treatment with bortezomib and celecoxib in p53-degraded cancer
cells induces apoptosis through p38 MAPK-dependent cell cycle
arrest rather than ER stress.
A previous clinical trial showed that the
combination of bortezomib and celecoxib can be used to treat
several types of advanced solid tumors without dose-limiting
toxicities, although disease progression was reported in most of
the patients (46). Sequential
treatment with bortezomib and celecoxib demonstrated the most
effective antitumor effects in vivo study. Unfortunately,
tumor growth was not completely inhibited by sequential treatment
with bortezomib and celecoxib. Based on these results, further
studies are needed to clarify related apoptotic pathways and new
target molecules for the development of a combined anticancer
chemotherapy.
The present study supports the concept that
apoptosis induced by ER stress is enhanced by treatment with a
combination of bortezomib and celecoxib. In addition, our results
suggest that the sequential administration of bortezomib and
celecoxib may be an effective strategy for inducing apoptosis in
p53-degraded cancer cells via p38-mediated cell cycle
regulation.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(NRF-2013-R1A1A2-010668, NRF-2012-R1A1A3-013468).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar
|
|
2
|
ZurHausen H and de Villiers EM: Human
papillomaviruses. Annu Rev Microbiol. 48:27–47. 1994.
|
|
3
|
Maher DM, Bell MC, O’Donnell EA, Gupta BK,
Jaggi M and Chauhan SC: Curcumin suppresses human papillomavirus
oncoproteins, restores p53, Rb, and PTPN13 proteins and inhibits
benzo[a]pyrene-induced upregulation of HPV E7. Mol Carcinog.
50:47–57. 2011.PubMed/NCBI
|
|
4
|
Werness BA, Levine AJ and Howley PM:
Association of human papillomavirus types 16 and 18 E6 proteins
with p53. Science. 248:76–79. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alani RM and Munger K: Human
papillomaviruses and associated malignancies. J Clin Oncol.
16:330–337. 1998.PubMed/NCBI
|
|
6
|
Ortega S, Malumbres M and Barbacid M:
Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim
Biophys Acta. 1602:73–87. 2002.PubMed/NCBI
|
|
7
|
Sherr CJ and Roberts JM: Living with or
without cyclins and cyclin-dependent kinases. Genes Dev.
18:2699–2711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bahnassy AA, Zekri AR, Alam El-Din HM,
Aboubakr AA, Kamel K, El-Sabah MT and Mokhtar NM: The role of
cyclins and cyclin inhibitors in the multistep process of
HPV-associated cervical carcinoma. J Egypt Natl Canc Inst.
18:292–302. 2006.PubMed/NCBI
|
|
10
|
Lin KY, Guarnieri FG, Staveley-O’Carroll
KF, Levitsky HI, August JT, Pardoll DM and Wu TC: Treatment of
established tumors with a novel vaccine that enhances major
histocompatibility class II presentation of tumor antigen. Cancer
Res. 56:21–26. 1996.PubMed/NCBI
|
|
11
|
Tseng CW, Monie A, Wu CY, Huang B, Wang
MC, Hung CF and Wu TC: Treatment with proteasome inhibitor
bortezomib enhances antigen-specific CD8+
T-cell-mediated antitumor immunity induced by DNA vaccination. J
Mol Med. 86:899–908. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Malara N, Focà D, Casadonte F, Sesto MF,
Macrina L, Santoro L, Scaramuzzino M, Terracciano R and Savino R:
Simultaneous inhibition of the constitutively activated nuclear
factor kappaB and of the interleukin-6 pathways is necessary and
sufficient to completely overcome apoptosis resistance of human
U266 myeloma cells. Cell Cycle. 7:3235–3245. 2008. View Article : Google Scholar
|
|
13
|
Wen J, Feng Y, Huang W, Chen H, Liao B,
Rice L, Preti HA, Kamble RT, Zu Y, Ballon DJ and Chang CC: Enhanced
antimyeloma cytotoxicity by the combination of arsenic trioxide and
bortezomib is further potentiated by p38 MAPK inhibition. Leuk Res.
34:85–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eberhart CE, Coffey RJ, Radhika A,
Giardiello FM, Ferrenbach S and DuBois RN: Upregulation of
cyclooxygenase-2 gene expression in human colorectal adenomas and
adenocarcinomas. Gastroenterology. 107:1183–1188. 1994.PubMed/NCBI
|
|
15
|
Kulkarni S, Rader JS, Zhang F, Liapis H,
Koki AT, Masferrer JL, Subbaramaiah K and Dannenberg AJ:
Cyclooxygenase-2 is overexpressed in human cervical cancer. Clin
Cancer Res. 7:429–434. 2011.
|
|
16
|
Kim K, Jeon YT, Park IA, Kim JW, Park NH,
Kang SB, Lee HP and Song YS: Cyclooxygenase-2 expression in
cervical intraepithelial neoplasia. Ann NY Acad Sci. 1171:111–115.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Subbaramaiah K and Dannenberg AJ:
Cyclooxygenase-2 transcription is regulated by human papillomavirus
16 E6 and E7 oncoproteins: evidence of a corepressor/coactivator
exchange. Cancer Res. 67:3976–3985. 2007. View Article : Google Scholar
|
|
18
|
Ferrandina G, Ranelletti FO, Legge F,
Lauriola L, Salutari V, Gessi M, Testa AC, Werner U, Navarra P,
Tringali G, Battaglia A and Scambia G: Celecoxib modulates the
expression of cyclooxygenase-2, ki67, apoptosis-related marker, and
microvessel density in human cervical cancer: a pilot study. Clin
Cancer Res. 9:4324–4331. 2003.PubMed/NCBI
|
|
19
|
Sareddy GR, Geeviman K, Ramulu C and Babu
PP: The nonsteroidal anti-inflammatory drug celecoxib suppresses
the growth and induces apoptosis of human glioblastoma cells via
the NF-κB pathway. J Neurooncol. 106:99–109. 2011.PubMed/NCBI
|
|
20
|
Kardosh A, Golden EB, Pyrko P, Uddin J,
Hofman FM, Chen TC, Louie SG, Petasis NA and Schönthal AH:
Aggravated endoplasmic reticulum stress as a basis for enhanced
glioblastoma cell killing by bortezomib in combination with
celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib. Cancer
Res. 68:843–851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mukhopadhyay I, Sausville EA, Doroshow JH
and Roy KK: Molecular mechanism of adaphostin-mediated G1 arrest in
prostate cancer (PC-3) cells: signaling events mediated by
hepatocyte growth factor receptor, c-Met, and p38 MAPK pathways. J
Biol Chem. 281:37330–37344. 2006. View Article : Google Scholar
|
|
22
|
Fujita T, Doihara H, Washio K, Ino H,
Murakami M, Naito M and Shimizu N: Antitumor effects and drug
interactions of the proteasome inhibitor bortezomib (PS341) in
gastric cancer cells. Anticancer Drugs. 18:677–686. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang X, Morham SG, Langenbach R and Young
DA: Malignant transformation and antineoplastic actions of
nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null
embryo fibroblasts. J Exp Med. 190:451–459. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nawrocki ST, Carew JS, Pino MS, Highshaw
RA, Dunner K Jr, Huang P, Abbruzzese JL and McConkey DJ: Bortezomib
sensitizes pancreatic cancer cells to endoplasmic reticulum
stress-mediated apoptosis. Cancer Res. 65:11658–11666. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Obeng EA, Carlson LM, Gutman DM,
Harrington WJ Jr, Lee KP and Boise LH: Proteasome inhibitors induce
a terminal unfolded protein response in multiple myeloma cells.
Blood. 107:4907–4916. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fribley A and Wang CY: Proteasome
inhibitor induces apoptosis through induction of endoplasmic
reticulum stress. Cancer Biol Ther. 5:745–748. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hanif R, Pittas A, Feng Y, Koutsos MI,
Qiao L, Staiano-Coico L, Shiff SI and Rigas B: Effects of
nonsteroidal anti-inflammatory drugs on proliferation and on
induction of apoptosis in colon cancer cells by a
prostaglandin-independent pathway. Biochem Pharmacol. 52:237–245.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kardosh A, Blumenthal M, Wang WJ, Chen TC
and Schönthal AH: Differential effects of selective COX-2
inhibitors on cell cycle regulation and proliferation of
glioblastoma cell lines. Cancer Biol Ther. 3:9–16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arico S, Pattingre S, Bauvy C, Gane P,
Barbat A, Codogno P and Ogier-Denis E: Celecoxib induces apoptosis
by inhibiting 3-phosphoinositide-dependent protein kinase-1
activity in the human colon cancer HT-29 cell line. J Biol Chem.
277:27613–27621. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kawano M, Hirano T, Matsuda T, Taga T,
Horii Y, Iwato K, Asaoku H, Tang B, Tanabe O, Tanaka H, Kuramoto A
and Kishimoto T: Autocrine generation and requirement of BSF-2/IL-6
for human multiple myelomas. Nature. 332:83–85. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen J, Wang J, Lin L, He L, Wu Y, Zhang
L, Yi Z, Chen Y, Pang X and Liu M: Inhibition of STAT3 signaling
pathway by nitidine chloride suppressed the angiogenesis and growth
of human gastric cancer. Mol Cancer Ther. 11:277–287. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang J, Li X, Lu X and Pi L: The
regulation of stat3 signal transduction pathway to G1 to S phase of
laryngocarcinoma cell. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za
Zhi. 22:699–703. 2008.(In Chinese).
|
|
34
|
Ishii Y, Pirkmaier A, Alvarez JV, Frank
DA, Keselman I, Logothetis D, Mandeli J, O’Connell MJ, Waxman S and
Germain D: Cyclin D1 overexpression and response to bortezomib
treatment in a breast cancer model. J Natl Cancer Inst.
98:1238–1247. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Harper JV and Brooks G: The mammalian cell
cycle: an overview. Methods Mol Biol. 296:113–153. 2005.PubMed/NCBI
|
|
36
|
Wang J, Wang Q, Cui Y, Liu ZY, Zhao W,
Wang CL, Dong Y, Hou L, Hu G, Luo C, Chen J and Lu Y: Knockdown of
cyclin D1 inhibits proliferation, induces apoptosis, and attenuates
the invasive capacity of human glioblastoma cells. J Neurooncol.
106:473–484. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shapiro GI: Cyclin-dependent kinase
pathways as targets for cancer treatment. J Clin Oncol.
24:1770–1783. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Munagala R, Kausar H, Munjal C and Gupta
RC: Withaferin A induces p53-dependent apoptosis by repression of
HPV oncogenes and upregulation of tumor suppressor proteins in
human cervical cancer cells. Carcinogenesis. 32:1697–1705. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mihailidou C, Papazian I, Papavassiliou AG
and Kiaris H: CHOP-dependent regulation of p21/waf1 during ER
stress. Cell Physiol Biochem. 25:761–766. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mori T, Hayashi T and Su TP: Compromising
sigma-1 receptors at the ER renders cytotoxicity to
physiologicallyrelevant concentrations of dopamine in a
NF-κB/Bcl-2-dependent mechanism: Potential relevance to Parkinson’s
disease. J Pharmacol Exp Ther. 341:663–671. 2012.PubMed/NCBI
|
|
41
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shimazawa M, Miwa A, Ito Y, Tsuruma K,
Aihara M and Hara H: Involvement of endoplasmic reticulum stress in
optic nerve degeneration following
N-methyl-D-aspartate-induced retinal damage in mice. J
Neurosci Res. 90:1960–1969. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang KH, Kuo KL, Chen SC, Weng TI, Chuang
YT, Tsai YC, Pu YS, Chiang CK and Liu SH: Down-regulation of
glucose-regulated protein (GRP) 78 potentiates cytotoxic effect of
celecoxib in human urothelial carcinoma cells. PLoS One.
7:e336152012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shimada Y, Kobayashi H, Kawagoe S, Aoki K,
Kaneshiro E, Shimizu H, Eto Y, Ida H and Ohashi T: Endoplasmic
reticulum stress induces autophagy through activation of p38 MAPK
in fibroblasts from Pompe disease patients carrying c.546G>T
mutation. Mol Genet Metab. 104:566–573. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee B, Kim CH and Moon SK: Honokiol causes
the p21WAF1-mediated G(1)-phase arrest of the cell cycle through
inducing p38 mitogen activated protein kinase in vascular smooth
muscle cells. FEBS Lett. 580:5177–5184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hayslip J, Chaudhary U, Green M, Meyer M,
Dunder S, Sherman C, Salzer S, Kraft A and Montero AJ: Bortezomib
in combination with celecoxib in patients with advanced solid
tumors: a phase I trial. BMC Cancer. 7:2212007. View Article : Google Scholar : PubMed/NCBI
|