Introduction
Breast cancer is one of the most common causes of
cancer-related mortality in women, and its incidence is steadily
increasing (1,2). It is considered a highly metastatic
cancer as a large portion of the patients frequently relapse with
systemic dissemination of cancer even after radical extensive
surgery (3). In addition, many
patients succumb to this disease each year worldwide although many
cancer therapeutic methods have been developed for the treatment of
breast cancer. Therefore, new treatment strategies are needed for
this disease.
Arsenic trioxide (As2O3) was
used in Chinese medicine for solid cancer treatment, and is now
being used as a standard treatment for refractory acute
promyelocytic leukemia (APL) (4,5).
Clinical trials with As2O3 were performed in
a certain type of solid cancers (6,7), but
failed to prove clinical efficacy due to serious toxicities
(8,9). Arsenic hexoxide
(As4O6) has been used as a Korean folk remedy
for cancer management since the late 1980s. There were scarce
toxicities at the doses where As4O6 was used
as a Korean folk remedy for the solid and hematologic malignancies.
However, few studies regarding the anticancer effects of
As4O6 have been performed. Only a few reports
showed that the anticancer effects of As4O6
were more potent than those of As2O3 in human
cancer cells in vitro, and that signaling pathways of
As4O6-induced cell death were different from
those of As2O3 (10). We previously demonstrated that
As4O6 induced both caspase-dependent
apoptosis and autophagic cell death in human cancer cells (11). In addition,
As4O6 has mostly been used for solid cancers
in Korea. The anecdotal cases show some marked responses even in
very advanced cancer.
Tumor necrosis factor-α (TNF-α) is a cytokine
involved in systemic inflammation and is produced chiefly by
activated macrophages. The primary role of TNF consists in the
regulation of immune cells. TNF is able to induce interleukin (IL)
production, apoptotic cell death and inflammation to inhibit
tumorigenesis and viral replication. TNF-α induces cell death
through the extrinsic pathway in some cancer cells (12). However, most cancer cells are
resistant to TNF-α-induced cell death by activation of nuclear
factor-κB (NF-κB) followed by the enhanced transcription of
anti-apoptotic proteins that interfere with cell death signaling
(13). High serum level of TNF-α is
more frequently observed in patients with advanced and metastatic
cancer than in those with early stage cancer (14), and is closely related to cancer
progression and patient quality of life (14,15).
In addition, NF-κB is involved in drug resistance as well as
metastasis (16). Therefore, NF-κB
is a suitable therapeutic target for cancer treatment. If NF-κB is
suppressed by less toxic drugs, TNF-α induces apoptosis of cancer
cells. This will be an alternative approach to treat the patients
with metastatic or advanced cancer without showing serious
side-effects. We also found the synergism between TNF-α and
As4O6. In the present study, we hypothesized
that As4O6 induced the synergism with TNF-α
by the inhibition of NF-κB. Therefore, we explored the anticancer
effects of As4O6 with a particular focus on
NF-κB and NF-κB-regulated gene products involved in cancer
metastasis, and on NF-κB-mediated cellular responses in breast
cancer cells.
Materials and methods
Cells and reagents
MCF-7 human breast cancer cells from the American
Type Culture Collection (Rockville, MD, USA) were cultured in
RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with
10% (v/v) fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY,
USA), 1 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml
streptomycin at 37°C in a humidified atmosphere of 95% air and 5%
CO2. As4O6 was provided by the
Chonjisan Institute (Seoul, Korea). Antibodies against
procaspase-3, procaspase-8, COX-2, cyclin D1, c-Myc, Bcl-2, Bcl-xL,
XIAP, cIAP-1, cIAP-2, MMP-2, MMP-9, VEGF and NF-κB (p65) were
purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
Antibodies against phospho-IκBα (Ser 32/36), and IκB, were
purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Antibodies against poly(ADP-ribose) polymerase (PARP), LC3 and
Beclin-1 were purchased from Pharmingen (San Diego, CA, USA). An
antibody against β-actin was from Sigma (Beverly, MA, USA).
Peroxidase-labeled donkey anti-rabbit and sheep anti-mouse
immunoglobulins, and an enhanced chemiluminescence (ECL) kit were
purchased from Amersham (Arlington Heights, IL, USA). All other
chemicals not specifically cited here were purchased from Sigma
Chemical Co. (St. Louis, MO, USA). All these solutions were stored
at −20°C. Stock solutions of DAPI (100 μg/ml) and propidium iodide
(PI; 1 mg/ml) were prepared in phosphate-buffered saline (PBS).
Cell viability assays
For the cell viability assay, the cells were seeded
onto 24-well plates at a concentration of 5×105
cells/ml, and then treated with the indicated concentration of
As4O6 for 24 or 48 h.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (0.5
mg/ml) was subsequently added to each well. After 3 h of additional
incubation, 100 μl of a solution containing 10% SDS (pH 4.8) plus
0.01 N HCl was added to dissolve the crystals. The absorption
values at 570 nm were determined with an ELISA plate reader.
Nuclear staining
After treatment with the indicated concentration of
As4O6, the cells were harvested, washed with
PBS and fixed with 3.7% paraformaldehyde in PBS for 10 min at room
temperature. Fixed cells were washed with PBS and stained with 2.5
μg/ml 4,6-diamidino-2-phenylindole (DAPI) solution for 10 min at
room temperature. The cells were washed twice with PBS and analyzed
under a fluorescent microscope.
Flow cytometry assay
The cells were plated at a concentration of
1×106 cells/well in 6-well plates. Reduced
(sub-G1) DNA content was measured by PI staining. The
DNA content in each cell nucleus was determined with a FACSCalibur
flow cytometer (Becton-Dickinson, San Jose, CA, USA). Three
independent experiments were performed (17).
Western blot analysis
Total cell lysates were obtained using lysis buffer
containing 0.5% SDS, 1% NP-40, 1% sodium deoxycholate, 150 mM NaCl,
50 mM Tris-Cl (pH 7.5) and protease inhibitors. The concentrations
of cell lysate proteins were determined by Bradford protein assay
(Bio-Rad Laboratories, Richmond, CA, USA) using bovine serum
albumin as the standard. To determine the protein expression of
NF-κB in the cytoplasm and the nuclei, we prepared separate
extracts. The cells were washed with ice-cold PBS (pH 7.4) and
lysed in buffer A [10 mM HEPES (pH 7.9), 1.5 mM MgCl2,
0.5 mM dithiothreitol (DTT), 5 μM leupeptin, 2 μM pepstatin A, 1 μM
aprotinin and 20 μM phenylmethylsulfonyl fluoride] by repeated
freezing and thawing. Nuclear and cytoplasmic fractions were
separated by centrifugation at 1,000 × g for 20 min. The
cytoplasmic extract (supernatant) was obtained. The pellets were
washed with buffer A, and resuspended in buffer B [10 mM Tris-Cl
(pH 7.5), 0.5% deoxycholate, 1% NP-40, 5 mM EDTA, 0.5 mM DTT, 5 μM
leupeptin, 2 μM pepstatin A, 1 μM aprotinin and 20 μM
phenylmethylsulfonyl fluoride]. The suspension was agitated for 30
min at 4°C and centrifuged at 10,000 × g for 20 min. The
supernatant fraction containing nuclear proteins was collected.
Molecular mass markers for proteins were obtained from Pharmacia
Biotech (Saclay, France). Thirty micrograms of the lysate proteins
were resolved by electrophoresis, electrotransferred to
polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA),
and then incubated with primary antibodies followed by secondary
antibody conjugated to peroxidase. Blots were developed with an ECL
detection system.
Transfection
NF-κB-luciferase constructs (consensus NF-κB binding
sequence was cloned into the pGL3 basic luciferase expression
vector) were kindly provided by Dr G. Koretzky (University of
Pennsylvania). Transient transfection was performed using
Lipofectamine (Gibco-BRL) according to the manufacturer’s
protocol.
Luciferase assay
After experimental treatments, cells were washed
twice with cold PBS, lysed in a passive lysis buffer provided in
the dual luciferase kit (Promega, Madison, WI, USA), and assayed
for luciferase activity using a TD-20/20 luminometer (Turner
Designs, Sunnyvale, CA, USA) according to the manufacturer’s
protocol. Data were presented as a ratio between Firefly and
Renilla luciferase activities.
Statistical analysis
Each experiment was performed in triplicate. The
results are expressed as means ± SD. Significant differences were
determined using the one-way ANOVA with post-test Neuman-Keuls in
the cases of at least three treatment groups and Student’s t-test
for two-group comparison. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of As4O6 on
cell growth in MCF-7 human breast cancer cells
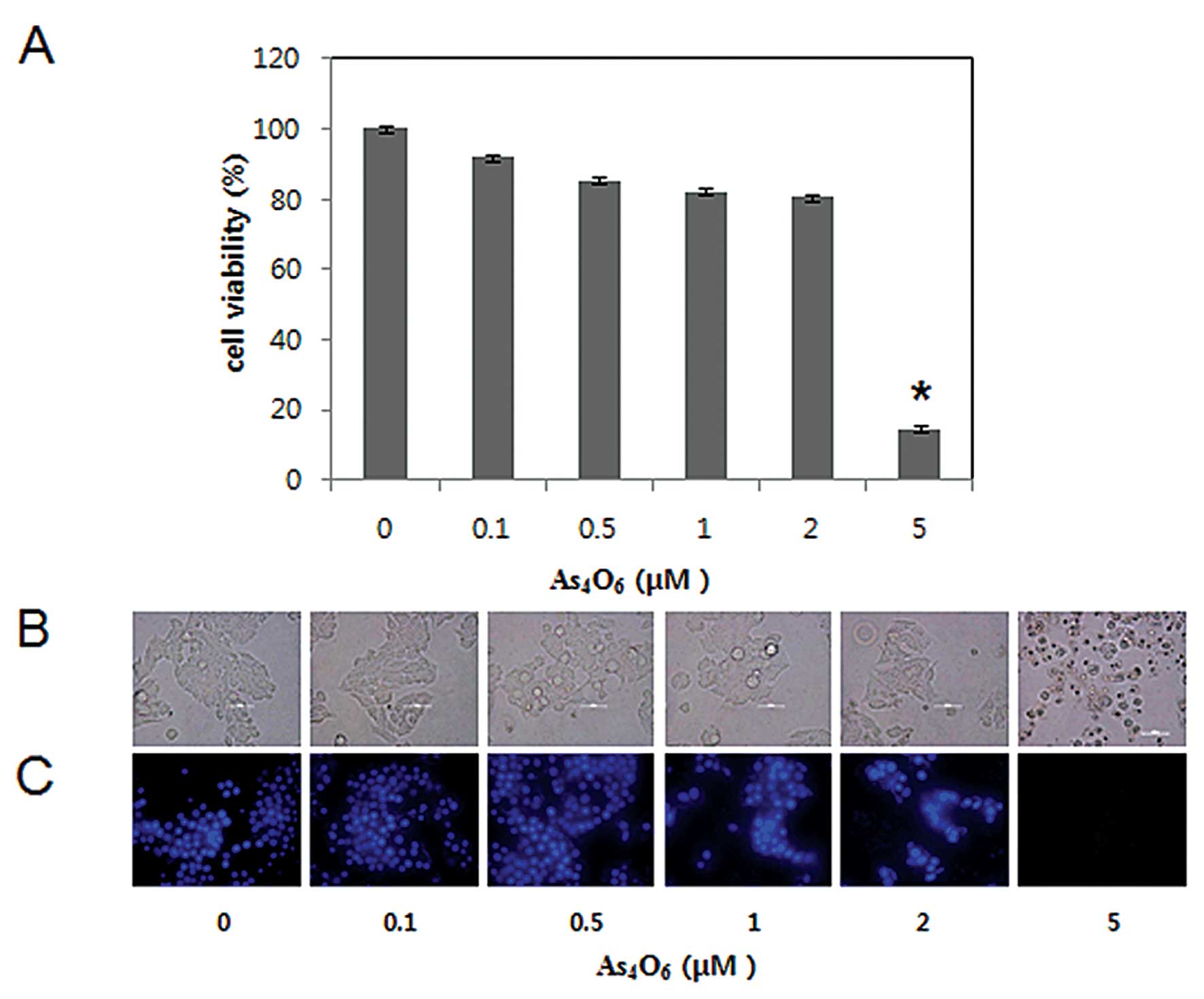
To investigate the antitumor activity of
As4O6 in MCF-7 cells, we performed MTT, light
microscopic observation, and DAPI staining. Cells were treated for
48 h with various concentrations of As4O6
(0.1–5 μM). The cell growth was assessed by MTT assay, which
revealed that As4O6 significantly inhibited
the growth of MCF-7 cells at the concentration of 5 μM, and the 50%
inhibition of cell growth (IC50) was >2 μM (Fig. 1A). To determine whether the decrease
in cell growth of MCF-7 cells was related to induction of cell
death and which type of cell death, we assessed the changes in
nuclear morphology of As4O6-treated cells
under microscopy with DAPI staining. The DAPI staining revealed
that the condensed and fragmented nuclei observed at a
concentration of ≥2 μM, and the amount of fragmented nuclei was
substantially increased at the concentration of 5 μM (Fig. 1B). The present study suggests that
As4O6 induced cell death at the
concentrations of >2 μM.
Effects of As4O6 on
NF-κB and the IκBα phosphorylation
NF-κB comprises a heterotrimer of p50, p65 and IκBα
in the cytoplasm; when activated, the heterodimer of p50 and p65 is
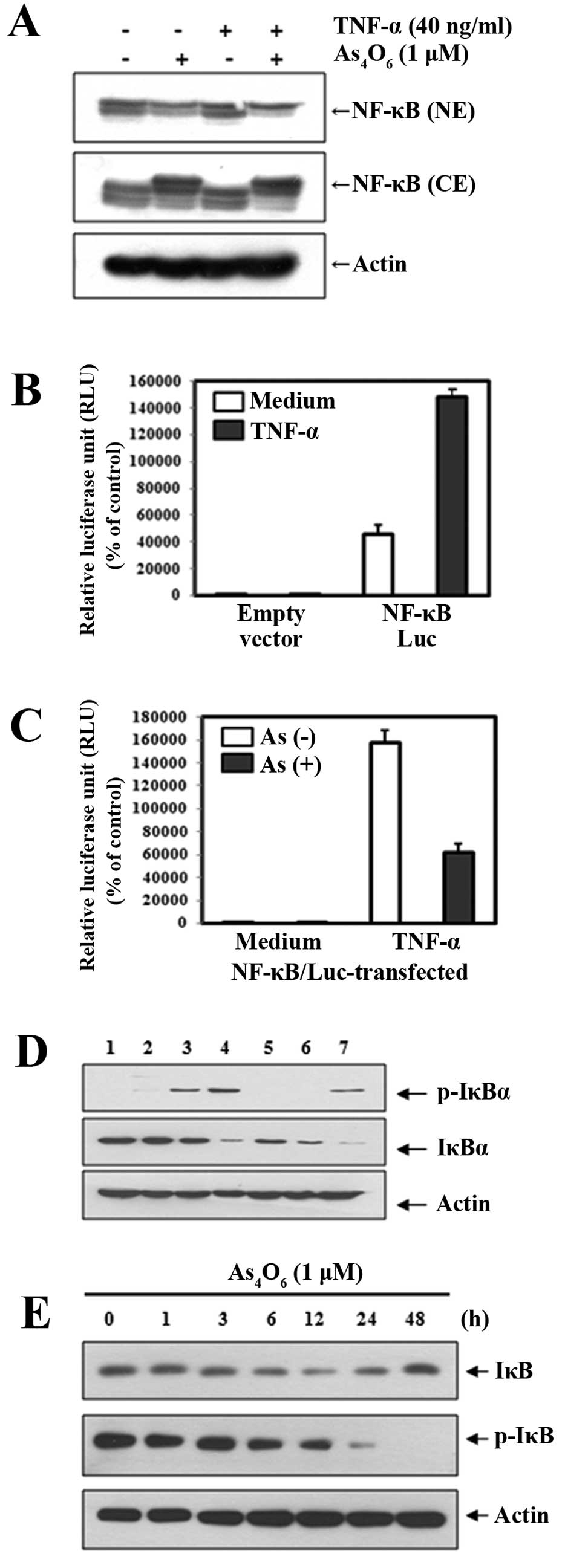
translocated into the nucleus. Using western blot analysis, we
determined whether As4O6 inhibited NF-κB
activation at the 1 μM where no cytotoxicity was observed. We used
TNF-α as an NF-κB stimulant to clearly demonstrate the effects of
As4O6 on NF-κB. Western blot analysis
revealed that treatment with As4O6 inhibited
nuclear NF-κB (p65) activity whether TNF-α was co-treated or not.
This finding indicated that As4O6 had clear
inhibitory effects on NF-κB activation (Fig. 2A). To confirm this finding, we
performed the luciferase assay for NF-κB. As shown in Fig. 2B, the NF-κB-luciferase activity was
augmented by TNF-α, which indicated that the NF-κB gene was
successfully transfected into the cells. The NF-κB-luciferase
activity induced by TNF-α was suppressed by
As4O6 (Fig.
2C).
NF-κB activation is known to require the degradation
of inhibitory κBα (IκBα) through phosphorylation by kinases. Next,
we tested whether As4O6 suppressed
TNF-α-induced phosphorylation of IκBα. The degradation of IκBα
through phosphorylation was observed as early as 5 min after adding
TNF-α, and 1-h pretreatment with As4O6
delayed the TNF-α-induced phosphorylation of IκBα in MCF-7 cells
(Fig. 2D). We also observed over 48
h the effects of As4O6 on IκBα
phosphorylation with the TNF-α-treated cells. We found that
As4O6 suppressed TNF-α-induced
phosphorylation of IκBα and the effects became prominent 24 h after
As4O6 treatment (Fig. 2E). These findings suggest that
As4O6 inhibits NF-κB at least in part through
suppression of the IκBα phosphorylation.
As4O6 suppresses
NF-κB-related cellular responses
We found that As4O6 clearly
inhibited NF-κB activity in both TNF-α-treated cells and control
cells. TNF-α is known to be an NF-κB activator and to bind two
receptors, TNF receptor 1 (TNF-R1) and TNF receptor 2 (TNF-R2).
Most information of TNF signaling regards TNF-R1 as TNF-R2 is only
expressed in immune cells and the mechanism is barely understood.
When TNF-α binds to TNF-R1, this binding leads to the adaptor
protein TRADD to bind to the death domain. With this binding, three
pathways can be initiated: NF-κB, MAPK and death signaling
(18,19). The first one is involved in cell
survival pathway and the others in pro-apoptotic or death pathway.
The former pathway is related to NF-κB activation. Similar to all
death-domain-containing members of the TNF receptor (TNFR)
superfamily, TNF-R1 is also involved in death signaling at the same
time (20). Therefore, the final
results of TNF-α treatment are expressed as the sum of strength
regarding TNF-induced cell death and NF-κB-related anti-apoptotic
effects (Fig. 3). In the present
study, we tested the combination effects of TNF-α and
As4O6 to observe whether
As4O6 augments the anticancer effects of
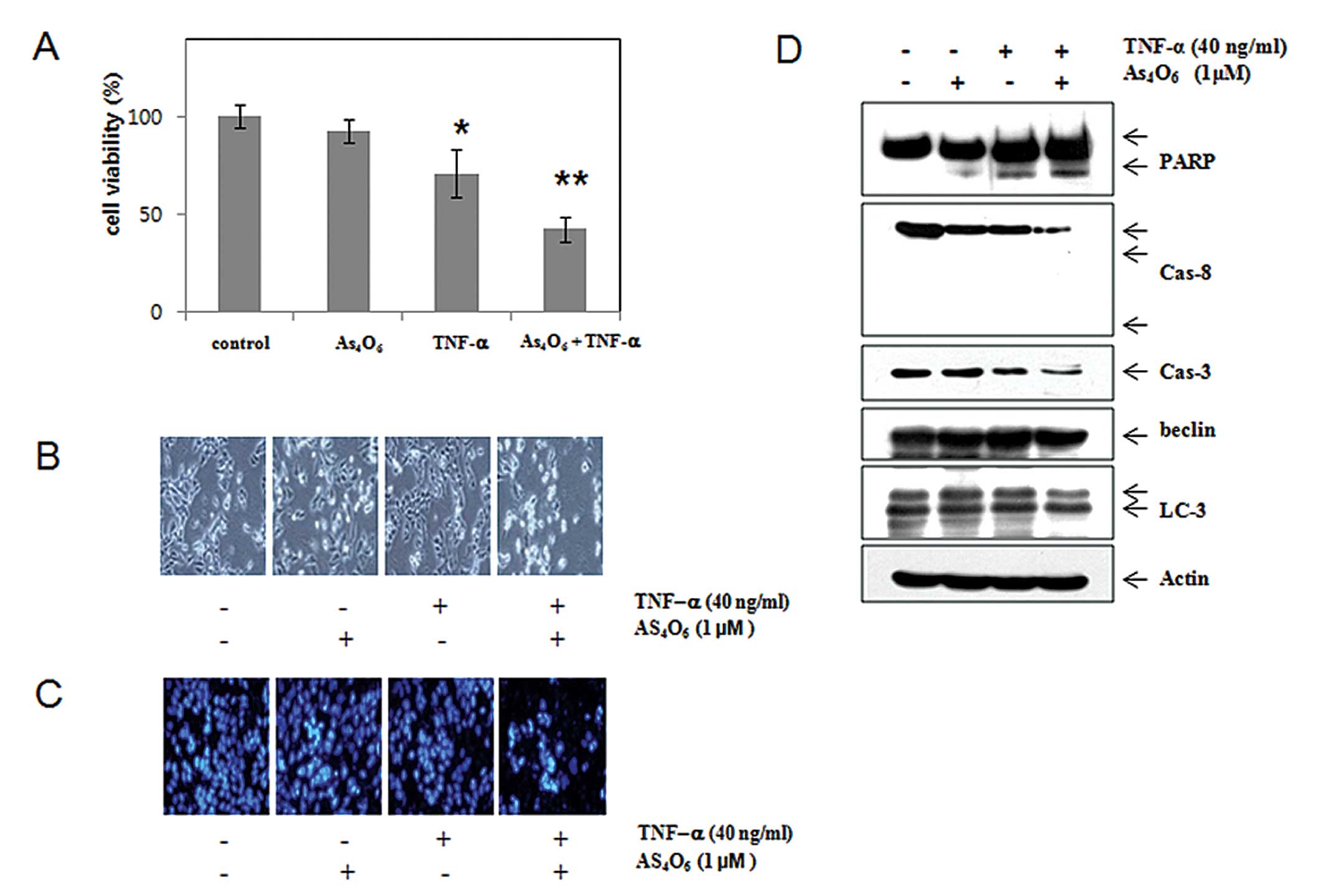
TNF-α by suppressing NF-κB activity. As shown in Fig. 4A, As4O6
potentiated the effects of TNF-α induced cell death. We then
assessed the effects of As4O6 on caspases and
their substrates (PARP). TNF-α in combination with
As4O6 significantly decreased the expression
levels of procaspase-3, procaspase-8. With the decrease of
procaspases, the cleavages of PARP were prominent in the
combination treatment group (Fig.
4D). We also assessed the expression of LC-3 (a marker for
autophagy) and Beclin-1 to examine whether the augmented cell death
by As4O6 is involved in type II programmed
cell death, autophagy. Western blotting revealed that
As4O6 induced LC3 conversion (increase in the
ratio of LC3-II/LC3-I), but that the combination with TNF-α did not
augment LC3 conversion (Fig. 4D).
These findings suggest that As4O6 augments
TNF-α-induced apoptosis through extrinsic pathways.
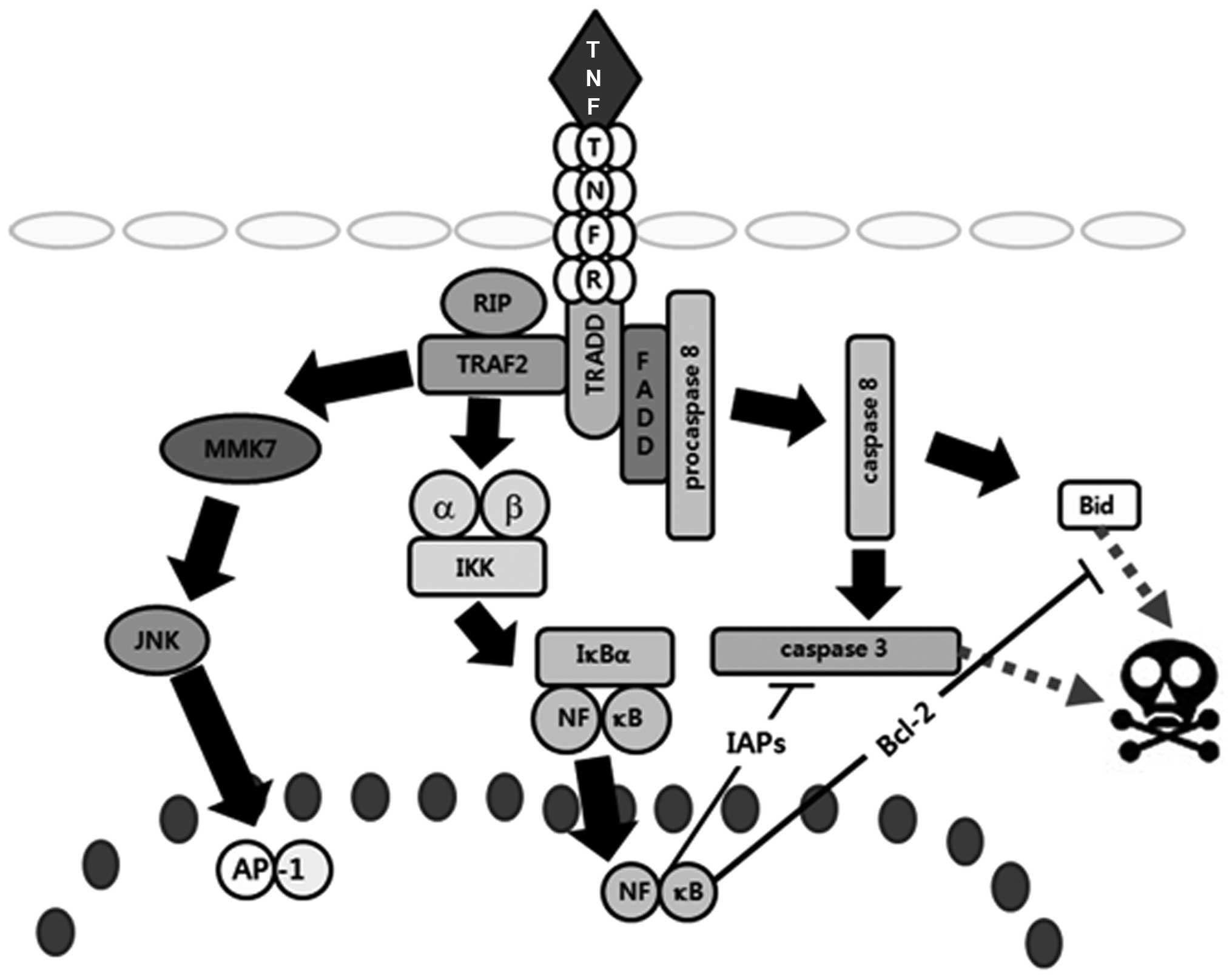 | Figure 3Schematic representation of TNF-R1
signaling pathways and the effects of As4O6
on MCF-7 human breast cancer cells. When TNF-α binds TNF receptor 1
(TNF-R1), inhibitory protein SODD from the intracellular death
domain is dissociated followed by the adaptor protein TRADD binding
to the death domain. Subsequently, three pathways can be activated:
the MAPK, the NF-κB and the death signaling pathway. Of the three
major MAPK cascades, TNF-α generally induces JNK pathways involved
in cell differentiation and proliferation. For activation of NF-κB
pathways, TNF-α recruits TRAF2 and RIP through TRADD to activate
IKK, then IκBα is phosphorylated by IKK, and finally activates
NF-κB, which induces anti-apoptotic proteins (Bcl-2, Bcl-xL, XIAP,
cIAP1 and cIAP2) to inhibit the death signaling pathways. For the
induction of death signaling, FADD recruits procaspase-8 by binding
to TRADD, and then procaspase-8 is activated. The activated
caspase-8 leads to apoptosis through activation of caspase-3 and
Bid. Data suggested that As4O6 may augment
TNF-α-induced death signaling pathways by suppressing IκBα
phosphorylation. |
As4O6 suppresses
NF-κB-regulated proteins involved in anti-apoptosis, proliferation,
invasion and angiogenesis
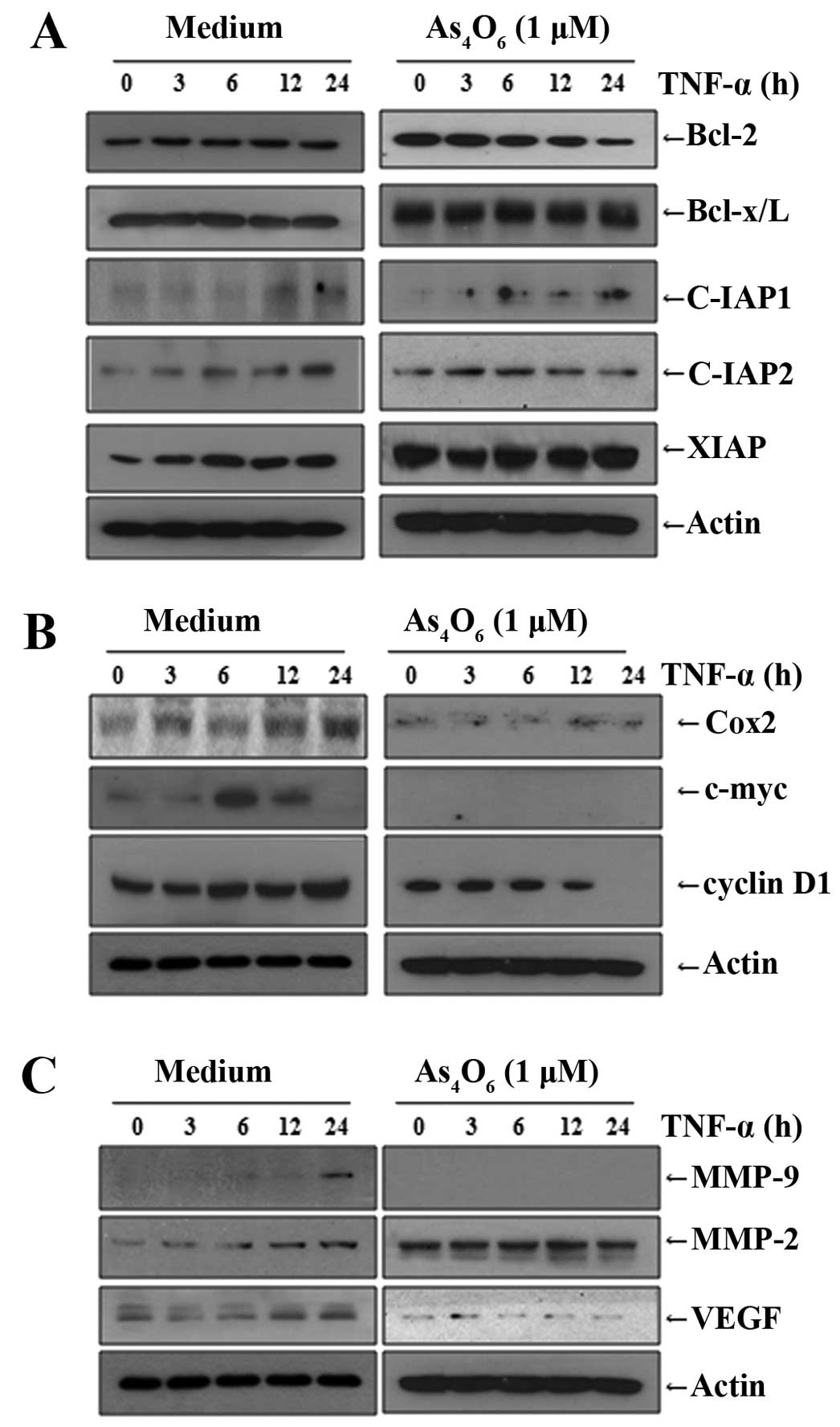
We also observed that As4O6
inhibited NF-κB-related cellular response. NF-κB also regulates
several genes involved in cancer metastasis. We investigated the
effects of As4O6 on the NF-κB-regulated
proteins involved in anti-apoptosis, proliferation, invasion and
angiogenesis. We found that TNF-α stimulated the NF-κB-regulated
proteins involved in anti-apoptosis (c-IAP1, c-IAP2, XIAP, Bcl-2
and Bcl-xL) (Fig. 5A), cancer cell
proliferation (COX-2, c-Myc and cyclin D1) (Fig. 5B), and invasion and angiogenesis
(MMP-2, MMP-9 and VEGF) (Fig. 5C).
As4O6 suppressed the TNF-α-induced
NF-κB-regulated proteins involved in anti-apoptosis, proliferation,
invasion and angiogenesis in MCF-7 cells (Fig. 5). These findings also support that
As4O6 suppresses NF-κB activity, and that it
has anticancer properties especially in conditions of advanced or
metastatic cancer where TNF-α is highly secreted from activated
macrophages or cancer cells themselves.
Discussion
With the assumption that As4O6
inhibits NF-κB at a safe concentration, showing anticancer effects
without serious side-effects in advanced cancer where TNF-α is
highly secreted, we investigated the anticancer effects of
As4O6 with a particular focus on NF-κB
pathway, NF-κB-regulated gene products, and NF-κB-mediated cellular
responses in human breast cancer cells. In the present study,
As4O6 inhibited NF-κB activity and
NF-κB-regulated proteins at the concentrations where no
cytotoxicity was observed, indicating that it can induce anticancer
effects without definite side-effects and may be used for
maintenance therapy. In addition, As4O6 can
be used in combination with conventional chemotherapeutics without
increasing toxicity since the activation of NF-κB is one of the
drug resistance mechanisms. Bortezomib is a good example; it is a
proteasome inhibitor that has inhibitory effects on NF-κB by
suppressing IκBα degradation (21);
it can be used in combination with conventional agents and for
maintenance therapy.
As4O6 also augmented the
TNF-α-induced cell death in MCF-7 cells. The evidence supports that
arsenic compounds could suppress NF-κB activation (22,23).
However, As4O6 showed the inhibitory effect
on NF-κB at a concentration showing no cytotoxicity. This is the
first report regarding anti-NF-κB effects of
As4O6.
TNF-α induces cell death through the extrinsic
pathway in some cells, such as MCF-7 cells (12), but most cancer cell lines are
resistant to TNF-α-induced cell death as its death-inducing ability
is weak and often masked by NF-κB activation followed by the
enhanced transcription of anti-apoptotic proteins (13). In the present study, we demonstrated
that As4O6 enhanced the anticancer effects of
TNF-α by inhibiting NF-κB. In TNF-α-resistant cells,
As4O6 in combination with TNF-α showed
synergism (data not shown). TNF-α is generally increased in
patients with advanced and metastatic cancer and is associated with
cancer progression and patient quality of life (14,15).
Furthermore, TNF-α is abundantly released in chronic inflammatory
disorders including rheumatoid arthritis as well, and a TNF-α
inhibitor is used to control the chronic inflammatory disorders
(24). However, the direct
inhibition of TNF-α raises some suspicion that it may cause cancer
development in chronic inflammatory disorders (24). The present study suggests that TNF-α
can be used as a therapeutic tool by inhibiting NF-κB activation in
advanced and metastatic cancers.
In the present study, we also investigated the
inhibitory effects of As4O6 on MMP-2 and
MMP-9 expression in TNF-α-treated cells. MMP-2 and MMP-9 are key
molecules in cancer cell invasion (25,26)
which have been targets for drug development against cancer
invasion (27). We also found that
As4O6 suppressed COX-2, cyclin D1 and c-Myc
involved in cell proliferation. COX-2 is overexpressed in a variety
of cancers and mediates cancer cell proliferation (13,28,29)
and c-Myc is also involved in cancer cell proliferation (30). In addition, VEGF is an angiogenic
factor (13). Both are important in
metastasis and are regulated by NF-κB (13,28).
The IκB family consists of IκBα, IκBβ, IκBɛ and Bcl-3. Among them,
IκBα is the most extensively studied and major IκB protein. NF-κB
activation is initiated by the degradation of IκBα protein which is
an inhibitor of NF-κB. The degradation of IκBα occurs through the
activation of IκB kinase (IKK). When activated by signals, the IκB
kinase phosphorylates two serine residues located in an IκBα
regulatory domain. When phosphorylated IκBα at serines 32 and 36,
the IκBα is degraded by ubiquitination (31). Here, we found that
As4O6 suppressed phosphorylation of IκBα.
This finding suggests that the anti-NF-κB activities of
As4O6 are contributed by suppression of IκBα
phosphorylation.
In conclusion, the present study demonstrated that
As4O6 has anticancer properties by inhibiting
NF-κB activation and NF-κB-regulated proteins at least in part
through the inhibition of IκB phosphorylation, especially in
conditions of advanced or metastatic cancer where TNF-α is highly
secreted (Fig. 3). The present
study provides evidence that As4O6 may have
anticancer effects through inhibiting NF-κB activity in human
breast cancer.
Acknowledgements
The present study was supported by a grant from the
National Research Foundation of Korea (NRF) funded by the Korea
government (MEST) (no. 20120002631).
References
|
1
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar
|
|
2
|
Jung KW, Park S, Kong HJ, Won YJ, Lee JY,
Seo HG, et al: Cancer statistics in Korea: incidence, mortality,
survival, and prevalence in 2009. Cancer Res Treat. 44:11–24. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nguyen DX and Massague J: Genetic
determinants of cancer metastasis. Nat Rev Genet. 8:341–352. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM,
Qiu QY, et al: Use of arsenic trioxide
(As2O3) in the treatment of acute
promyelocytic leukemia (APL): II. Clinical efficacy and
pharmacokinetics in relapsed patients. Blood. 89:3354–3360.
1997.PubMed/NCBI
|
|
5
|
Niu C, Yan H, Yu T, Sun HP, Liu JX, Li XS,
et al: Studies on treatment of acute promyelocytic leukemia with
arsenic trioxide: remission induction, follow-up, and molecular
monitoring in 11 newly diagnosed and 47 relapsed acute
promyelocytic leukemia patients. Blood. 94:3315–3324. 1999.
|
|
6
|
Munshi NC, Tricot G, Desikan R, Badros A,
Zangari M, Toor A, et al: Clinical activity of arsenic trioxide for
the treatment of multiple myeloma. Leukemia. 16:1835–1837. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin YC, Li DR and Lin W: Relationship
between radiotherapy enhancing effect of arsenic trioxide and the
proliferation and apoptosis of related protein in nasopharyngeal
carcinoma patients. Zhongguo Zhong Xi Yi Jie He Za Zhi. 27:704–707.
2007.(In Chinese).
|
|
8
|
Welch JS, Klco JM, Gao F, Procknow E, Uy
GL, Stockerl-Goldstein KE, et al: Combination decitabine, arsenic
trioxide, and ascorbic acid for the treatment of myelodysplastic
syndrome and acute myeloid leukemia: a phase I study. Am J Hematol.
86:796–800. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beer TM, Tangen CM, Nichols CR, Margolin
KA, Dreicer R, Stephenson WT, et al: Southwest Oncology Group phase
II study of arsenic trioxide in patients with refractory germ cell
malignancies. Cancer. 106:2624–2629. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang HS, Bae SM, Kim YW, Kwak SY, Min HJ,
Bae IJ, et al: Comparison of diarsenic oxide and tetraarsenic oxide
on anticancer effects: Relation to the apoptosis molecular pathway.
Int J Oncol. 30:1129–1135. 2007.PubMed/NCBI
|
|
11
|
Han MH, Lee WS, Lu JN, Yun JW, Kim G, Jung
JM, et al: Tetraarsenic hexoxide induces Beclin-1-induced
autophagic cell death as well as caspase-dependent apoptosis in
U937 human leukemic cells. Evid Based Complement Alternat Med.
2012:2014142012.PubMed/NCBI
|
|
12
|
Messmer UK, Pereda-Fernandez C,
Manderscheid M and Pfeilschifter J: Dexamethasone inhibits
TNF-α-induced apoptosis and IAP protein downregulation in MCF-7
cells. Br J Pharmacol. 133:467–476. 2001.
|
|
13
|
Aggarwal BB: Nuclear factor-κB: the enemy
within. Cancer Cell. 6:203–208. 2004.
|
|
14
|
Correia M, Cravo M, Marques-Vidal P,
Grimble R, Dias-Pereira A, Faias S, et al: Serum concentrations of
TNF-alpha as a surrogate marker for malnutrition and worse quality
of life in patients with gastric cancer. Clin Nutr. 26:728–735.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tas F, Duranyildiz D, Argon A, Oguz H,
Camlica H, Yasasever V, et al: Serum levels of leptin and
proinflammatory cytokines in advanced-stage non-small cell lung
cancer. Med Oncol. 22:353–358. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guttridge DC, Albanese C, Reuther JY,
Pestell RG and Baldwin AS Jr: NF-κB controls cell growth and
differentiation through transcriptional regulation of cyclin D1.
Mol Cell Biol. 19:5785–5799. 1999.
|
|
17
|
Cui ZG, Hong NY, Guan J, Kang HK, Lee DH,
Lee YK, et al: cAMP antagonizes ERK-dependent antiapoptotic action
of insulin. BMB Rep. 44:205–210. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wajant H, Pfizenmaier K and Scheurich P:
Tumor necrosis factor signaling. Cell Death Differ. 10:45–65. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen G and Goeddel DV: TNF-R1 signaling: a
beautiful pathway. Science. 296:1634–1635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gaur U and Aggarwal BB: Regulation of
proliferation, survival and apoptosis by members of the TNF
superfamily. Biochem Pharmacol. 66:1403–1408. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Demchenko YN and Kuehl WM: A critical role
for the NFκB pathway in multiple myeloma. Oncotarget. 1:59–68.
2010.
|
|
22
|
Kerbauy DM, Lesnikov V, Abbasi N, Seal S,
Scott B and Deeg HJ: NF-κB and FLIP in arsenic trioxide
(ATO)-induced apoptosis in myelodysplastic syndromes (MDSs). Blood.
106:3917–3925. 2005.
|
|
23
|
Han SS, Kim K, Hahm ER, Park CH, Kimler
BF, Lee SJ, et al: Arsenic trioxide represses constitutive
activation of NF-κB and COX-2 expression in human acute myeloid
leukemia, HL-60. J Cell Biochem. 94:695–707. 2005.
|
|
24
|
Jamnitski A, Levels JH, Oever IA and
Nurmohamed MT: High-density lipoprotein profiling changes in
patients with rheumatoid arthritis treated with tumor necrosis
factor inhibitors: a cohort study. J Rheumatol. 40:825–830. 2013.
View Article : Google Scholar
|
|
25
|
Davies B, Waxman J, Wasan H, Abel P,
Williams G, Krausz T, et al: Levels of matrix metalloproteases in
bladder cancer correlate with tumor grade and invasion. Cancer Res.
53:5365–5369. 1993.PubMed/NCBI
|
|
26
|
Bogenrieder T and Herlyn M: Axis of evil:
molecular mechanisms of cancer metastasis. Oncogene. 22:6524–6536.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vihinen P and Kahari VM: Matrix
metalloproteinases in cancer: prognostic markers and therapeutic
targets. Int J Cancer. 99:157–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gilmore TD: Introduction to NF-κB:
players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
|
|
29
|
Chun KS and Surh YJ: Signal transduction
pathways regulating cyclooxygenase-2 expression: potential
molecular targets for chemoprevention. Biochem Pharmacol.
68:1089–1100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schmidt EV: The role of c-myc in
regulation of translation initiation. Oncogene. 23:3217–3221.
2004.
|
|
31
|
Chen ZJ, Parent L and Maniatis T:
Site-specific phosphorylation of IκBα by a novel
ubiquitination-dependent protein kinase activity. Cell. 84:853–862.
1996.
|