Introduction
Urinary bladder cancer is the fifth most common
malignancy in the US. In 2012, there were an estimated 73,510 cases
and 14,880 people succumbed to this disease (1). More than 90% of these bladder cancers
are urothelial carcinoma (UC), followed by squamous cell carcinoma
(5%) and adenocarcinoma (2%) (2).
Approximately 20–30% of bladder cancer patients present with an
aggressive tumor that invades the muscle, and more than half of
these patients develop distant metastases (3).
Cisplatin (CDDP), the first and most widely used
platinum-based chemotherapy drug, is the cornerstone of
chemotherapy for metastatic UC (4,5). CDDP
binds to purine DNA bases to form inter- and intra-strand
crosslinks and causes DNA damage that leads to cell death via the
activation of the apoptotic pathway (6). However, the efficacy of CDDP is often
reduced due to the drug resistance of cancer cells. Multiple
mechanisms underlying this drug resistance have been identified and
broadly classified into cancer cell drug efflux pumps,
intracellular antioxidants, DNA repair pathway modulations and
enhanced anti-apoptotic signaling (6).
Docetaxel (DTX) belongs to the taxane class of
medications and has demonstrated activity in various solid tumors
(7–9). DTX, a close molecular relative of
paclitaxel, promotes the intracellular bundling of microtubules,
subsequently inhibits microtubule depolymerization and results in
cell cycle arrest and cell death (10). A previous study reported that DTX
would be a promising first-line agent for
non-chemotherapy-pretreated patients with metastatic UC (11). Another study suggested that DTX
could be an option for patients with relapsed UC, with a 13.3%
major response rate (12).
Gemcitabine (GEM) is a deoxycytidine analog that
exerts its chemotherapeutic effect by incorporating itself into DNA
to block replication, which results in apoptotic cell death
(13). Based on its low toxicity
and good tolerability and response, GEM has been described as the
single most effective agent for bladder cancer (14).
Currently, the standard treatment for advanced or
metastatic urothelial bladder cancer is the combination
chemotherapy with GEM and CDDP (4,15).
Although this chemotherapy combination initially produces high
response rates, the disease ultimately recurs in most patients and
the majority of patients die shortly after recurrence (16). Previous reports suggested that DTX
shows a significant antitumor effect in combination with other
drugs to treat advanced or metastatic UC (17,18),
but the anticancer effect of this regimen has not completely
satisfied. Accordingly, it is imperative to develop more optimal
anticancer regimens by incorporating novel targeted agents to
improve the survival outcomes and quality of life in advanced or
metastatic bladder cancer patients.
The heat shock protein (HSP) 90, which has emerged
as an important target in cancer therapy, is a well-known molecular
chaperone that maintains the correct conformational folding,
cellular localization and stabilization of numerous client proteins
involved in cell proliferation, differentiation, survival and
various signal pathways (19).
Although HSP90 exists in almost all living organisms, HSP90 is
typically highly expressed and activated in cancer cells (20). The HSP90 inhibitor
17-N-allylamino-17-demethoxygeldanamycin (17-AAG), a derivative of
geldanamycin (GA), has been found to significantly reduce the
toxicity and maintain the positive effects of HSP90 inhibition in
comparison with GA (21). By
targeting the N-terminal ATPase of HSP90, 17-AAG potently disrupts
its function and induces the degradation of client proteins such as
Akt, PLK, ERBB2, EGFR, ERK1/2 and p53 (19,22).
HSP90 derived from cancer cells has a 100-fold higher binding
affinity for 17-AAG compared with HSP90 obtained from normal cells
(20). 17-AAG has been under phase
I and II clinical trials for various solid tumors (23–25).
Moreover, previous studies suggested that 17-AAG enhances the
cytotoxic effects of CDDP in non-small cell lung cancer (26) and colon cancer cell lines (27). Other reports suggest that 17-AAG
also enhanced the effect of paclitaxel in breast cancer cells
(28) and the effect of GEM in
ovarian cancer and cervical cancer cell lines (29).
On the other hand, recent reports have suggested
that HSP70, which is an anti-apoptotic chaperone that aids protein
recovery before proteosomal degradation, would be overexpressed
with the suppression of HSP90 (22,30,31).
HSP70 is potentially a key molecule in resistance to HSP90-targeted
therapy (31); thus, we
hypothesized that the dual targeting of HSP90 and HSP70 could
induce an intense anticancer effect and would enhance the effects
of chemotherapeutic agents.
Therefore, in the present study, we initially
investigated the synergistic effect of a HSP90 inhibitor and
chemotherapeutic agent (CDDP, DTX or GEM). Next, we examined the
expression of HSP70 after the administration of the HSP90
inhibitor. Finally, we tested the effect of the HSP70 inhibitor in
combination with the HSP90 inhibitor and a chemotherapeutic agent
using human bladder cancer cell lines.
Materials and methods
Cell culture
The present study was performed using five human
bladder cancer cell lines: T24 (grade 3), KK47 (grade 1), 5637
(grade 2), 1376 (grade 3) and RT4 (grade 1). The T24, 5637, 1376
and RT4 cell lines were obtained from the American Type Culture
Collection (Manassas, VA, USA), and KK47 was generously provided by
Dr Naito (Kyushu University, Fukuoka, Japan). The T24 and KK47
cells were cultured in minimum essential medium (MEM) with Earle’s
salts, L-glutamine (Invitrogen, Carlsbad, CA, USA), 10% newborn
calf serum (Equitech-Bio, Inc., Kerrville, TX, USA) and 1%
penicillin (PC) streptomycin (SM; Gibco, Grand Island, NY, USA).
The 5637 and 1376 cells were incubated in RPMI-1640 medium
supplemented with 1% L-glutamine (Invitrogen), 10% fetal bovine
serum (FBS; Nichirei Biosciences Inc., Tokyo, Japan), 1% HEPES
(Gibco) and 1% PCSM. The RT4 cells were grown in modified McCoy’s
5A medium (Gibco), supplemented with 10% FBS and 1% PCSM. All cell
lines were maintained in a humidified incubator at 37°C and 5%
CO2.
Chemicals and antibodies
The HSP90 inhibitor 17-AAG was purchased from
InvivoGen (San Diego, CA, USA). CDDP was obtained from Nippon
Kayaku (Tokyo, Japan). DTX, GEM and pifithrin-μ (PFT-μ) were
supplied by Sigma-Aldrich (St. Louis, MO, USA). The HSP90 primers
were synthesized by Qiagen (Hilden, Germany). The antibodies
against HSP90, HSP70, phospho-Akt (Ser473), Akt, phospho-Stat3
(Tyr705), Stat3, phospho-p44/42 MAPK, p44/42 MAPK,
phospho-SAPK/JNK, SAPK/JNK, cleaved PARP (Asp214), phospho-Bad
(Ser136) and Bad, which were purchased from Cell Signaling
Technology Inc. (Hertfordshire, UK), were used for the western blot
analyses. Enhanced chemiluminescence (ECL) and ECL prime western
blotting detection reagents were obtained from GE Healthcare Life
Sciences (Buckinghamshire, UK). The HSP90 and HSP70 antibodies were
also used for immunohistochemistry.
Immunohistochemistry
The formalin-fixed and paraffin-embedded tissues of
urinary bladder cancer cells from patients who had undergone
radical cystectomy were used for immunostaining and for evaluating
the expression of HSP90 and HSP70. After incubation at 60°C for 10
min, 4-μm paraffin-embedded sections of the specimens were
deparaffinized in xylene and rehydrated in different concentrations
of alcohol. Slides were pretreated with 10 mM citrate buffer (pH
6.0) at 105°C for 10 min in a microwave oven for antigen retrieval.
To inhibit the endogenous peroxidase, slides were then incubated
with 0.3% H2O2 for 10 min. After blocking the
non-specific binding for 10 min with 10% goat serum, the slides
were incubated with primary antibodies against HSP90 (dilution
1:100) and HSP70 (dilution 1:500) at 4°C in a humidified chamber
overnight. The following day, after incubating for 30 min with a
horseradish peroxidase-labeled secondary antibody at room
temperature, the color was developed using 3,3-diaminobenzidine
tetrahydrochloride. Finally, the sections were counterstained with
hematoxylin. Negative controls were treated with 1% bovine serum
albumin and 0.01 M phosphate-buffered saline which replaced the
primary antibody. The intensity of the immunostaining activity was
classified into four grades and was determined by R.S. blindly
without any information about patient background or clinical
status.
Quantitative real-time
reverse-transcription polymerase chain reaction (real-time
RT-PCR)
Total RNA from the five bladder cancer cell lines
was extracted using TRIzol reagent (Invitrogen), according to the
manufacturer’s instructions. Total RNA (1 μg) was used for
first-strand cDNA synthesis at a final volume of 20 μl, using the
Thermoscript RT-PCR System (Invitrogen). The cDNA was amplified by
PCR using a LightCycler FastStart DNA Master SYBR-Green I reaction
mix (Roche Molecular Biochemicals, Mannheim, Germany) on a
LightCycler System (Roche Diagnostics, Indianapolis, IN, USA); this
included each cycle of denaturation at 95°C for 15 sec, annealing
at 55°C for 5 sec and polymerization at 72°C for 10 sec. The
primers for HSP90 and HSP70 were obtained from Qiagen (Hilden,
Germany) and the quantification was normalized by actin
(Qiagen).
Cell growth inhibition assay
Exponentially growing cells were plated at a density
of 2×105 cells/well on 6-well plates. The cells were
then treated with various drug concentrations and counted at 24, 48
or 72 h following drug treatments using a hemocytometer. The
average number of cells was calculated in triplicate for each
concentration.
Flow cytometry
T24 cells were seeded on 6-well plates at a density
of 2×105 cells/well. Following the indicated drug
treatments for 48 or 72 h, the cells were collected using
trypsin-EDTA, fixed with 70% ethanol and stored at −20°C overnight.
The next day, the fixed cells were incubated with 10 μg/ml
ribonuclease A (Sigma-Aldrich, St. Louis, MO, USA) for 30 min and
were stained with 25 μg/ml propidium iodide for 30 min. The cell
cycles were analyzed with a FACSCalibur flow cytometer and the
results were processed with the CellQuest software
(Becton-Dickinson, San Jose, CA, USA).
Western blot analysis
The bladder cancer cells were collected by a
scraper, and the proteins were extracted using the Mammalian
Protein Extraction Reagent supplemented with a protease inhibitor
cocktail (Pierce Biotechnology, Rockford, IL, USA). Approximately
20 μg of the total protein preparations was loaded onto a 12% or
4–20% SDS-polyacrylamide gel and was electrotransferred to
nitrocellulose membranes. After blocking with Blocking One-P
(Nacalai Tesque Inc., Kyoto, Japan), the membranes were incubated
with the appropriate primary antibodies at 4°C overnight under
constant shaking conditions. On the next day, the membranes were
incubated with the suitable anti-mouse or anti-rabbit
HPR-conjugated secondary antibody at room temperature for 1 h with
constant shaking. Anti-β-tubulin (Millipore, Temecula, CA, USA) was
used as the loading control. Protein expressions were visualized
through an ECL or ECL prime protein detection system according to
the manufacturer’s instructions.
Caspase-3/7 luminometric assay
For measuring the activities of caspase-3 and -7,
the Caspase-Glo®-3/7 assay system (Promega, Madison, WI,
USA) was used according to the manufacturer’s instructions. T24
cells (2×105) which were plated in each well of the
6-well plates were exposed to the indicated drug treatments for 24
or 48 h. After incubation with 50 μl cell culture lysis reagent
overnight at −70°C, the cells were then incubated with an equal
volume of Caspase-Glo®-3/7 assay reagent at room
temperature for 1 h. Subsequently, the luminescence was determined
using a luminometer.
Transmission electron microscopy
(TEM)
The T24 cells were seeded on 10-cm plates at a
density of 2×106 cells/plate. These cells were treated
with 500 nM of 17-AAG, 10 μM of PFT-μ or 17-AAG + PFT-μ for 48 h.
After collection by a scraper and centrifugation at 1,500 rpm for 5
min, the cells were then fixed in a solution containing 2.5%
glutaraldehyde and 2% paraformaldehyde in a 0.1 M cacodylate buffer
(pH 7.2) for 30 min. The cells were post-fixed in 2%
OsO4 for 1 h at 4°C, dehydrated in a graded series of
ethanol and embedded in epoxy resin. Ultrathin sections (80–90 nm)
were stained with uranyl acetate and lead citrate and examined
under a transmission electron microscope (Hitachi H7650, Tokyo,
Japan).
Results
HSP90 is highly expressed in bladder
cancer
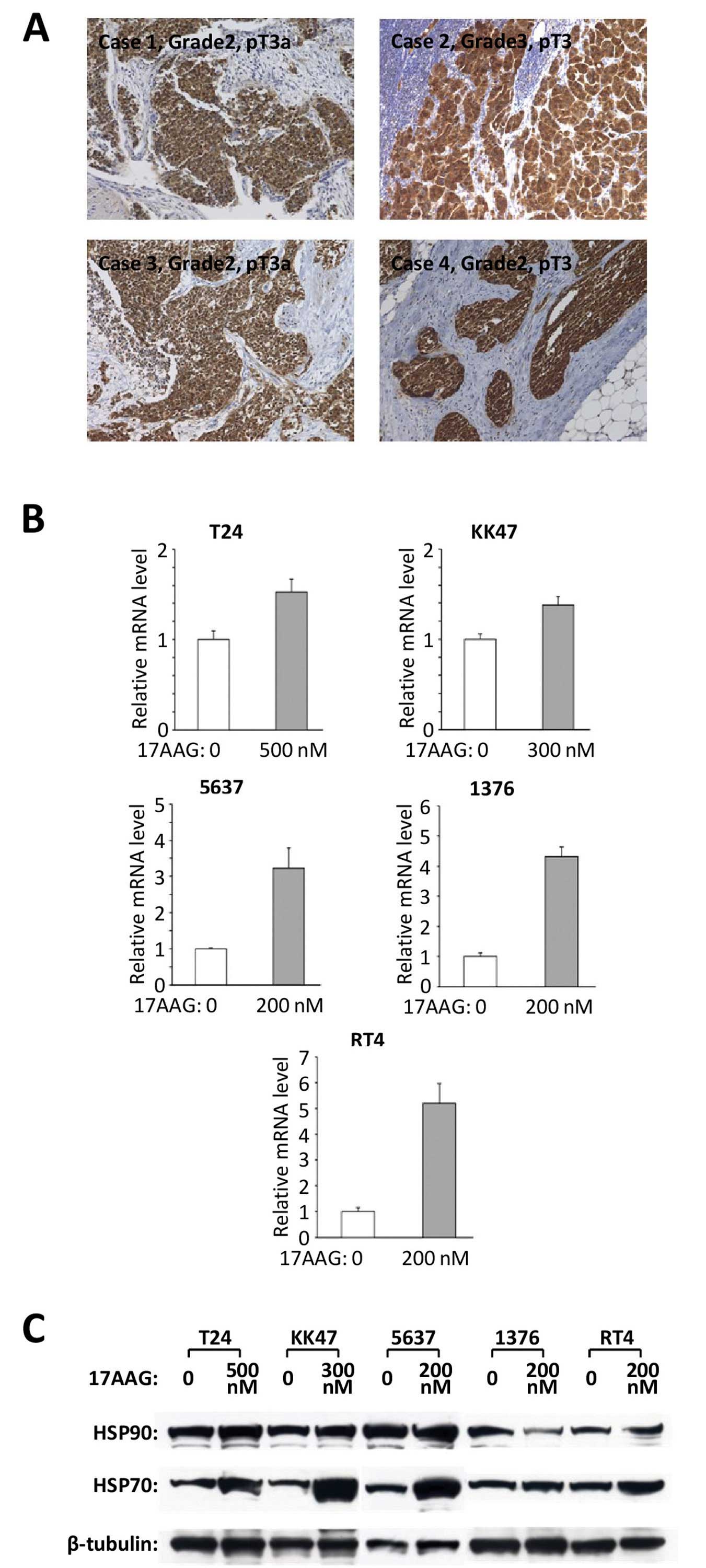
We first examined the expression of HSP90 in human
bladder cancer. Tissues obtained from 33 bladder cancer patients
who underwent radical cystectomy were used for immunohistochemistry
to investigate the expression of HSP90. All cases had specific
positive expressions of HSP90 in cancer cells (Fig. 1A), while the normal bladder tissues
had negative immunoreactions. The intensity of HSP90
immunoreactivity did not have an evident correlation with the grade
and pathological T stage of the bladder cancer. This result
suggests that there is a high level of expression of HSP90 in
bladder cancers when compared with that in normal bladder tissues.
We also examined the expression of HSP90 in the five specific
bladder cancer cell lines using real-time RT-PCR and western
blotting. The results revealed that all of the cell lines expressed
HSP90 at the mRNA (Fig. 1B) and
protein levels (Fig. 1C).
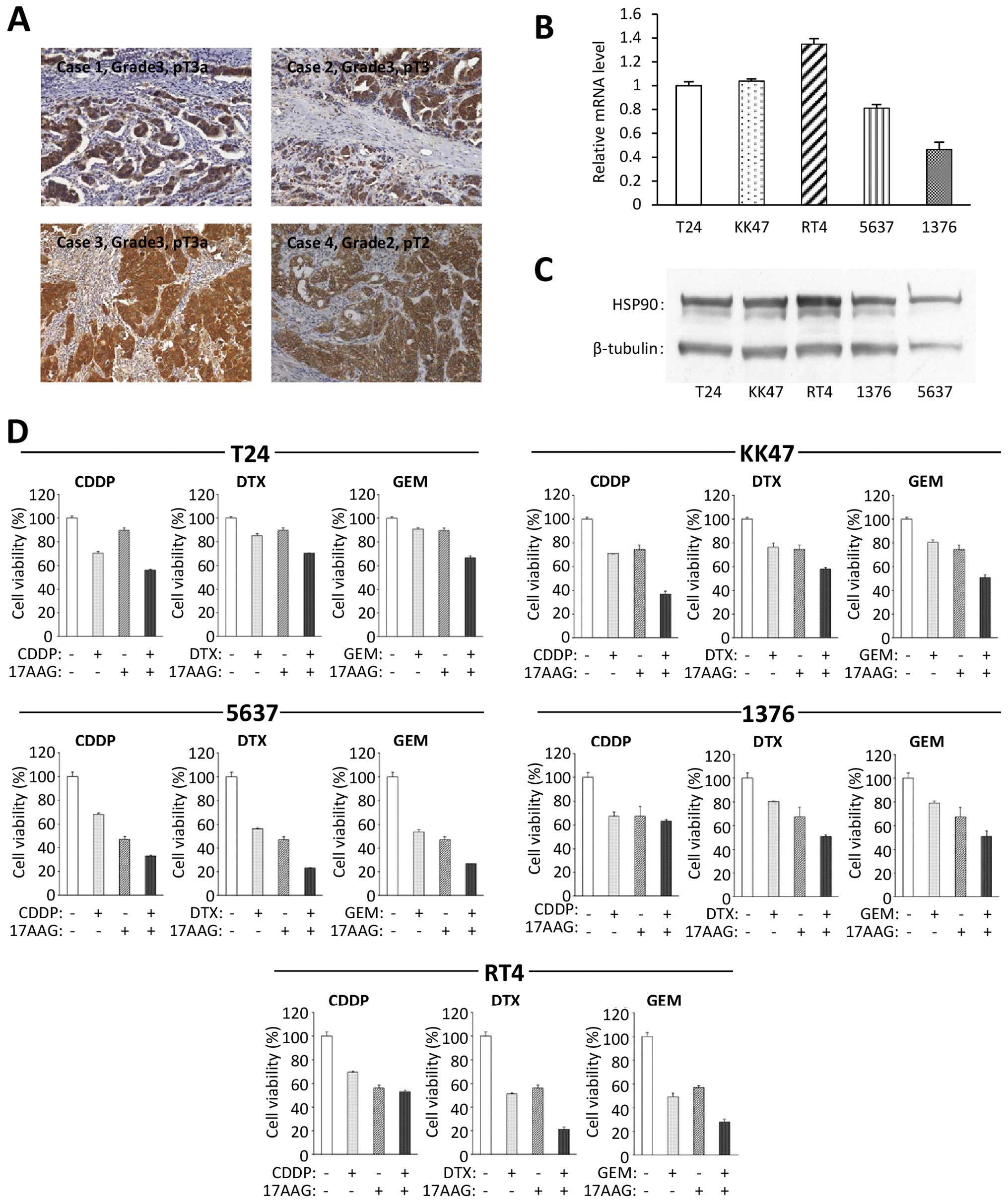 | Figure 1HSP90 expression and the combined
effects of an HSP90 inhibitor and a chemotherapeutic agent in
bladder cancer cell lines. (A) HSP90 immunoreactivity in human
bladder cancer tissues was investigated. All 33 cases had specific
positive reactions for HSP90 and the representative four cases are
shown here. Original magnification, ×200. The HSP90 expressions of
these five cell lines were examined at the (B) mRNA and (C) protein
levels and by real-time RT-PCR and western blot analyses. (D) Cell
viability was analyzed in the indicated bladder cancer cell lines
which were treated with the indicated antitumor chemicals for 48 h
by cell count assay. Values represent the means ± SD of 3
independent experiments. The concentration of each chemical to
cancer cells: T24: 17-AAG, 200 nM; CDDP, 500 nM; DTX, 30 nM; GEM,
100 nM. KK47: 17-AAG, 100 nM; CDDP, 600 nM; DTX, 20 nM; GEM, 40 nM.
5637: 17-AAG, 100 nM; CDDP, 500 nM; DTX, 10 nM; GEM, 20 nM. 1376:
17-AAG, 100 nM; CDDP, 700 nM; DTX, 25 nM; GEM, 30 nM. RT4: 17-AAG,
75 nM (when 17-AAG was combined with GEM, the concentration of
17-AAG was 50 nM); CDDP, 500 nM; DTX, 10 nM; GEM, 10 nM. |
17-AAG sensitizes bladder cancer cells to
chemotherapeutic agents
To examine the cytotoxic effects of the HSP90
inhibitor in combination with a chemotherapeutic agent, we used a
cell count assay for all five bladder cancer cell lines. Each
chemical concentration was determined using IC50 values.
The treatment of 17-AAG combined with CDDP, DTX or GEM had more
cytotoxic effects than a single drug treatment in all cell lines
(Fig. 1D). These findings indicated
that there was a synergistic antiproliferative effect of 17-AAG
with each chemotherapeutic agent.
Further investigations were performed using the T24
bladder cancer cell line since that line had the most aggressive
features (grade 3).
Combination treatment with 17-AAG and a
chemotherapeutic agent induces more cell apoptosis during cell
cycles
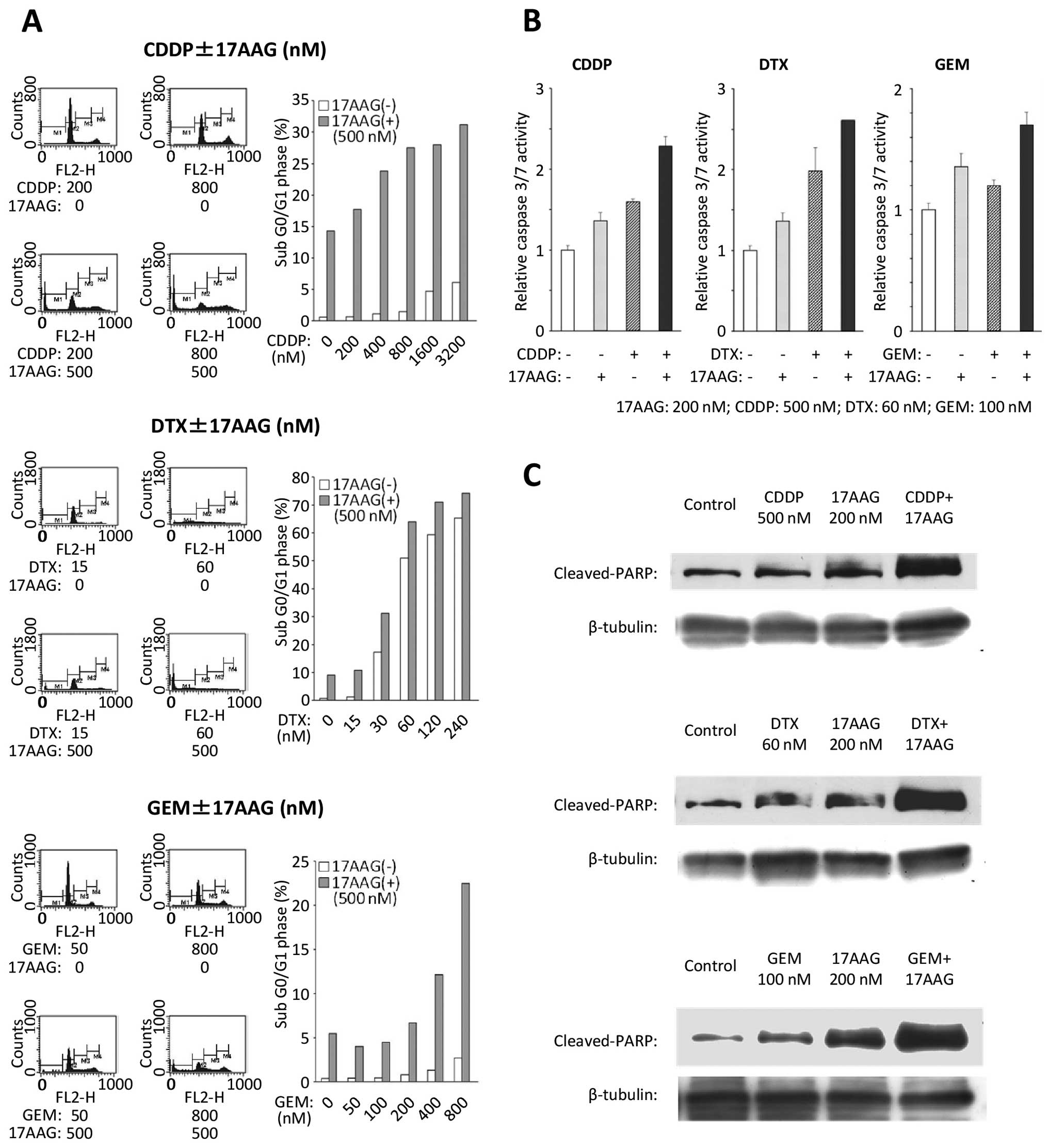
To examine the inhibitory effects on cell cycle
progression, flow cytometry was conducted on the T24 cell line
(Fig. 2A). With the combination
treatments of 17-AAG and chemotherapeutic agents, the population in
the sub G0/G1 phase increased significantly when compared with the
population subjected to single drug treatments.
17-AAG combined with a chemotherapeutic
agent induces activation of caspase-dependent death processes
To examine the impact on the apoptosis of the T24
cell line following each combination treatment of 17-AAG with
chemotherapeutic agents, a caspase-3/7 assay was performed. The T24
cells that were exposed to 17-AAG combined with a chemotherapeutic
agent showed an increased caspase-3/7 activity compared with those
exposed to single drug treatments (Fig.
2B). Furthermore, increased cleaved PARP expressions were
induced by the combined treatments with 17-AAG and chemotherapeutic
agents in comparison with a single agent (Fig. 2C).
17-AAG induces the downregulation of
critical targets in the cell survival pathway and the upregulation
of HSP70
To investigate the major intracellular signaling
associated with cell survival, western blotting was performed in
the T24 cell line to evaluate the changes in Akt, JNK, MAPK and
Stat3, which were the client proteins of HSP90. 17-AAG induced the
downregulation of phospho-Akt (P-Akt), Akt and JNK; in contrast,
17-AAG also induced the upregulation of HSP70 in a time-dependent
manner (Fig. 3A). The upregulation
of HSP70 was also induced by 17-AAG in a dose-dependent manner
(Fig. 3B). On the other hand, none
of the chemotherapeutic agents (CDDP, DTX or GEM) demonstrated a
specific influence on these protein expressions (Fig. 3A).
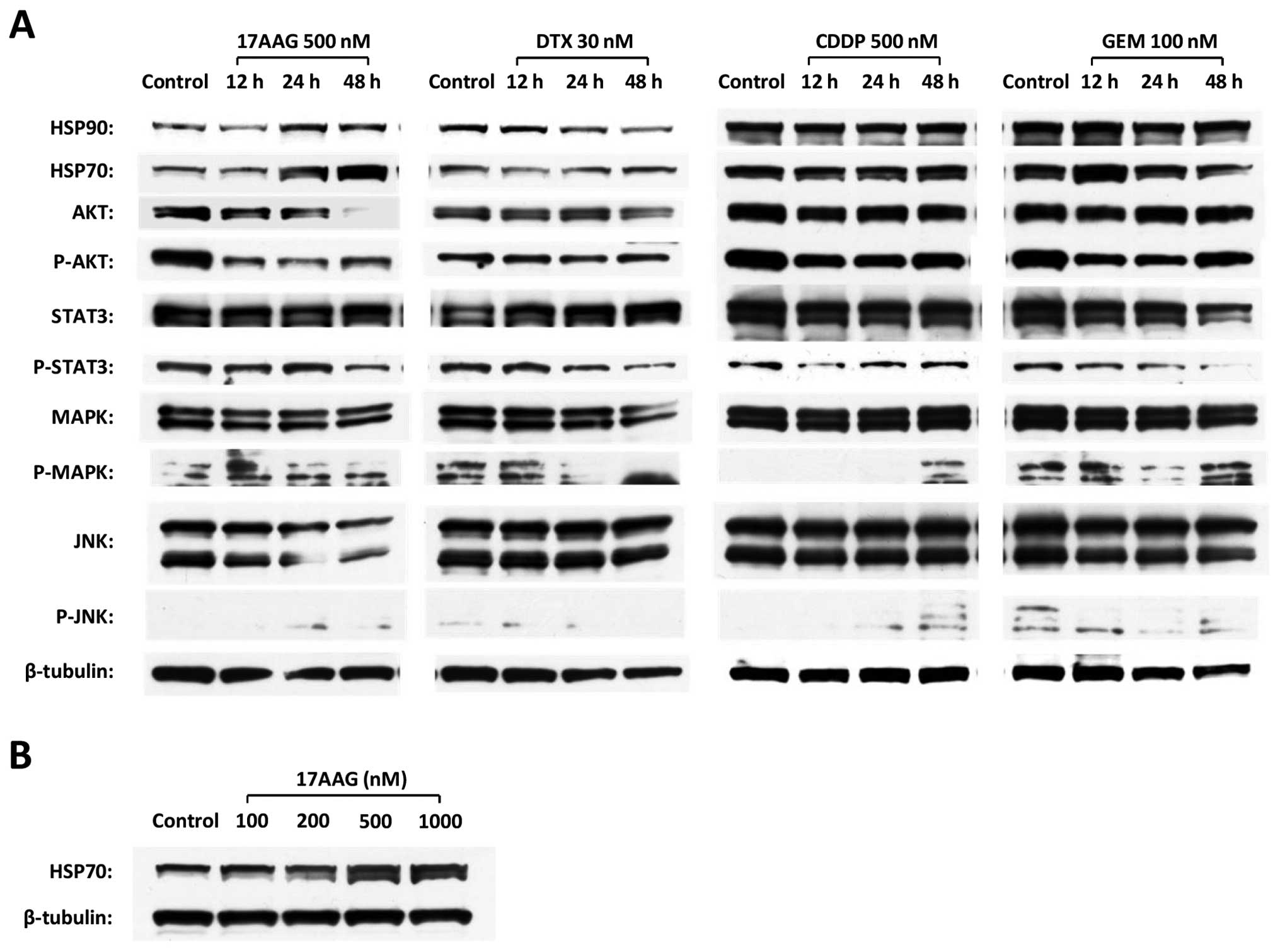 | Figure 3The expression changes of key
proteins that contribute to anti-apoptotic signal transduction and
HSP70 in the T24 cell line. (A) The alterations in HSP90, HSP70,
Akt, P-Akt, Stat3, P-Stat3, MAPK, P-MAPK, JNK and P-JNK expressions
were investigated by western blot analyses in a time course after
individual chemical administration. (B) The expression changes of
HSP70 were examined by western blot analyses in the presence of
17-AAG at the concentrations of 100, 200, 500 and 1,000 nM for 48
h. |
HSP70 has high expression in bladder
cancer
Twenty-eight bladder cancer tissues were chosen and
used for HSP90 immunohistochemistry; all had a specific positive
expression of HSP70 (Fig. 4A). The
intensity of HSP70 immunoreactivity did not have a clear
correlation with the grade and pathological T stage of bladder
cancer. Real-time RT-PCR and western blot analyses revealed that
all five cell lines had expressions of HSP70 at the mRNA (Fig. 4B) and protein levels (Fig. 4C), respectively.
The HSP70 inhibitor PFT-μ enhances the
cytotoxic effect of 17-AAG and a chemotherapeutic agent in the T24
cell line
PFT-μ, which is an HSP70 inhibitor that targets the
C-terminal substrate binding domain of HSP70, disrupts the
associations of client proteins and causes protein aggregation and
autophagic dysfunction (32). A
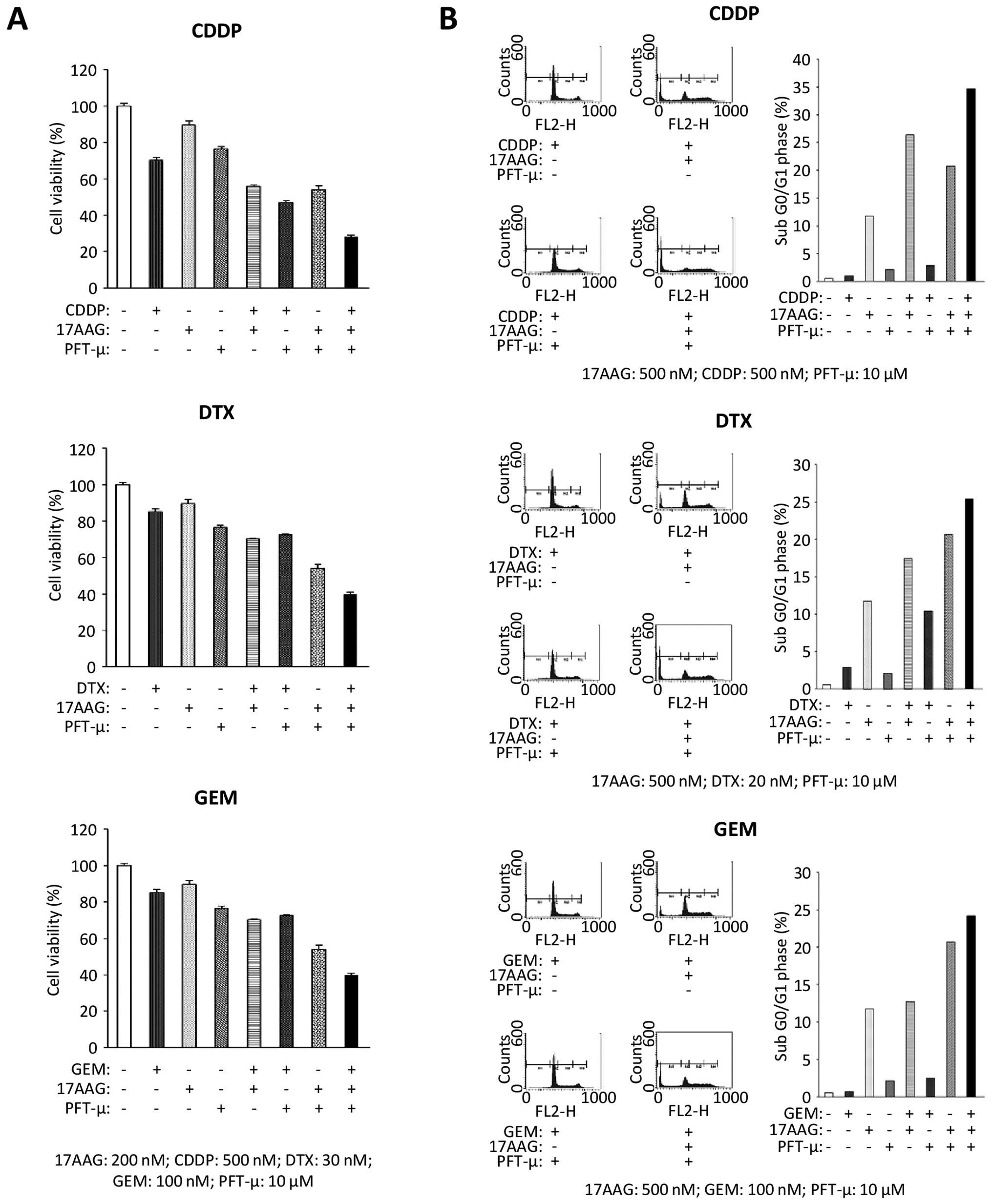
cell count assay was used to evaluate the inhibition of cell
proliferation with the addition of a chemotherapeutic agent, 17-AAG
or PFT-μ, in a T24 cell line. The treatment with PFT-μ alone
induced a limited suppression of cell viability, but the dual
targeted treatment of HSP90 and HSP70 using 17-AAG and PFT-μ showed
the prominent suppression of cell proliferation. The triple drug
combination of PFT-μ, 17-AAG, and each chemotherapeutic agent
showed the most significant cytotoxic effect on the T24 cancer
cells when compared with the dual therapies of 17-AAG and each
chemotherapeutic agent (Fig.
5A).
Using flow cytometry, the triple drug treatments of
PFT-μ, 17-AAG, and each chemotherapeutic agent induced cell
accumulation in the sub G0/G1 phase compared with the single or
dual drug treatments (Fig. 5B).
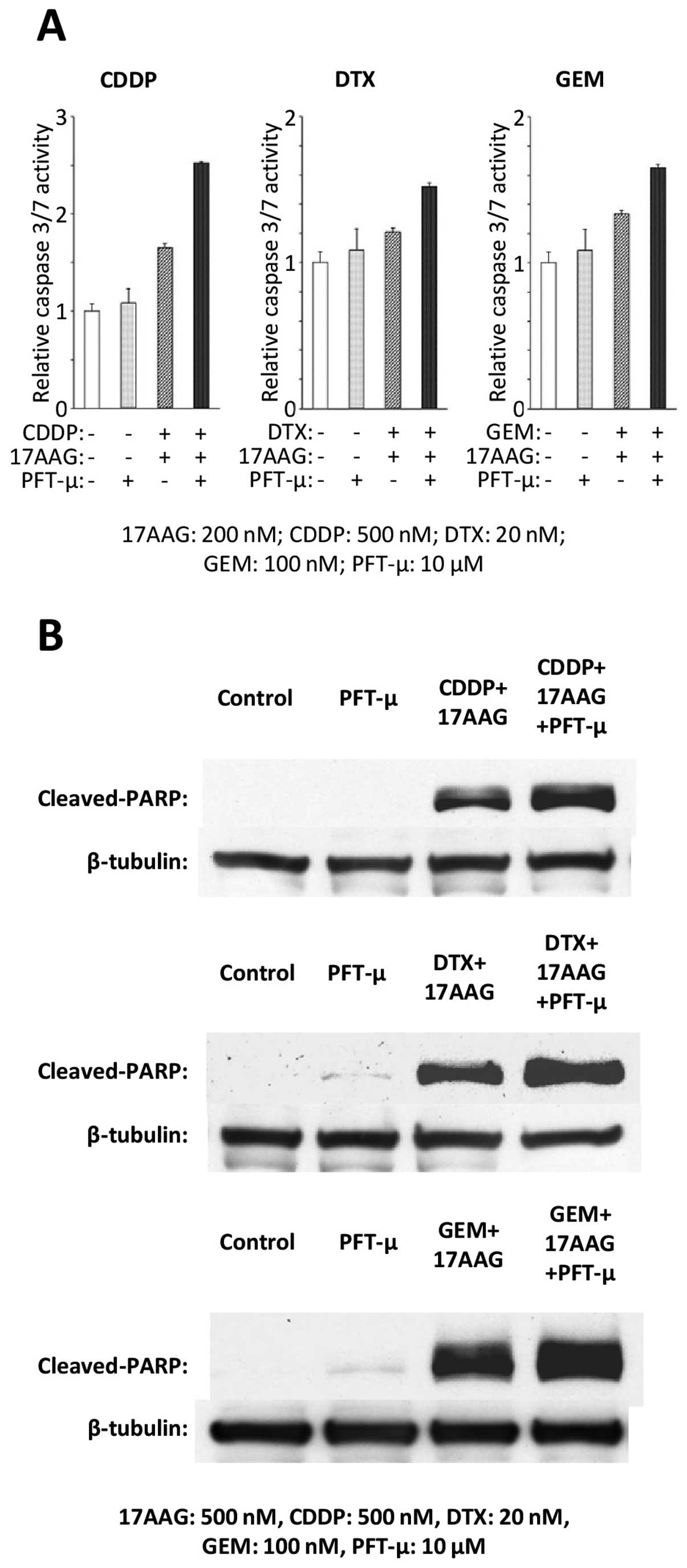
To examine the impact on apoptosis in the T24 cell
line after the administration of PFT-μ or triple treatments with
PFT-μ, 17-AAG and a chemotherapeutic agent, the caspase-3/7 assay
and western blot analyses of the cleaved PARP were performed. PFT-μ
alone did not induce obvious changes in the caspase-3/7 activity,
but PFT-μ enhanced the activation of the caspase-3/7 that was
induced by the dual treatments of 17-AAG and CDDP, DTX or GEM
(Fig. 6A). Also, PFT-μ did not
induce obvious changes in the cleaved PARP expression, but PFT-μ
did enhance the expression of the cleaved PARP that was induced by
the dual treatments of 17-AAG and CDDP, DTX or GEM (Fig. 6B).
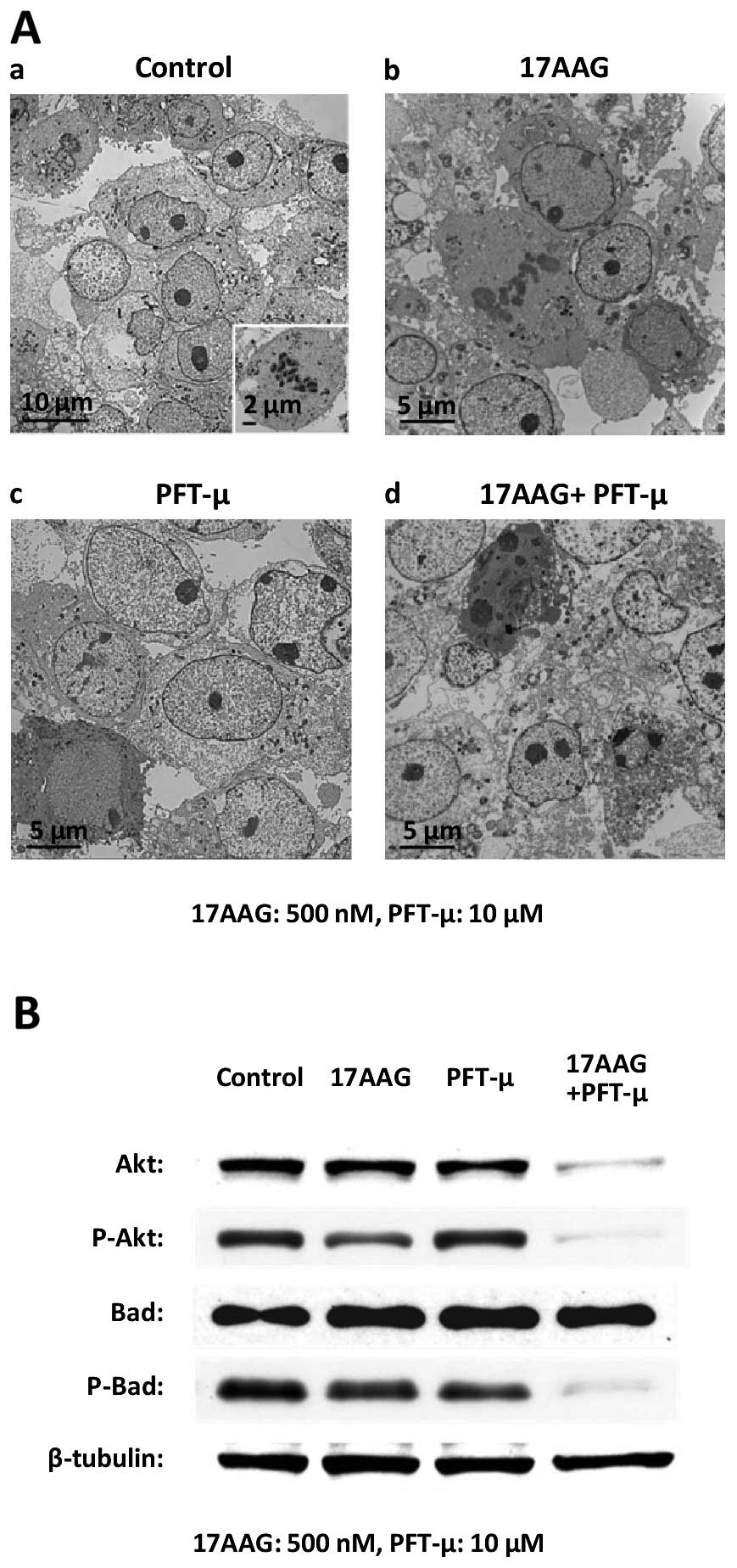
The ultrastructural alterations of the cancer cells
were observed by TEM following the treatment with 17-AAG, PFT-μ or
17-AAG + PFT-μ for 48 h. Compared with the control (Fig. 7Aa), most cells treated with 17-AAG
(Fig. 7Ab) or PFT-μ (Fig. 7Ac) showed relatively similar
features. We found intact and oval nuclei with a clear nucleolus,
Golgi apparatus, free ribosomes and cell division in many cells.
However, a small amount of atrophy of nuclei and cytoplasm and
several small vesicles were observed in a few cells. Meanwhile, the
cells treated with the combined treatment of 17-AAG and PFT-μ
presented with clear ultrastructural alterations. Many cells showed
moderate shrinkage, and their cytoplasm had numerous vacuoles of
various sizes as well as lysosomes. In addition, a considerable
amount of cell atrophy appearing as an electron dense image was
observed (Fig. 7Ad). No cell
division was found. The present morphological findings demonstrated
that the combined treatment with 17-AAG and PFT-μ induced more
apoptotic cell death than either 17-AAG or PFT-μ treatment.
Dual targeting of HSP90 and HSP70 using
17-AAG and PFT-μ induces the notable downregulation of p-Akt and
p-Bad
Finally, we examined the expressions of the proteins
Akt and Bad following the treatment with PFT-μ and 17-AAG for 48 h.
The treatment with 17-AAG or PFT-μ alone decreased the expression
of each indicated protein to some extent. The dual treatment of
PFT-μ combined with 17-AAG markedly reduced the expression of
P-Akt, Akt and P-Bad (Fig. 7B).
Discussion
The inhibition of HSP90 functions could lead to the
simultaneous disruption of its client proteins that have critical
roles in cancer proliferation and survival (33), making HSP90 an attractive molecular
target for advanced malignant diseases with drug resistance
(34). Thus, using the HSP90
inhibitor 17-AAG, we initially investigated a strategy to
potentiate its antitumor effect and to enhance the effect of
chemotherapeutic agents on human urinary bladder cancer cell
lines.
17-AAG enhances the cytotoxic effect of CDDP through
the downregulation of ERK1/2 and Akt activations in non-small cell
lung cancer cells (26). It had
also been reported that 17-AAG could effectively inhibit the
PI3k/Akt signaling pathway, thus enhancing paclitaxel-induced
apoptosis in breast cancer cell lines (28). Another report noted that by
disrupting the activation of Chk1, which is one of the client
proteins of HSP90 and has several functions in promoting cell
survival, 17-AAG sensitized HeLa, OVCAR3 and ML-1 cells to GEM
(29). In bladder cancer cell
lines, a low-dose of the HSP90 inhibitor could sensitize human
bladder cancer cells to CDDP in the setting of chemoradiotherapy
(35). Another report also
suggested that HSP90 inhibitors efficiently enhanced the anticancer
effect of CDDP on bladder cancer-initiating cells which were
isolated based on their CD44 expression status (36). To the best of our knowledge, this is
the first report to investigate the synergistic anticancer effects
of 17-AAG combined with DTX or GEM in bladder cancer cell lines.
Akt, a critical regulatory component in multiple signaling
pathways, is one of the major key proteins that mediate tumor cell
survival and the escape from apoptosis (35). The activation of Akt in tumor cells
leads to deregulation of growth and to desensitization to
pro-apoptotic stimuli (37). The
present study suggested that the anticancer effect of 17-AAG was at
least partly through the inhibition of Akt activity. This
inhibitory effect on Akt activity induced by the HSP90 inhibitor
has been supported in various cellular models (22,28,34).
Moreover, the HSP90 inhibitor accelerated the effect of
chemotherapeutic agents and increased the activity of the
caspase-3/7 and cleaved PARP. It was also reported that myocardial
calpain, the activation of which could lead to the cleavage of
HSP90, induced myocardial caspase-3 activation and apoptosis in
septic mice through the downregulation of Akt activation (38). A previous study also suggested that
the HSP90 inhibition by 17-AAG initiated the activation of a
caspase-induced cell death program in bladder cancer cell lines
(22). Thus, it was suggested that
the combined treatments of 17-AAG and each of the chemotherapeutic
agents CDDP, DTX or GEM presented more cytotoxic effects to the
bladder cancer cells compared with those presented after the
administration of each drug alone.
The major aims of the presents study were to confirm
the changes in HSP70 expression, to verify the inhibitory effect
for HSP70 under the condition of HSP90 inhibition, and to explore
the clinical applicability of this novel strategy of dual targeting
of HSP90 and HSP70 by concomitantly administering existing
anticancer drugs. HSP70, the second major HSP, has a structural
similarity to HSP90 and frequently interacts with HSP90. HSP70 is
an ATP-dependent chaperone that assists protein folding and
prevents the intracellular accumulation of misfolded or damaged
proteins (39). HSP70
overexpression, which is thought to provide a survival advantage to
cancer cells, has been shown to increase resistance to
chemotherapeutic agents, such as imatinib, etoposide, CDDP and
MG-132 (40,41). These results suggested that HSP70
would be a potential target in combination with other
chemotherapeutic agents in cancer therapy. Notably, several recent
reports have suggested that the upregulation of HSP70 was induced
by HSP90 inhibition in multiple malignancies including bladder
cancer (22,30,31).
In the present study, HSP70 was upregulated by 17-AAG in all five
bladder cancer cell lines we tested. Although this upregulation of
HSP70 under HSP90 suppression may be universal for malignant cells
to a certain extent, we do not have enough data on this matter. The
upregulation of HSP70 may be one of the reasons that 17-AAG single
treatment only induces insufficient cytotoxic effects on cancer
cells (30,31). The possible mechanism may be that
17-AAG disrupts the associations between HSP90 and the
transcription factor heat shock factor-1 (HSF-1), thereby promoting
the nuclear localization and activation of HSF-1; this results in
the induction of HSP70 synthesis (42). The simultaneous inhibition of HSP70
and HSP90 was previously reported in some malignancies (30,31,43).
Previous studies showed that co-treatment with HSP90 inhibitor
17-AAG and an HSP70 inhibitor or an HSP70-targeted siRNA induced a
synergistic antiproliferation effect in leukemia, myeloma (31) and breast cancer cells (43). Ghoshal et al reported that
the downregulation of HSP70 improved the effects on another HSP90
inhibitor, 17-DMAG on Stat3 activity (30). According to these reports, the
simultaneous inhibition of HSP90 and HSP70 could be more effective
than the inhibition of either HSP90 or HSP70 alone in cancer
therapy. To our knowledge, this is the first report to evaluate the
inhibitory effect of HSP70 along with HSP90 inhibition in bladder
cancer cells. We also showed new evidence regarding the dual
targeting of HSP90 and HSP70 concomitantly with a conventional
anticancer drug. Overexpression of HSP70, which has been found to
block the activation of caspase-3, is thought to provide a survival
advantage for cancer cells (39).
The most prominent induction of caspase-3/7 activity and cleaved
PARP expression by the dual targeting HSP90 and HSP70 using 17-AAG
and PFT-μ with a chemotherapeutic agent was evident in this study;
these results suggested that this trimodal anticancer treatment
could induce strong activation of the caspase-dependent apoptosis
pathway and exhibit more cytotoxicity to cancer cells. Although it
was reported that the increased expression of HSP70 did not affect
the downregulation of Akt proteins induced by 17-AAG (42), another report suggested that HSP70
would selectively bind the dephosphorylated species of Akt via the
unphosphorylated turn motif, thus stabilizing the protein and
allowing re-phosphorylation of Akt (44). In the present study, the targeting
of HSP70 with PFT-μ itself did not have a significant effect on Akt
activity but could enhance the Akt inactivation effect of the HSP90
inhibitor 17-AAG. The dual targeting of HSP90 and HSP70 markedly
reduced not only P-Akt, but also P-Bad expression. The
phosphorylation of Bad, which is controlled by multiple pathways,
could suppress cell apoptosis and promote cell survival (45). This is important since Akt can
phosphorylate Bad at serine 136, resulting in the inactivation of
Bad and reducing cell apoptosis (46).
In conclusion, the present study demonstrated the
synergistic anticancer effects of 17-AAG in combination with CDDP,
DTX or GEM in bladder cancer. Moreover, HSP70 was suggested to be a
key molecule to overcome the resistance to targeted therapy for
HSP90 or combination therapy with an HSP90 inhibitor and a
chemotherapeutic agent. These results also suggested a potential
therapeutic strategy for advanced or metastatic bladder cancer;
this should be further investigated in an in vivo setting
and in clinical trials in the near future.
Acknowledgements
This study was supported by a Grant-in-Aid for
Scientific Research (C) 25462489 (F. Sato) from the Japan Society
for the Promotion of Science. We gratefully acknowledge Ms. N.
Hamamatsu, Ms. S. Kato (Oita University) and Ms. Y. Ito (Ueo Breast
Surgical Hospital) for their excellent technical assistance in the
experiment. The authors would like to thank Enago (www.enago.jp) for the English language review.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2
|
Kaufman DS, Shipley WU and Feldman AS:
Bladder cancer. Lancet. 374:239–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitra AP, Datar RH and Cote RJ: Molecular
staging of bladder cancer. BJU Int. 96:7–12. 2005. View Article : Google Scholar
|
|
4
|
Bambury RM and Rosenberg JE: Advanced
urothelial carcinoma: overcoming treatment resistance through novel
treatment approaches. Front Pharmacol. 4:32013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo T, Yu J, Nguyen J, et al: Electron
transfer-based combination therapy of cisplatin with
tetramethyl-p-phenylenediamine for ovarian, cervical, and
lung cancers. Proc Natl Acad Sci USA. 109:10175–10180. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Drayton RM and Catto JW: Molecular
mechanisms of cisplatin resistance in bladder cancer. Expert Rev
Anticancer Ther. 12:271–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martin M, Pienkowski T, Mackey J, et al:
Adjuvant docetaxel for node-positive breast cancer. N Engl J Med.
352:2302–2313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tannock IF, de Wit R, Berry WR, et al:
Docetaxel plus prednisone or mitoxantrone plus prednisone for
advanced prostate cancer. N Engl J Med. 351:1502–1512. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fossella F, Pereira JR, von Pawel J, et
al: Randomized, multinational, phase III study of docetaxel plus
platinum combinations versus vinorelbine plus cisplatin for
advanced non-small-cell lung cancer: the TAX 326 study group. J
Clin Oncol. 21:3016–3024. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McKiernan JM, Masson P, Murphy AM, et al:
Phase I trial of intravesical docetaxel in the management of
superficial bladder cancer refractory to standard intravesical
therapy. J Clin Oncol. 24:3075–3080. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
de Wit R, Kruit WH, Stoter G, de Boer M,
Kerger J and Verweij J: Docetaxel (Taxotere): an active agent in
metastatic urothelial cancer; results of a phase II study in
non-chemotherapy-pretreated patients. Br J Cancer. 78:1342–1345.
1998.PubMed/NCBI
|
|
12
|
McCaffrey JA, Hilton S, Mazumdar M, Sadan
S, Kelly WK, Scher HI and Bajorin DF: Phase II trial of docetaxel
in patients with advanced or metastatic transitional-cell
carcinoma. J Clin Oncol. 15:1853–1857. 1997.PubMed/NCBI
|
|
13
|
Toschi L, Finocchiaro G, Bartolini S,
Gioia V and Cappuzzo F: Role of gemcitabine in cancer therapy.
Future Oncol. 1:7–17. 2005. View Article : Google Scholar
|
|
14
|
Bellmunt J, Albiol S, de Olano AR, Pujadas
J and Maroto P; Spanish Oncology Genitourinary Group (SOGUG).
Gemcitabine in the treatment of advanced transitional cell
carcinoma of the urothelium. Ann Oncol. 17(Suppl 5): v113–v117.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cognetti F, Ruggeri EM, Felici A, et al:
Adjuvant chemotherapy with cisplatin and gemcitabine versus
chemotherapy at relapse in patients with muscle-invasive bladder
cancer submitted to radical cystectomy: an Italian, multicenter,
randomized phase III trial. Ann Oncol. 23:695–700. 2012. View Article : Google Scholar
|
|
16
|
Kim JJ: Recent advances in treatment of
advanced urothelial carcinoma. Curr Urol Rep. 13:147–152. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Neri B, Vannini L, Giordano C, et al:
Gemcitabine plus docetaxel as first-line biweekly therapy in
locally advanced and/or metastatic urothelial carcinoma: a phase II
study. Anticancer Drugs. 18:1207–1211. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boukovinas I, Androulakis N, Vamvakas L,
et al: Sequential gemcitabine and cisplatin followed by docetaxel
as first-line treatment of advanced urothelial carcinoma: a
multicenter phase II study of the Hellenic Oncology Research Group.
Ann Oncol. 17:1687–1692. 2006. View Article : Google Scholar
|
|
19
|
Maloney A and Workman P: HSP90 as a new
therapeutic target for cancer therapy: the story unfolds. Expert
Opin Biol Ther. 2:3–24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kamal A, Thao L, Sensintaffar J, Zhang L,
Boehm MF, Fritz LC and Burrows FJ: A high-affinity conformation of
Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature.
425:407–410. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schulte TW and Neckers LM: The
benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds
to HSP90 and shares important biologic activities with
geldanamycin. Cancer Chemother Pharmacol. 42:273–279. 1998.
View Article : Google Scholar
|
|
22
|
Karkoulis PK, Stravopodis DJ, Margaritis
LH and Voutsinas GE: 17-Allylamino-17-demethoxygeldanamycin induces
downregulation of critical Hsp90 protein clients and results in
cell cycle arrest and apoptosis of human urinary bladder cancer
cells. BMC Cancer. 10:4812010. View Article : Google Scholar
|
|
23
|
Barluenga S, Fontaine JG, Wang C, et al:
Inhibition of HSP90 with pochoximes: SAR and structure-based
insights. Chembiochem. 10:2753–2759. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barluenga S, Wang C, Fontaine JG, et al:
Divergent synthesis of a pochonin library targeting HSP90 and in
vivo efficacy of an identified inhibitor. Angew Chem Int Ed Engl.
47:4432–4435. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Solit DB, Osman I, Polsky D, et al: Phase
II trial of 17-allylamino-17-demethoxygeldanamycin in patients with
metastatic melanoma. Clin Cancer Res. 14:8302–8307. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weng SH, Tseng SC, Huang YC, Chen HJ and
Lin YW: Inhibition of thymidine phosphorylase expression by using
an HSP90 inhibitor potentiates the cytotoxic effect of cisplatin in
non-small-cell lung cancer cells. Biochem Pharmacol. 84:126–136.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vasilevskaya IA, Rakitina TV and O’Dwyer
PJ: Quantitative effects on c-Jun N-terminal protein kinase
signaling determine synergistic interaction of cisplatin and
17-allylamino-17-demethoxygeldanamycin in colon cancer cell lines.
Mol Pharmacol. 65:235–243. 2004. View Article : Google Scholar
|
|
28
|
Solit DB, Basso AD, Olshen AB, Scher HI
and Rosen N: Inhibition of heat shock protein 90 function
down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer
Res. 63:2139–2144. 2003.PubMed/NCBI
|
|
29
|
Arlander SJ, Eapen AK, Vroman BT, McDonald
RJ, Toft DO and Karnitz LM: Hsp90 inhibition depletes Chk1 and
sensitizes tumor cells to replication stress. J Biol Chem.
278:52572–52577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghoshal S, Rao I, Earp JC, Jusko WJ and
Wetzler M: Down-regulation of heat shock protein 70 improves
arsenic trioxide and 17-DMAG effects on constitutive signal
transducer and activator of transcription 3 activity. Cancer
Chemother Pharmacol. 66:681–689. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang RE: Targeting heat shock proteins
70/90 and proteasome for cancer therapy. Curr Med Chem.
18:4250–4264. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leu JI, Pimkina J, Frank A, Murphy ME and
George DL: A small molecule inhibitor of inducible heat shock
protein 70. Mol Cell. 36:15–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Newman B, Liu Y, Lee HF, Sun D and Wang Y:
HSP90 inhibitor 17-AAG selectively eradicates lymphoma stem cells.
Cancer Res. 72:4551–4561. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang H and Burrows F: Targeting multiple
signal transduction pathways through inhibition of Hsp90. J Mol
Med. 82:488–499. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoshida S, Koga F, Tatokoro M, et al:
Low-dose Hsp90 inhibitors tumor-selectively sensitize bladder
cancer cells to chemoradiotherapy. Cell Cycle. 10:4291–4299. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tatokoro M, Koga F, Yoshida S, Kawakami S,
Fujii Y, Neckers L and Kihara K: Potential role of Hsp90 inhibitors
in overcoming cisplatin resistance of bladder cancer-initiating
cells. Int J Cancer. 131:987–996. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Page C, Lin HJ, Jin Y, Castle VP, Nunez G,
Huang M and Lin J: Overexpression of Akt/AKT can modulate
chemotherapy-induced apoptosis. Anticancer Res. 20:407–416.
2000.PubMed/NCBI
|
|
38
|
Li X, Luo R, Jiang R, Meng X, Wu X, Zhang
S and Hua W: The role of the Hsp90/Akt pathway in myocardial
calpain-induced caspase-3 activation and apoptosis during sepsis.
BMC Cardiovasc Disord. 13:82013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Evans CG, Chang L and Gestwicki JE: Heat
shock protein 70 (Hsp70) as an emerging drug target. J Med Chem.
53:4585–4602. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pocaly M, Lagarde V, Etienne G, et al:
Overexpression of the heat-shock protein 70 is associated to
imatinib resistance in chronic myeloid leukemia. Leukemia.
21:93–101. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gabai VL, Budagova KR and Sherman MY:
Increased expression of the major heat shock protein Hsp72 in human
prostate carcinoma cells is dispensable for their viability but
confers resistance to a variety of anticancer agents. Oncogene.
24:3328–3338. 2005. View Article : Google Scholar
|
|
42
|
Guo F, Rocha K, Bali P, et al: Abrogation
of heat shock protein 70 induction as a strategy to increase
antileukemia activity of heat shock protein 90 inhibitor
17-allylamino-demethoxy geldanamycin. Cancer Res. 65:10536–10544.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Håvik B and Bramham CR: Additive
viability-loss following hsp70/hsc70 double interference and
Hsp90 inhibition in two breast cancer cell lines. Oncol Rep.
17:1501–1510. 2007.
|
|
44
|
Gao T and Newton AC: The turn motif is a
phosphorylation switch that regulates the binding of Hsp70 to
protein kinase C. J Biol Chem. 277:31585–31592. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu H, Zhang T, Chen R, McConkey DJ, Ward
JF and Curley SA: Multiple kinase pathways involved in the
different de novo sensitivity of pancreatic cancer cell lines to
17-AAG. J Surg Res. 176:147–153. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pommier Y, Sordet O, Antony S, Hayward RL
and Kohn KW: Apoptosis defects and chemotherapy resistance:
molecular interaction maps and networks. Oncogene. 23:2934–2949.
2004. View Article : Google Scholar : PubMed/NCBI
|