Introduction
Chronic myelogenous leukemia (CML) is a
hematopoietic stem cell disease characterized by a reciprocal
translocation between chromosomes 9 and 22, resulting in the
formation of the Philadelphia (Ph) chromosome. This translocation
(9;22) results in the head-to-tail fusion of the breakpoint cluster
region (BCR) gene on chromosome 22 at band q11 with the Abelson
murine leukemia (ABL) gene located on chromosome 9 at band q34. The
product of the BCR-ABL1 fusion gene encodes a 210-kDa oncoprotein
designated as p210BCR-ABL1, which demonstrates
constitutive tyrosine kinase activity (1,2).
p210BCR-ABL1 plays a key role in the pathogenesis of CML
via its interaction with numerous molecules regulating cell
survival, proliferation and differentiation (3). Typically, CML passes through three
different clinicopathological phases: a chronic phase (CP), an
accelerated phase (AP) and a blastic phase (BP) (4). Untreated chronic phase CML (CP-CML)
patients eventually progress to advanced phases (AP and BP) within
3–5 years (5). Tyrosine kinase
inhibitor (TKI) resistance or blastic transformation has been found
in ~15–20% of patients with CML, although the molecular mechanisms
remain poorly understood (3,6–8).
However, recent studies have demonstrated that the bulk of the
genetic changes associated with progression occur in the
transformation from CP to AP or BP (9).
SHP-1 or PTPN6 (also previously referred to as
SH-PTP1, PTP1C, HCP or Hcph) is a non-receptor protein tyrosine
phosphatase (PTP) that is predominantly expressed in hematopoietic
cells and plays an important role in the negative regulation of
growth-promoting signaling molecules, such as JAKs/STATs and
PI3K/AKT/mTOR (10–12). Mice deficient in SHP-1
(SHP-1−/− and SHP-1+/−; motheaten and viable
motheaten mice, respectively) demonstrate marked myeloid
proliferation (13,14). Moreover, SHP-1 promoter methylation
causes loss of SHP-1 expression in hematological malignancies,
resulting in the activation of the JAK/STAT pathway (15–17).
SHP-1 also accounts for resistance to imatinib (IM) treatment in
patients with CML (18), as it
appears to be physically associated with BCR-ABL1 and is able to
block BCR-ABL-dependent and -independent signaling pathways
(19,20). In the present study, we demonstrated
that: i) the expression levels of SHP-1 are markedly decreased in
patients with BP-CML or AP-CML compared with CP-CML; ii) SHP-1
reduces p210BCR-ABL1 protein expression and activity and
negatively regulates the AKT, MAPK, MYC and JAK2/STAT5 signaling
pathways; and iii) abnormal methylation of the SHP-1 promoter
region occurs in K562 cells and cells from patients with advanced
CML. Therefore, low levels of SHP-1 caused by aberrant promoter
hypermethylation may be related to CML blastic transformation by
dysregulating the BCR-ABL1, AKT, MAPK, MYC and JAK2/STAT5 signaling
pathways.
Materials and methods
Patients and healthy donors
Between December 2010 and June 2013, bone marrow
(BM) aspirates or peripheral blood (PB) cells were collected from
94 consecutive patients with CML treated at the Second Hospital,
Hebei Medical University and 11 healthy donors under an approved
institutional protocol. The definitions of CP, AP and BP
corresponded to those defined in the European Leukemia Net (ELN).
The present study was approved by the Medical Ethics Committee of
the Second Hospital of Hebei Medical University.
Tumor cell line
The BCR-ABL-positive K562 cell line (maintained in
our laboratory) was maintained in RPMI-1640 medium supplemented
with 10% fetal bovine serum (FBS) in a humidified 5% CO2
atmosphere at 37°C. The cells were passed or the medium was renewed
every 2 to 3 days, and the cells were prepared for experimental
procedures when they reached log-phase growth and 80%
confluency.
Reagents and antibodies
Imatinib mesylate (IM) and nilotinib were kindly
provided by Novartis Pharma Stein AG (Basel, Switzerland).
Puromycin 2HCL was purchased from Beijing Solarbio (Beijing,
China). SHP-1, Crk-L and GAPDH antibodies were obtained from Santa
Cruz Biotechnology (Santa Cruz, CA, USA); JAK2 and phospho-JAK2
(pTyr1007/1008) antibodies were obtained from Abcam Inc.,
(Cambridge, UK); STAT5, phospho-STAT5 (pTyr694), AKT, phospho-AKT
(pSer473), MAPK and phospho-MAPK p44/42 (Erk1/2) (Thr202/Tyr204)
antibodies were purchased from Cell Signaling Technology (Danvers,
MA, USA); Alexa Fluor® 488 mouse anti-CrkL (pY207), the
isotype control antibody, anti-normal-rabbit immunoglobulins were
obtained from BD Biosciences (San Diego, CA, USA).
Assessment of apoptosis
The rate of apoptosis was evaluated using an Annexin
V/PI apoptosis kit (MultiSciences Biotech Co., Ltd., Hangzhou,
China) according to the manufacturer’s instructions. For each
sample, 1–5×105 cells were analyzed on a BD FACSCalibur
(Becton-Dickinson, San Jose, CA, USA). Annexin V/PI discriminates
intact cells (Annexin V−/PI−) from early
apoptotic cells (Annexin V+/PI−) and late
apoptotic/necrotic cells (Annexin
V+/PI+).
Cell cycle analysis
One million cells were centrifuged at 1,000 × g for
3 min, washed twice with ice-cold PBS, resuspended in 70% ice-cold
ethanol and fixed at 4°C overnight or stored at −20°C. The cells
were collected by centrifugation and washed twice with precooled
PBS to remove the ethanol. PI (10 μl) was then added into 500 μl of
binding buffer, and the cells were incubated at 4°C in the dark for
30 min. Samples were analyzed on a FACSCalibur (Becton-Dickinson)
using ModFit Lt3.0 software (Verity Software House).
Cell line viability and proliferation
assays
Cell line viability and proliferation were assessed
using a Cell Counting Kit-8 CCK-8/WST-8 kit (Dojindo Laboratories,
Kumamoto, Japan), respectively, according to the manufacturer’s
instructions.
Immunoblotting
Cells (107) were harvested, washed twice
with ice-cold PBS and lysed in 100 μl of SDS lysis buffer [50 mM
DTT, 2% SDS, 62.5 mM Tris (pH 6.8), 10% glycerin]. Lysates were
centrifuged (12,000 × g, 15 min, 4°C), denatured (10 min, 100°C),
subjected to 8–12% SDS-PAGE and electrotransferred to
polyvinylidene fluoride (PVDF) membranes (0.45 μm; Millipore,
Billerica, MA, USA). The membranes were blocked in 10% bovine serum
albumin (BSA) at 4°C overnight or at room temperature for 2 h and
immunoblotted with antibodies (1:1,000 dilution) at room
temperature for 40 min. The membranes were then incubated with
horseradish peroxidase (HRP)-labeled immunoglobulin (Santa Cruz
Biotechnology) at room temperature for 30 min at a 1:2,000 dilution
followed by reaction using an enhanced chemiluminescence (ECL) kit
(Amersham Pharmacia Biotech Inc., Piscataway, NJ, USA). The
membranes were exposed to X-ray film (Kodak, Rochester, NY USA),
and protein expression was expressed as densitometric units after
normalization to GAPDH levels (ImageJ).
SYBR-Green-based qRT-PCR
Mononuclear cells were isolated from the BM
aspirates or peripheral blood by centrifugation on a Ficoll-Hypaque
gradient. Total RNA was extracted using the TRIzol reagent
(Invitrogen-Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. Total RNA (1 μg) was reverse
transcribed in 20 μl using an All-in-One First-Strand cDNA
Synthesis kit (GeneCopoeia Inc., Guangzhou, China), and cDNA (1 μl)
was amplified using a SYBR-Green-based quantitative PCR (qPCR)
reaction using an All-in-One™ qPCR Mix kit (GeneCopoeia Inc.). The
qPCR conditions consisted of 35 cycles at 94°C for 10 sec, 65°C for
45 sec and 72°C for 10 sec. The primer sequences are documented in
Table I. The qPCR reactions were
performed in triplicate using a Three-Step-with-Melt PCR system.
β-actin was monitored as the housekeeping control gene for equal
amplification. SHP-1 mRNA was normalized to β-actin levels to
obtain the relative threshold cycle (ΔCt), and the resulting number
was then related to the ΔCt of the control gene to obtain the
relative expression level (2−ΔΔCt).
 | Table IMSP and SYBR-Green-based qRT-PCR:
primer sequences and products. |
Table I
MSP and SYBR-Green-based qRT-PCR:
primer sequences and products.
| Gene | Sense primer
(5′-3′) | Antisense primer
(5′-3′) | Products (bp) |
|---|
| β-actin |
gagctacgagctgcctgac |
ggtagtttcgtggatgccacag | 121 |
| SHP-1 |
aacagccgtgtcatcgtcat |
atcaggtctccattgtccagc | 191 |
| SHP-1M-MSP |
gaacgttattatagtatagcgttc |
tcacgcatacgaacccaaacg | 158 |
| SHP-1U-MSP |
tcacgcatacgaacccaaacg |
ttcacacatacaaacccaaacaat | 158 |
CrkL phosphorylation assay
Phosphorylated CrkL (pCrkL) analysis by flow
cytometry was performed as previously described (21,22).
One million cells were resuspended in 500 μl of 2% paraformaldehyde
and fixed for 10 min at 37°C, then chilled on ice for 1 min. The
cells were harvested by centrifugation (770 × g, 3 min), and 500 μl
of 90% methanol was added. The cells were vortexed briefly and
incubated on ice for 30 min. The cells were then washed (throughout
with 1 ml of incubation buffer containing phosphate-buffered saline
and 0.5% bovine serum albumin), harvested and resuspended in 25 μl
of incubation buffer and incubated at room temperature for 10 min.
Alexa Fluor® 488 mouse anti-CrkL (pY207) was added (3
μl), and the cells were incubated at room temperature in the dark
for 30 min. The cells were washed twice and then analyzed by flow
cytometry (Becton-Dickinson) using CellQuest Pro Software
(Becton-Dickinson) for data analysis. The amount of pCrkL in the
sample was determined as the geometric mean fluorescence intensity
(MFI) minus the MFI value of the isotype control.
Methylation-specific polymerase chain
reaction (MSP)
Genomic DNA was isolated, sodium bisulfite-modified
and purified with the EZ DNA Methylation-Direct™ kit (Zymo
Research, Irvine, CA, USA) according to the manufacturer’s
protocol. Primers (Table I) were
designed to detect the methylated and unmethylated sequence of the
promoter region for exon 1b of the SHP-1 gene (15). Initial denaturation at 94°C for 3
min was followed by 40 cycles of denaturation at 94°C for 30 sec,
an annealing step at 55°C for 1 min, an extension step at 72°C for
1 min and a final extension step at 72°C for 10 min. The products
were separated by electrophoresis on 2% agarose gels.
Tumor cell line transfection
The lentiviral expression plasmids,
pEX-SHP-1-puro-Lv105 and pEX-EGFP-puro-Lv105, were constructed and
packaged by GeneCopoeia Inc. pEX-SHP-1-puro-Lv105 contains the open
reading frame (ORF) of human SHP-1 (National Center for
Biotechnology Information NM_002831.4) under the control of the CMV
promoter. K562 cells were infected with the specified vector and
the mock vectors according to the manufacturer’s protocol.
Statistical analysis
Data values represent the mean ± SD of at least 3
independent experiments. Data were analyzed statistically using SAS
9.1.3 (SAS Institute Inc., Cary, NC, USA; 5695502). Data were
tested for normal distribution. One-way ANOVA analysis (LSD),
independent-samples t-test, non-parametric Mann-Whitney test,
Bonferroni-correction non-parametric test, RC contingency table
Chi-square test, or Fisher’s exact test were applied; P-values
<0.05 were considered to indicate statistically significant
results.
Results
Decreased expression of SHP-1 is
associated with progression of CML
The mRNA levels of SHP-1 in BM aspirates or
peripheral blood mononuclear cells (PBMCs) from 94 (CP, 42; AP, 22;
BP, 30) highly heterogeneous CML patients were assessed using SYBR
Green-based relative quantitative RT-PCR. The main clinical and
hematologic features of the 3 groups of patients are summarized in
Table II, and there were no
significant differences in the main features among the groups.
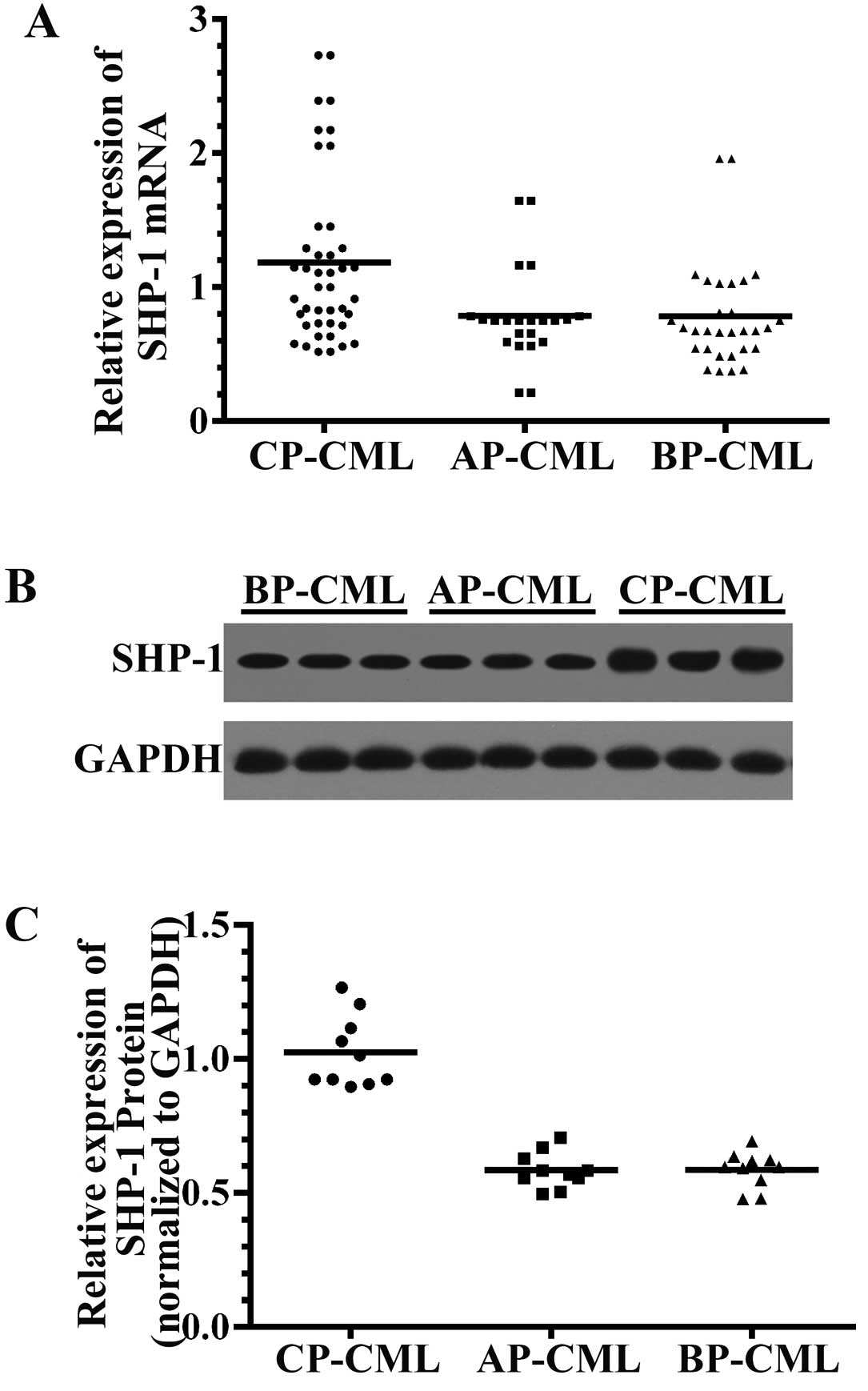
SHP-1 mRNA transcripts were significantly lower (P<0.01) in the
samples from the AP and BP patients (0.79±0.36 and 0.78±0.40,
respectively) compared to those from the CP patients (1.18±0.64).
We observed no significant difference in SHP-1 mRNA levels between
AP-CML and BP-CML patients (P=0.987) (Fig. 1A).
| Table IICharacteristics of the patients
included in the study. |
Table II
Characteristics of the patients
included in the study.
| CP-CML (n=42) | AP-CML (n=22) | BP-CML (n=30) | P-valuea |
|---|
| Age (years), median
(range) | 45 (16–72) | 45 (33–69) | 50 (8–68) | 0.37 |
| Male/female,
(n/n) | 24/18 | 14/8 | 16/14 | 0.87 |
| WBCs,
×109/l, median (range) | 199 (33–427) | 199 (60–373) | 236 (60–356) | 0.48 |
| Hemoglobin level
(g/dl) | 95 (62–138) | 88 (68–138) | 89 (62–117) | 0.32 |
| PLT count
×109/l, median (range) | 470 (39–998) | 350 (87–1147) | 308 (54–1208) | 0.10 |
We detected the expression levels of the SHP-1
protein in BM aspirate mononuclear cells from 10 cases each in
three different stages of CML using immunoblotting assays;
similarly, the results showed that the levels of SHP-1 protein were
lower in AP-CML (0.59±0.07) and BP-CML (0.59±0.07) patient-derived
mononuclear BM cells compared to CP-CML (1.02±0.13) (P<0.05)
(Fig. 1B).
Effects of SHP-1 on the biological
characteristics of K562 cells
To assess the effects of SHP-1 on K562 cells, we
stably expressed SHP-1 in K562 cells and puromycin-selected
K562EGFP and K562SHP-1 cells. After
transfection, SHP-1 protein was re-expressed in the
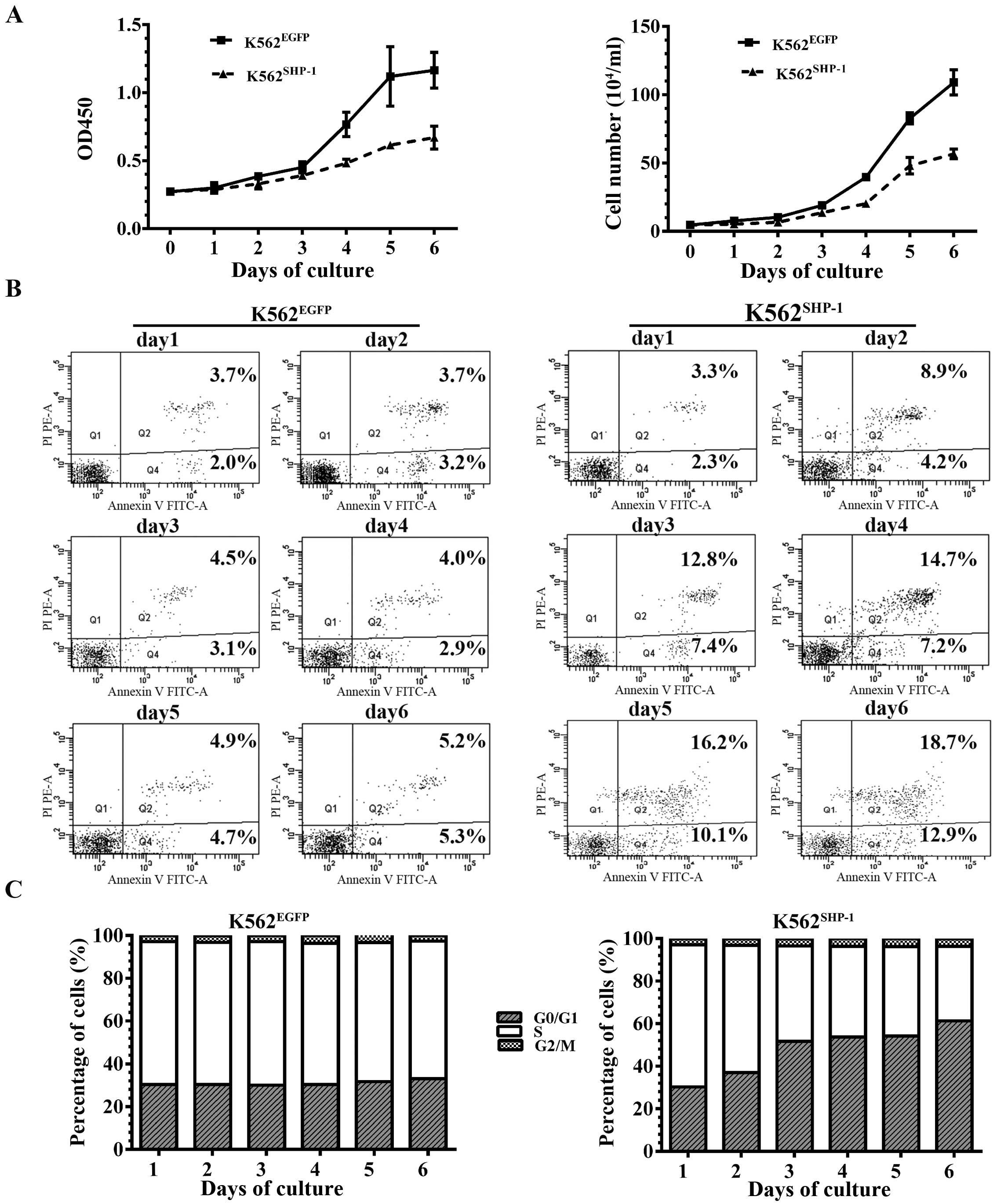
K562SHP-1 cells. CCK-8 assays and cell counts showed
that the cell proliferation rate was sharply reduced in the
K562SHP-1 cells compared to the K562EGFP
control cells from the third day of culture (Fig. 2A).
We also observed a sharp increase in early and late
stage apoptosis among the K562SHP-1 cells, as measured
by Annexin V/PI staining and flow cytometry, and the percentage of
apoptotic K562SHP-1 cells was significantly higher than
that of apoptotic K562EGFP control cells from the third
day of culture (Fig. 2B).
Notably, an accumulation of cells in the G0/G1 phase
and a reduction of cells in the S phase were observed from the
third day of culture for the K562SHP-1 and
K562EGFP cells, whereas no difference was found in G2/M
cell populations for K562SHP-1 compared to
K562EGFP cells (Fig.
2C).
Overexpression of SHP-1 in K562 cells
reduces p210BCR-ABL1 expression and activity
To establish the effects of SHP-1 on BCR-ABL1, we
first detected the expression of BCR-ABL1 in K562SHP-1
and K562EGFP cells 72 h after transfection with
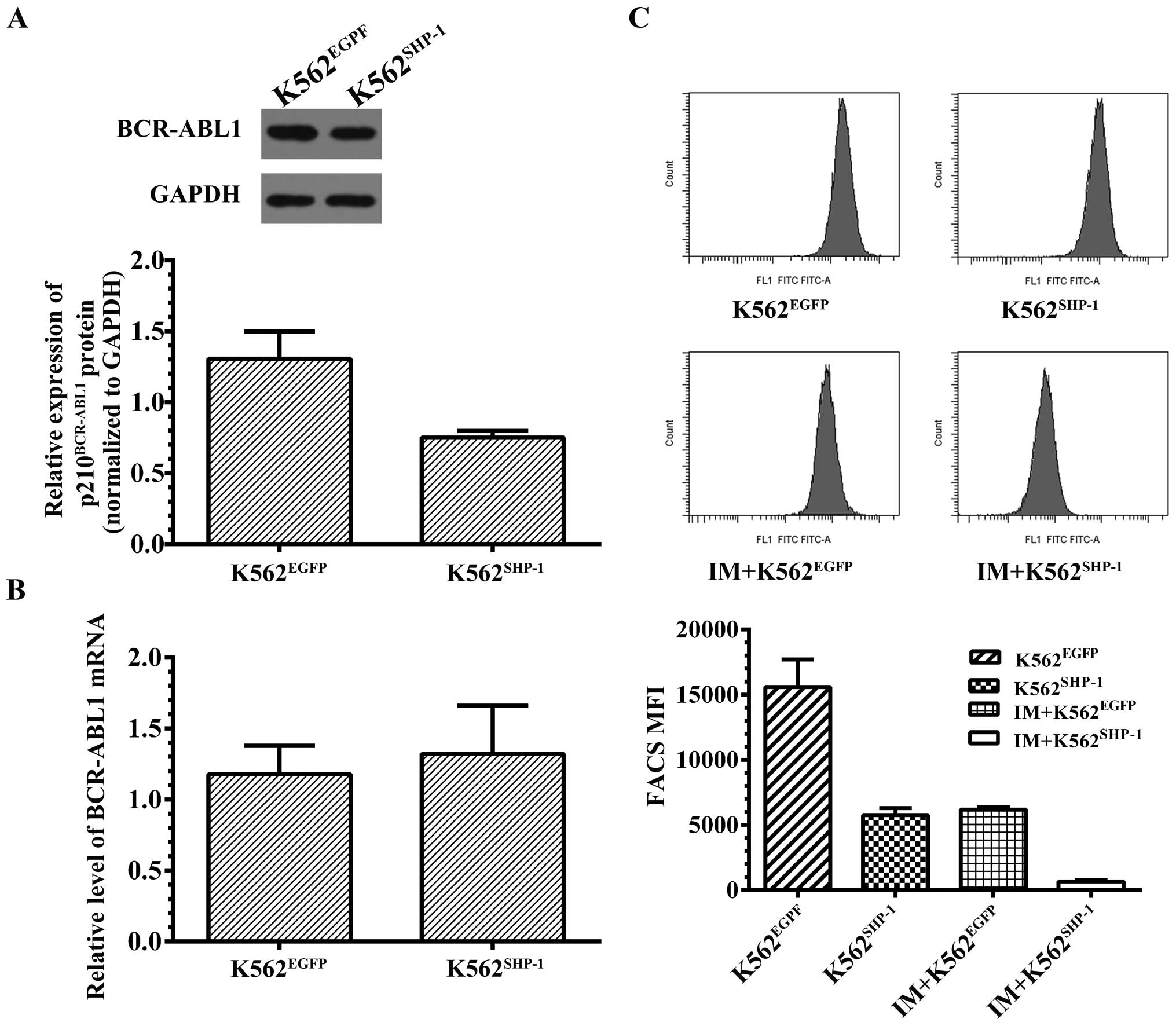
pEX-SHP-1-puro-Lv105. We found that SHP-1 controlled BCR-ABL1
protein levels, as SHP-1 overexpression caused a slight decrease in
p210BCR-ABL1 protein in K562SHP-1 cells
(0.78±0.15) compared to the mock control K562EGFP cells
(1.27±0.24) (P=0.038) (Fig. 3A).
However, the levels of BCR-ABL1 mRNA were not affected in the
K562SHP-1 cells (1.32±0.34) compared to the
K562EGFP cells (1.18±0.20) (P=0.64) (Fig. 3B).
The CrkL protein is a downstream signaling substrate
of p210BCR-ABL1, and the tyrosine phosphorylation of
CrkL (pCrkL) has been identified as a surrogate marker of
p210BCR-ABL1 tyrosine kinase activity in CML cells
(21). We, therefore, examined the
effects of SHP-1 on p210BCR-ABL1 activity by measuring
the level of pCrkL by flow cytometry. The results showed that pCrkL
was decreased by 63.1% following transfection of SHP-1 into K562
cells (P<0.01). Since previous research has demonstrated that
treatment of K562 cells with IM results in a dose-dependent
decrease in pCrkL (21), we also
examined the effect of IM treatment (50 nM) for 8 h on CrkL
phosphorylation in K562EGFP and K562SHP-1
cells. The results revealed a significant decrease in pCrkL between
K562SHP-1 and K562EGFP cells (P<0.01)
(decreased by 88.2 and 60.3%, respectively) following treatment
with IM (50 nM) for 8 h (Fig.
3C).
Effects of SHP-1 on BCR-ABL1-independent
signaling pathways in K562 cells
The ability of p210BCR-ABL1 to induce and
sustain CML depends on the modulation of the expression and/or
activity of downstream signaling molecules, such as those involved
in the RAS/MAPK, PI3K/AKT, MYC and JAK/STAT pathways. Thus, we
investigated whether SHP-1 affects the expression and/or activity
of these molecules controlling cell growth, survival and
differentiation.
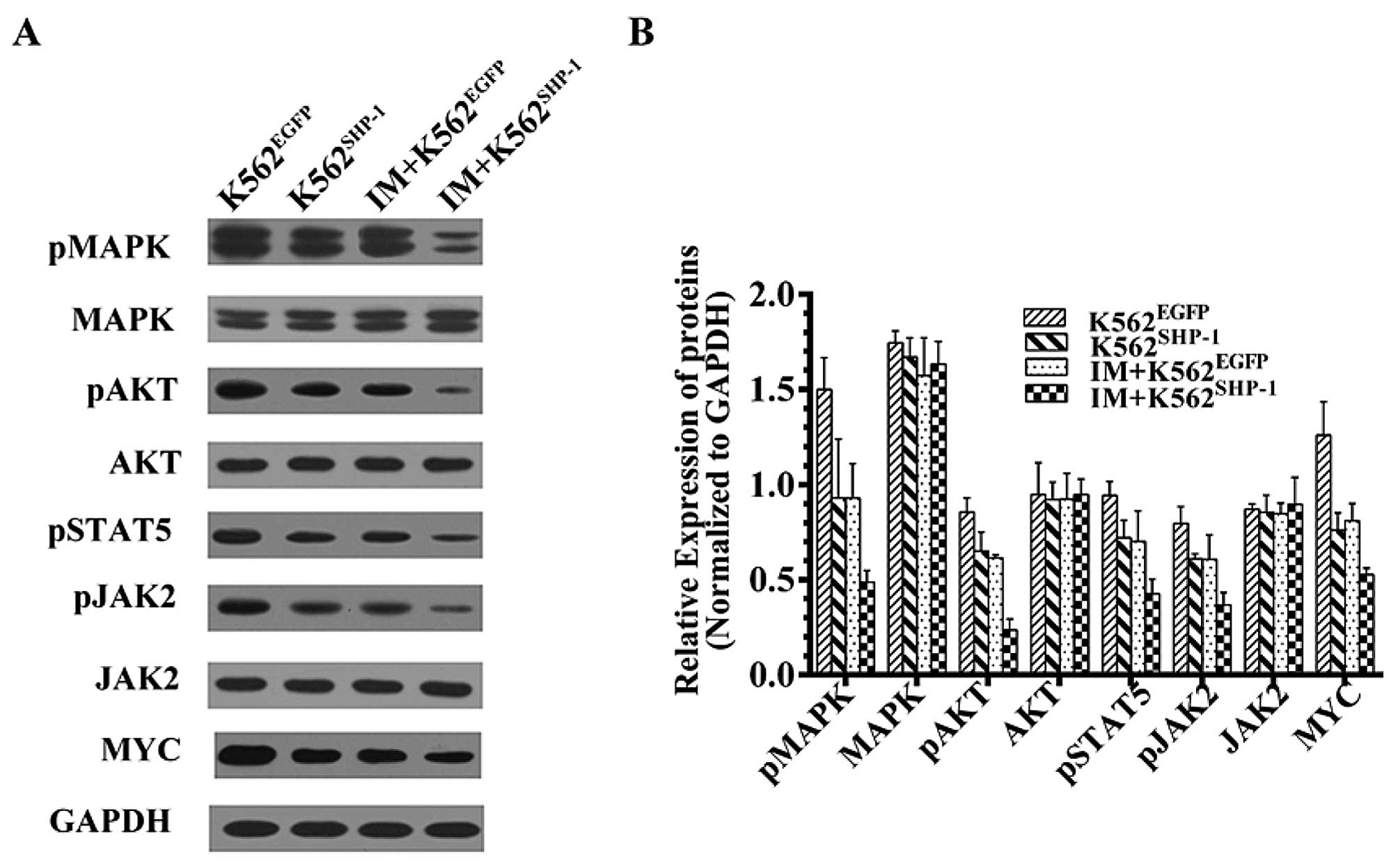
At 48 h after transfection with
pEX-SHP-1-puro-Lv105, the transfected cells were collected, and
western blotting was performed using various specific antibodies.
Expression was estimated by dividing the intensity of the bands
normalized to the GAPDH loading controls by the normalized
intensity of the bands from K562EGFP control cells.
Overexpression of SHP-1 resulted in a marked decrease in MYC and
the phosphorylated forms of JAK2, STAT5, AKT and MAPK (P<0.01);
however, the unphosphorylated forms of these molecules were not
significantly affected (P>0.05) (Fig. 4). These results indicate that AKT,
MAPK, MYC and JAK2/STAT5 are downstream of SHP-1 and that SHP-1
negatively regulates AKT, MAPK, Myc and JAK2/STAT5 signaling.
To further investigate the SHP-1-mediated negative
regulation of these molecules using a BCR-ABL1-independent model,
we stably expressed SHP-1 in K562 cells and treated
puromycin-selected K562EGFP and K562SHP-1
cells with 50 nM IM for 8 h. The results revealed a decrease in
phosphorylated AKT, MAPK and JAK2/STAT5 in K562SHP-1
cells in comparison to K562EGFP cells (P<0.01)
(Fig. 4).
Decreased expression of SHP-1 due to
aberrant CpG methylation of the SHP-1 promoter in both K562 cells
and cells from patients with advanced CML
To assess the function of SHP-1 in K562 cells, we
initially assessed SHP-1 mRNA levels in K562 cells compared to
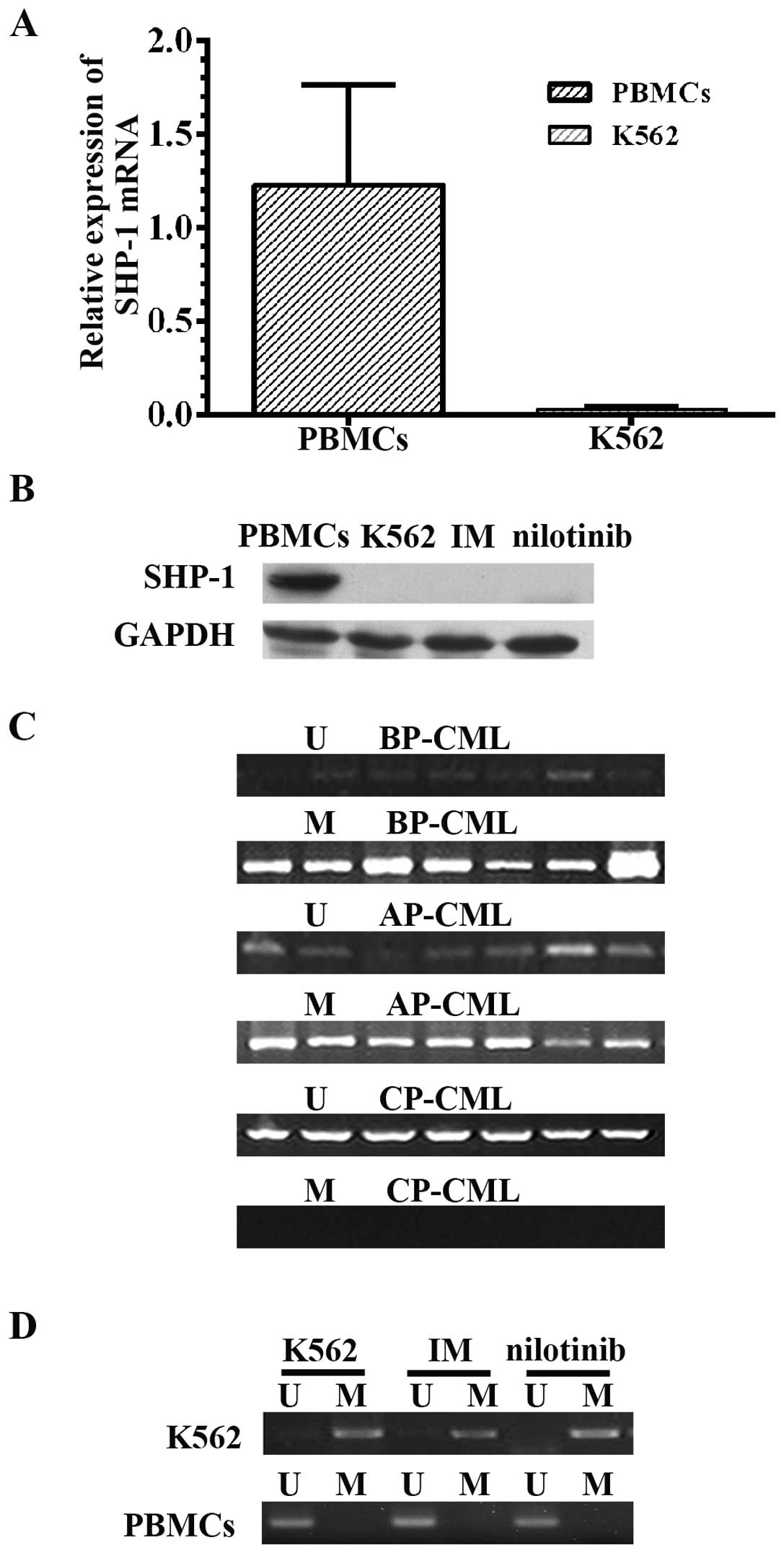
PBMCs from healthy donors. qRT-PCR analysis indicated that SHP-1
mRNA was significantly reduced by ~98% in K562 cells (0.03±0.01)
compared to normal PBMCs (1.23±0.53) (P=0.005), as previously
reported (Fig. 5A). Consistent with
these results, SHP-1 protein was expressed in PBMCs from healthy
donors but was not detected in the K562 cells by western blot
analysis. In addition, SHP-1 was not detected after treatment with
IM (200 nM for 72 h) or nilotinib (150 nM for 72 h) (Fig. 5B).
We also analyzed whether epigenetic modifications
could explain the decrease in SHP-1 levels observed in cells from
patients with advanced CML. Genomic DNA obtained from primary BM
samples or PBMCs from 94 CML patients (Table II) was analyzed using MSP. The
frequency of SHP-1 DNA promoter methylation at selected loci in 42
CP-CML samples was 23.8% (n=10), and methylated regions were
detected in all advanced CML samples with lower SHP-1 expression
(P=0.001) (Fig. 5C).
Previous studies in malignant lymphoma and multiple
myeloma have implicated SHP-1 gene methylation as responsible for
the marked decrease in SHP-1 levels. Our MSP analysis revealed that
the SHP-1 promoter was unmethylated in all 11 normal human samples
but was methylated in K562 cells. As TKIs have become first-line
therapy in CML, we also investigated whether the levels of SHP-1
were affected by TKI treatment. However, treatment with IM (200 nM
for 72 h) or nilotinib (150 nM for 72 h) did not modify the
methylation status of SHP-1, and no SHP-1 protein was detected
(Fig. 5D).
Discussion
CML accounts for 15% of adult leukemias. The median
age of disease onset is 50–60 years, although CML can occur in all
age groups (23). The oncogenic
potential of p210BCR-ABL1 has been demonstrated by the
ability of this protein to transform hematopoietic progenitor cells
both in vitro and in vivo. The
p210BCR-ABL1 protein becomes constitutively active as a
protein tyrosine kinase and increases proliferation, affects
differentiation and blocks apoptosis. CML occurs in 3 distinct
phases (CP, AP and BP). Before the advent of BCR-ABL1 TKIs, all
patients with CP-CML progressed spontaneously to the advanced
phases of CML, and patients with BP-CML demonstrate a median
survival time of approximately 6 months. Some patients progress
directly to BP without an intermediate stage. Although TKI therapy
alone or in combination with conventional chemotherapy followed by
allogeneic hematopoietic stem cell transplantation has improved
prognosis to some degree, these responses are not durable in BP-CML
(24). Currently, the biological
basis of BP is poorly understood, and reliable predictive markers
that can identify those at risk of progression from CP-CML to
advanced phases of CML are lacking. Gene expression profiling has
shown a close correlation in the gene expression profile between
CP-CML and advanced-phase CML (9).
In the present study, we found that the level of the SHP-1
phosphatase was significantly decreased in cells from patients with
advanced-phase CML as well as K562 cells, and this reduction in
SHP-1 levels was related to the methylation status of the SHP-1
promoter.
The SHP-1 phosphatase is considered a negative
regulator of cell proliferation and differentiation through its
activity in dephosphorylating growth factors and cytokine
receptors, including JAK/STAT and PI3K-AKT signaling components
(25,26). Indeed, constitutive activation of
the JAK/STAT pathway contributes to the development and progression
of CML (27–31). SHP-1 expression has also been shown
to be important for K562 differentiation in response to various
differentiating inducers (19).
Thus, we suggest that loss of SHP-1 function may play a key role in
the progression to blast crisis in CML. Indeed, as our previous
research and studies by others have shown that SHP-1 gene mutation
is infrequent in acute leukemia and mutations are not observed in
CML cell lines or clinical samples (32,33),
it is tempting to speculate that epigenetic abnormality of the
SHP-1 gene may contribute to blastic transformation in CML.
Previously, decreased expression of SHP-1 in the progression of CML
was reported in a study that assessed expression in the BM from 30
patients with CML using RT-PCR and immunohistochemistry (33). In the present study, we evaluated
the expression of SHP-1 mRNA and protein using SYBR-Green-based
qRT-PCR and western blot methods, respectively, in 94 patients with
CML at different stages and explored the possible role of SHP-1 in
CML progression.
When we examined BM or PB samples from patients with
CML, we found that SHP-1 mRNA levels were significantly decreased
in AP and BP samples compared to CP samples (0.78±0.40 and
0.79±0.36 in AP and BP, respectively, vs. 1.18±0.64 in CP), which
is similar to the results of a previous study (33). Furthermore, the expression of SHP-1
protein was lower in advanced CML compared to CP-CML. These
findings suggest that with CML progression, the levels of SHP-1
mRNA and protein are markedly decreased. In agreement with our
observations in the advanced stages of CML, previous studies have
shown that SHP-1 mRNA and protein are markedly decreased in
aggressive lymphomas, and reduced SHP-1 levels are involved in the
evolution of undetermined significance into multiple myeloma
(34–36). DNA methylation is the most common
mechanism of SHP-1 gene silencing in several types of hematopoietic
tumors (5,15,16,35,36).
Therefore, we analyzed the frequency of SHP-1 DNA methylation in
cells from patients in different phases of CML and in K562 cells by
MSP and UMSP. We detected SHP-1 methylation in 23.8% (10/42) of
CP-CML cells, all of the AP-CML, BP-CML and K562 cells and none of
the healthy donor cells, indicating that SHP-1 promoter methylation
correlates with the reduced expression of SHP-1 observed during CML
progression.
To further investigate the potential mechanism by
which the SHP-1 gene functions in the progression to blast crisis
in CML, we stably expressed SHP-1 in the K562 cell line and showed
that transfection of SHP-1 into K562 cells could result in growth
suppression, enhanced apoptosis and accumulation of cells in the
G0/G1 phase. p210BCR-ABL1 transforms hematopoietic
progenitor cells and plays a key role in the progression of CML,
although specific PTPs that antagonize constitutively active
BCR-ABL1 have not been sufficiently investigated. SHP-1 has been
shown to be physically associated with BCR-ABL1 (19,20),
and our results demonstrated that forced expression of SHP-1
slightly decreased the level of p210BCR-ABL1 protein in
K562SHP-1 cells; however, no changes were observed for
BCR-ABL1 mRNA. We also showed that the overexpression of SHP-1
inhibited the tyrosine kinase activity of BCR-ABL1, according to
the levels of pCrkL. Thus, our results indicate that SHP-1
suppresses oncogenic BCR-ABL1 kinase activity without influencing
its transcription. A previous study indicated that PP2A, another
phosphatase and tumor suppressor, makes BCR-ABL1 prone to
proteasome-dependent proteolysis (37). Therefore, we propose that SHP-1 may
also promote the degradation of BCR-ABL1 via the proteasome to
decrease the level of p210BCR-ABL1.
The tumor-suppressing activity of SHP-1 depends on
its ability to interact with and dephosphorylate several molecules
implicated in cell cycle progression, proliferation, survival and
differentiation (38). Remarkably,
several targets are shared by the BCR-ABL1 kinase and the SHP-1
phosphatase. Among these, the expression and/or activity of the
SHP-1 substrates JAK/STAT, RAS/MAPK, PI3K/AKT and MYC are either
essential for BCR-ABL1 leukemogenesis or have been found to be
altered in CML progression (15,22,28,30–32,36,38,39).
Our findings showed that forced expression of SHP-1 in K562 cells
led to the inhibition of JAK2, STAT5, MAPK and AKT phosphorylation
and decreased MYC expression. Moreover, by treating
K562SHP-1 and K562EGFP cells with 50 nM IM
(far lower than the IC50 concentration), we provide
evidence that in CML, SHP-1 likely downregulates JAK2/STAT5,
RAS/MAPK and PI3K/AKT signaling independently of BCR-ABL1 during
disease progression.
In conclusion, our data demonstrate a novel
BCR-ABL1-independent mechanism of CML progression and reveal the
correlation between SHP-1 expression and different phases of CML.
SHP-1 most likely plays an important role in modulating the
activation state of BCR-ABL1-dependent and -independent pathways as
well as CML blastic transformation. Thus, the analysis of SHP-1
levels and the methylation status may be useful as an indicator of
CML progression, although our results should be validated in a
larger cohort of patients. Together, our findings suggest that
SHP-1 may be used for the early assessment of blastic
transformation in patients with CML and may also better tailor
targeted therapy.
Acknowledgements
The authors thank Novartis Pharma AG for providing
imatinib and nilotinib. The present study was supported by the
Industry Special Research Fund of the Health Ministry of China (no.
201202017).
Abbreviations:
|
CML
|
chronic myelogenous leukemia
|
|
CP
|
chronic phase
|
|
AP
|
accelerated phase
|
|
BP
|
blastic phase
|
|
PBMCs
|
normal peripheral blood mononuclear
cells
|
|
U
|
amplified products used as primers for
the unmethylated sequence
|
|
M
|
amplified products used as primers for
the methylated sequence
|
|
IM
|
imatinib mesylate
|
|
MSP
|
methylation-specific polymerase chain
reaction
|
References
|
1
|
Shtivelman E, Lifshitz B, Gale R and
Canaani E: Fused transcript of abl and bcr genes in chronic
myelogenous leukaemia. Nature. 315:550–554. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ben-Neriah Y, Daley G, Mes-Masson A, Witte
O and Baltimore D: The chronic myelogenous leukemia-specific P210
protein is the product of the BCR/ABL hybrid gene. Science.
233:212–214. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Quintas-Cardama A and Cortes J: Molecular
biology of bcr-abl1-positive chronic myeloid leukemia.
Blood. 113:1619–1630. 2009.
|
|
4
|
Baccarani M, Deininger MW, Rosti G, et al:
European LeukemiaNet recommendations for the management of chronic
myeloid leukemia: 2013. Blood. 122:872–884. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nash I: Chronic myeloid leukemia. N Engl J
Med. 341:7651999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Perrotti D, Jamieson C, Goldman J and
Skorski T: Chronic myeloid leukemia: mechanisms of blastic
transformation. J Clin Invest. 120:2254–2264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Y, Peng C, Li D and Li S: Molecular
and cellular bases of chronic myeloid leukemia. Protein Cell.
1:124–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rumpold H and Webersinke G: Molecular
pathogenesis of Philadelphia-positive chronic myeloid leukemia - is
it all BCR-ABL? Curr Cancer Drug Targets. 11:3–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Radich JP, Dai H, Mao M, et al: Gene
expression changes associated with progression and response in
chronic myeloid leukemia. Proc Natl Acad Sci USA. 103:2794–2799.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng G, Hui C and Pawson T: SH2-containing
phosphotyrosine phosphatase as a target of protein-tyrosine
kinases. Science. 259:1607–1611. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lorenz U: SHP-1 and SHP-2 in T cells: two
phosphatases functioning at many levels. Immunol Rev. 228:342–359.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Xue L, Hao H, Han Y, Yang J and Luo
J: Rapamycin provides a therapeutic option through inhibition of
mTOR signaling in chronic myelogenous leukemia. Oncol Rep.
27:461–466. 2012.PubMed/NCBI
|
|
13
|
Dong Q, Siminovitch KA, Fialkow L,
Fukushima T and Downey GP: Negative regulation of myeloid cell
proliferation and function by the SH2 domain-containing tyrosine
phosphatase-1. J Immunol. 162:3220–3230. 1999.PubMed/NCBI
|
|
14
|
Tapley P, Shevde N, Schweitzer P, et al:
Increased G-CSF responsiveness of bone marrow cells from
hematopoietic cell phosphatase deficient viable motheaten mice. Exp
Hematol. 25:122–131. 1997.PubMed/NCBI
|
|
15
|
Oka T, Ouchida M, Koyama M, et al: Gene
silencing of the tyrosine phosphatase SHP1 gene by aberrant
methylation in leukemias/lymphomas. Cancer Res. 62:6390–6394.
2002.PubMed/NCBI
|
|
16
|
Chim CS, Fung TK, Cheung WC, Liang R and
Kwong YL: SOCS1 and SHP1 hypermethylation in multiple myeloma:
implications for epigenetic activation of the Jak/STAT pathway.
Blood. 103:4630–4635. 2004. View Article : Google Scholar
|
|
17
|
Zhang Q, Raghunath P, Vonderheid E, Odum N
and Wasik M: Lack of phosphotyrosine phosphatase SHP-1 expression
in malignant T-cell lymphoma cells results from methylation of the
SHP-1 promoter. Am J Pathol. 157:1137–1146. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Esposito N, Colavita I, Quintarelli C, et
al: SHP-1 expression accounts for resistance to imatinib treatment
in Philadelphia chromosome-positive cells derived from patients
with chronic myeloid leukemia. Blood. 118:3634–3644. 2011.
View Article : Google Scholar
|
|
19
|
Bruecher-Encke B, Griffin J, Neel B and
Lorenz U: Role of the tyrosine phosphatase SHP-1 in K562 cell
differentiation. Leukemia. 15:1424–1432. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kharbanda S, Bharti A, Pei D, et al: The
stress response to ionizing radiation involoves c-Abl-dependent
phosphorylation of SHPTP1. Proc Natl Acad Sci USA. 93:6898–6901.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hamilton A, Elrick L, Myssina S, et al:
BCR-ABL activity and its response to drugs can be determined in
CD34+ CML stem cells by CrkL phosphorylation status
using flow cytometry. Leukemia. 20:1035–1039. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lucas C, Harris R, Giannoudis A, Knight K,
Watmough S and Clark R: BCR-ABL1 tyrosine kinase activity at
diagnosis, as determined via the pCrkL/CrkL ratio, is predictive of
clinical outcome in chronic myeloid leukaemia. Br J Haematol.
149:458–460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
24
|
Khoury H, Kukreja M, Goldman J, et al:
Prognostic factors for outcomes in allogeneic transplantation for
CML in the imatinib era: a CIBMTR analysis. Bone Marrow Transplant.
47:810–816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chong Z and Maiese K: The Src homology 2
domain tyrosine phosphatases SHP-1 and SHP-2: diversified control
of cell growth, inflammation, and injury. Histol Histopathol.
22:1251–1267. 2007.PubMed/NCBI
|
|
26
|
Rawlings JS, Rosler KM and Harrison DA:
The JAK/STAT signaling pathway. J Cell Sci. 117:1281–1283. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Warsch W, Kollmann K, Eckelhart E, et al:
High STAT5 levels mediate imatinib resistance and indicate disease
progression in chronic myeloid leukemia. Blood. 117:3409–3420.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie S, Wang Y, Liu J, et al: Involvement
of Jak2 tyrosine phosphorylation in Bcr-Abl transformation.
Oncogene. 20:6188–6195. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Samanta AK, Lin H, Sun T, Kantarjian H and
Arlinghaus RB: Janus kinase 2: a critical target in chronic
myelogenous leukemia. Cancer Res. 66:6468–6472. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Samanta A, Chakraborty S, Wang Y, et al:
Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1
pathway, causing apoptosis in drug-resistant cells from chronic
myelogenous leukemia patients. Oncogene. 28:1669–1681. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Samanta A, Perazzona B, Chakraborty S, et
al: Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid
leukemia. Leukemia. 25:463–472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Luo J, Liu Z, Hao H, Wang F, Dong Z and
Ryuzo O: Mutation analysis of hematopoietic cell phosphatase gene
in acute leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 12:128–132.
2004.(In Chinese).
|
|
33
|
Amin H, Hoshino K, Yang H, Lin Q, Lai R
and Garcia-Manero G: Decreased expression level of SH2
domain-containing protein tyrosine phosphatase-1 (Shp1) is
associated with progression of chronic myeloid leukaemia. J Pathol.
212:402–410. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chim CS, Liang R, Leung MH and Kwong YL:
Aberrant gene methylation implicated in the progression of
monoclonal gammopathy of undetermined significance to multiple
myeloma. J Clin Pathol. 60:104–106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang Q, Wang HY, Marzec M, Raghunath PN,
Nagasawa T and Wasik MA: STAT3- and DNA methyltransferase
1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor
suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci USA.
102:6948–6953. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han Y, Amin HM, Franko B, Frantz C, Shi X
and Lai R: Loss of SHP1 enhances JAK3/STAT3 signaling and decreases
proteosome degradation of JAK3 and NPM-ALK in ALK+
anaplastic large-cell lymphoma. Blood. 108:2796–2803. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Neviani P, Santhanam R, Trotta R, et al:
The tumor suppressor PP2A is functionally inactivated in blast
crisis CML through the inhibitory activity of the BCR/ABL-regulated
SET protein. Cancer Cell. 8:355–368. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu C, Sun M, Liu L and Zhou G: The
function of the protein tyrosine phosphatase SHP-1 in cancer. Gene.
306:1–12. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Radich JP: The biology of CML blast
crisis. Hematology. 2007:384–391. 2007. View Article : Google Scholar
|