Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignant tumors, and its incidence is increasing (1–3). HCC
is often diagnosed at advanced stages with local invasion and
remote metastasis, making surgical resection and liver
transplantation more difficult and less effective (4). Thus, studies are needed to identify
this type of invasive cancer cell, and the signaling molecules that
are specifically involved in tumor invasion.
Galectin-3 (Gal-3), a member of the
carbohydrate-binding protein family, plays an important and
multifaceted role in cancer pathogenesis (5–7). Bound
to oncogenic Ras proteins, Gal-3 activates V-raf-1 murine leukemia
viral oncogene homolog 1 (RAF1) and phosphatidylinositol 3-kinase
(PI3K), which in turn induces aberrant gene expression and specific
signaling cascades, resulting in the facilitation of tumor
transformation (7,8). Gal-3 has also been shown to modulate
tumor invasion and metastasis by binding to integrins or by
regulating their expression (9,10).
Additionally, Gal-3 secreted by tumors induces angiogenesis
(9,11,12).
These characteristics have made therapeutic targeting of Gal-3 an
attractive concept in cancer biology (13–18).
In HCC, Gal-3 overexpression is involved in tumor progression and
is related to prognosis (19).
However, the underlying molecular mechanism of Gal-3 in the
development of HCC remains unclear.
The urokinase plasminogen activator receptor (uPAR),
a multidomain glycoprotein tethered to the cell membrane with a
glycosylphosphotidylinositol (GPI) anchor, is elevated in many
human cancers, and is frequently associated with poor prognosis
(20–22). uPAR regulates proteolysis by binding
the extracellular protease urokinase-type plasminogen activator
(uPA) and also activates many intracellular signaling pathways via
interactions with membrane-bound integrins (23,24).
Coordination of extracellular matrix (ECM) proteolysis and cell
signaling by uPAR underlies its important function in cell
migration, proliferation and survival. These attributes make uPAR
an attractive therapeutic target in cancer treatment (25–27).
Previous studies have shown that induction of uPAR
in cancer is ERK-dependent (28–30).
In human hepatocarcinoma cells, ERK-dependent uPAR expression is
required for motility of tumor cells (31). Gal-3 also promotes cancer
progression by modulating the activity of ERK (32,33).
Therefore, it is possible that in HCC, Gal-3 regulates tumor
development via modulation of uPAR expression, as the
overexpression of Gal-3 and uPAR has been reported in HCC (19,34).
To investigate whether Gal-3 is related to uPAR in the development
of HCC, we knocked down the expression of Gal-3 in HepG2 cells and
assayed uPAR expression, and the proliferation, migration and
invasion of the cells.
Materials and methods
Cell culture
HepG2 and Huh7 cells were purchased from the Chinese
Academy of Sciences Cell Bank (Shanghai, China). Cells were
maintained at 37°C in Minimum Essential Medium (MEM) (Invitrogen,
USA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin
and 100 U/ml streptomycin under humidified conditions containing
95% air and 5% CO2.
siRNA transfection
siRNA was designed and synthesized by the Shanghai
GenePharma Co., Ltd. according to the galectin-3 gene sequence
(GenBank accession no. NM 002306.3) as listed in Table I. Transfection was carried out using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the
manufacturer’s instructions. The siRNA-transfected cells were
analyzed by RT-PCR and western blotting.
 | Table IsiRNA sequences. |
Table I
siRNA sequences.
| Name | Sense/antisense
siRNA (5′-3′) | Target |
|---|
| Gal-3-homo-422 | GCC ACU GAU UGU GCC
UUA UTT/AUA AGG CAC AAU CAG UGG CTT | 424–442 |
| Gal-3-homo-568 | CAC GCU UCA AUG AGA
ACA ATT/UUG UUC UCA UUG AAG CGU GTT | 570–588 |
| Gal-3-homo-746 | GUA CAA UCA UCG GGU
UAA ATT/UUU AAC CCG AUG AUU GUA CTT | 748–766 |
| Negative
control | UUC UCC GAA CGU GUC
ACG UTT/ACG UGA CAC GUU CGG AGA ATT | |
Reverse transcription polymerase-chain
reaction (RT-PCR)
Total RNAs were isolated with the TRIzol reagent
according to the manufacturer’s protocol (Invitrogen). cDNA was
then synthesized using the SuperScript First Strand Synthesis
System (Invitrogen) and amplified by polymerase chain reaction
(PCR). The primer sequences are listed in Table II. PCR conditions were as follows:
95°C for 3 min, followed by 28 cycles of 95°C for 30 sec, 55°C for
30 sec, and 72°C for 1 min, the final extension was at 72°C for 6
min. The PCR products were electrophoresed on 1% agarose.
| Table IIPrimers for RT-PCR. |
Table II
Primers for RT-PCR.
| Primers | Sequences
(5′-3′) |
|---|
| Gal-3-F |
ATGGCAGACAATTTTTCGCTCCA |
| Gal-3-R |
TATCATGGTATATGAAGCACTGG |
| uPAR-F |
TTACCGAGGTTGTGTGTGGG |
| uPAR-R |
GGGCATGTTGGCACATTGAG |
| GAPDH-F |
TGAACGGGAAGCTCACTGG |
| GAPDH-R |
TCCACCACCCTGTTGCTGTA |
Western blot analysis
All cells were harvested after the indicated control
and Gal-3 siRNA treatments. Protein was extracted in lysis buffer
(50 mmol/l Tris-HCl pH 7.5, 150 mmol/l NaCl, 1% NP-40, 0.5% sodium
deoxycholate, 0.1% SDS and protease inhibitors). Fifty micrograms
of protein was loaded on a 10% SDS-PAGE gel, followed by protein
separation and electroblotting onto a polyvinylidene difluoride
membrane. The membrane was labeled with the following primary
antibodies: anti-galectin-3 (Abcam, USA), goat anti-uPAR (Santa
Cruz Biotechnology, USA), anti-GAPDH antibody (Chemicon, USA),
anti-phospho-ERK, anti-ERK, anti-phospho-AKT and anti-AKT (Cell
Signaling Technology, USA). HRP-conjugated secondary antibodies
were incubated in 5% BSA in TBST buffer for 1.5 h at room
temperature. Immunoreactivity was detected using an enhanced
chemiluminescence detection system (Pierce, USA).
Colony-formation assay
Control and Gal-3 siRNA-treated HepG2 cells were
plated in duplicate on 6-well culture plates at a density of 3,000
cells/well. Culture medium was subsequently changed every 3 days.
After 2 weeks, the colonies were fixed and stained with 2% crystal
violet, and the number of colonies that consisted of more than 10
cells were counted.
Cell proliferation assay
Cell proliferation was measured by an MTT
tetrazolium assay. HepG2 cells (2.5×103 cells/well)
transfected with either control or Gal-3 siRNA were cultured in
96-well microtiter plates in a total volume of 100 μl/well for 3
days. Each day, 10 μl of MTT (5 mg/ml) in 100 μl of basic MEM per
well were added and incubated for 4 h. After removing MTT, 150 μl
of dimethyl sulfoxide (DMSO) was added and mixed vigorously.
Absorbance was measured at 490 nm using the Emax-precision
microtiter plate reader (Molecular Devices, USA).
In vitro migration and invasion
assays
Cell motility was measured using 48-well BioCoat
Cell Culture Inserts (BD Biosciences, USA). Fibronectin (5 mg/ml)
in serum-free medium was placed in each lower chamber, which was
separated from the upper chamber by a membrane with 8-μm pores. A
single-cell suspension of HepG2 cells (5×104) in
serum-free medium was placed in each upper chamber. After
incubation for 24 h at 37°C, the cells were fixed with methanol and
stained with 0.1% crystal violet. The cells on the upper surface of
the filter were wiped off with a cotton swab, and the number of
cells that migrated out to the lower surface of the membranes were
counted in 5 randomly selected fields. Invasion assays were
performed with Matrigel-coated chambers from the BioCoat Matrigel
Invasion Chamber kit (BD Biosciences) using the same method as
described above for the migration assays.
Wound healing assay
Cells were seeded in 96-well plates and allowed to
grow until 70% confluency. The cells were pretreated with mitomycin
C, which inhibits cell division, so that the difference in motility
was not affected by the difference in cell proliferation rates. The
cells were treated as above and wounding was performed by scraping
through the cell monolayer with a 10-μl pipette tip. After being
washed with PBS, images were captured immediately after scratching
for various periods of time in the same marked location of the
plate. All experiments were performed in triplicate.
Statistical analysis
Experiments were carried out at least in triplicate,
and the results were expressed as mean ± SD. The data were analyzed
using the Student’s t-test. Statistical significance was considered
at P<0.05.
Results
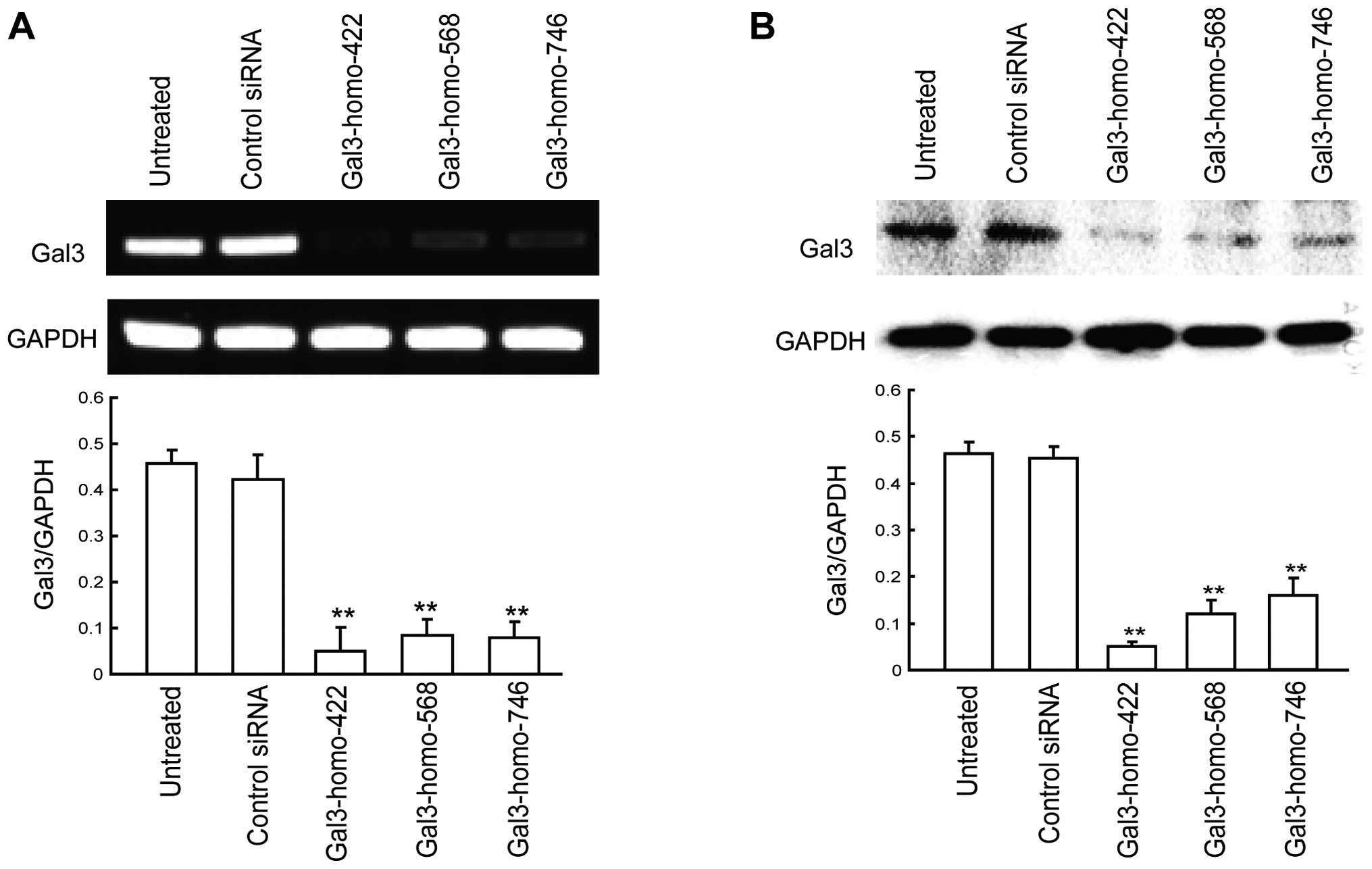
Downregulation of Gal-3 in HepG2 cells by
siRNA
To investigate the role of Gal-3 in HCC cells,
endogenous Gal-3 expression was inhibited by siRNA in HepG2 cells.
RT-PCR and western blot analysis were used to evaluate the ability
of different Gal-3 siRNAs to silence Gal-3 expression in
vitro. We chose three Gal-3 siRNA sequences (Gal-3-homo-422,
Gal-3-homo-568, and Gal-3-homo-746) based on previous research. The
suppression rate of Gal-3 mRNA expression was separately reached at
89.17, 61.61 and 82.56%, as measured by RT-PCR (Fig. 1A). The suppression rate of Gal-3
protein was separately reached at 89.26, 74.51 and 65.59% for each
Gal-3 siRNA, as measured by western blot analysis (Fig. 1B). The results indicated that
Gal-3-homo-422 was the most effective silencer. Thus,
Gal-3-homo-422 was chosen in the subsequence experiments for gene
knockdown. No differences were observed in regards to Gal-3 mRNA or
protein levels in HepG2 cells which were transfected with control
siRNA.
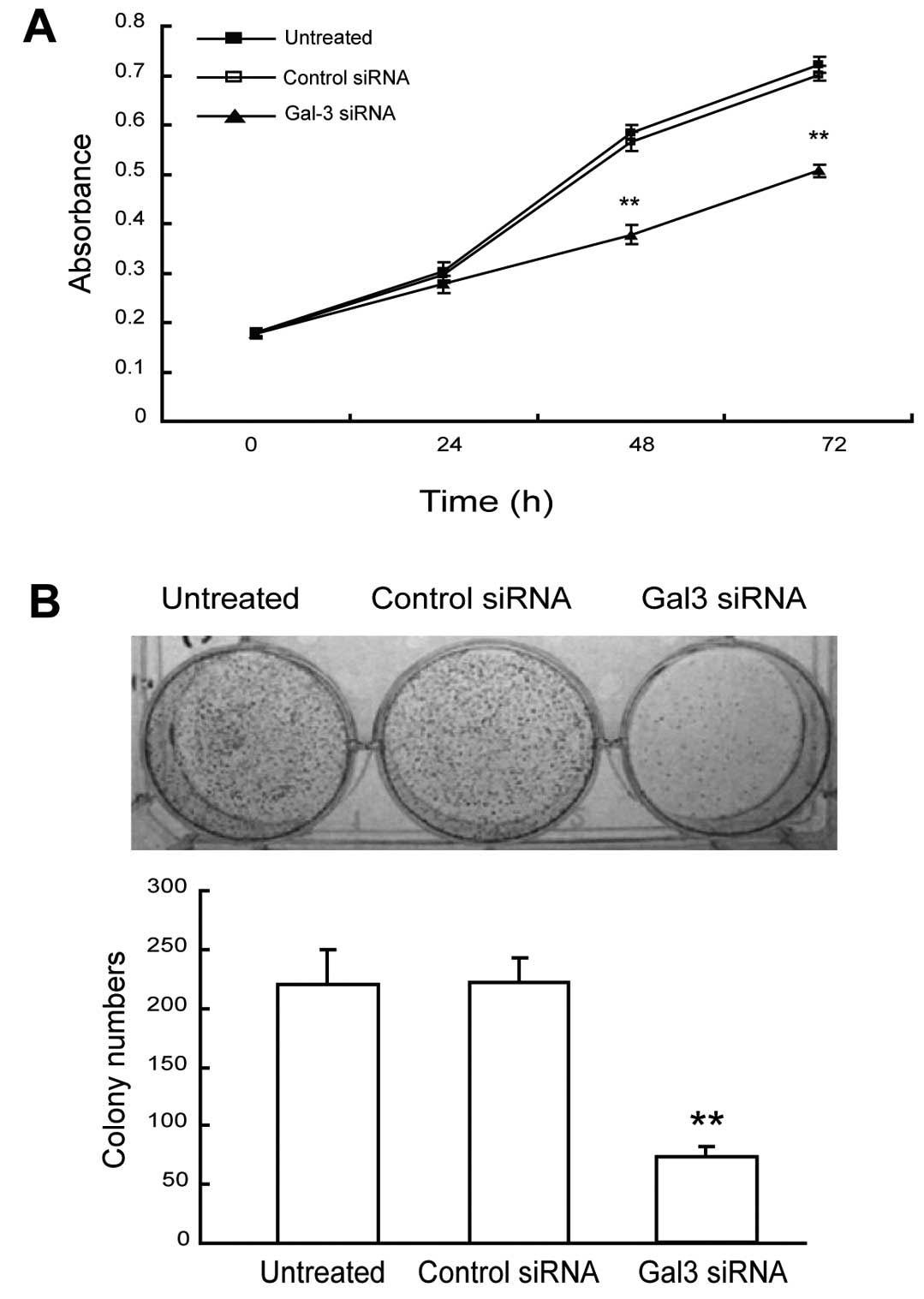
Downregulation of Gal-3 inhibits the
proliferation and colony formation of HepG2 cells
The roles of Gal-3 downregulation in cell
proliferation and tumorigenesis were evaluated by MTT and colony
formation assays, respectively. The HepG2 cells knocked down for
Gal-3 displayed significantly altered growth profiles when compared
to the control siRNA-transfected cells (Fig. 2A). While the growth curves of
Gal-3-knockdown cells reached a plateau at ~24 h following seeding,
the control siRNA transfectants displayed a steadily increasing
population level. Total cell number became significantly different
(P<0.05) between the two populations at 48 h. Additionally,
Gal-3-knockdown HepG2 cells displayed a significantly decreased
capacity to form colonies (P<0.05, Fig. 2B). While untransfected and control
siRNA-transfected HepG2 cells were able to form similar numbers of
colonies after 14 days (220±29.0 and 222±20.1, respectively),
Gal-3-knockdown HepG2 cells were only able to form an average of
73.7 (±8.5) colonies per well, representing a 67% reduction in
colony-forming capacity. These data indicate that Gal-3 plays an
important role in the proliferation and colony formation of HepG2
cells.
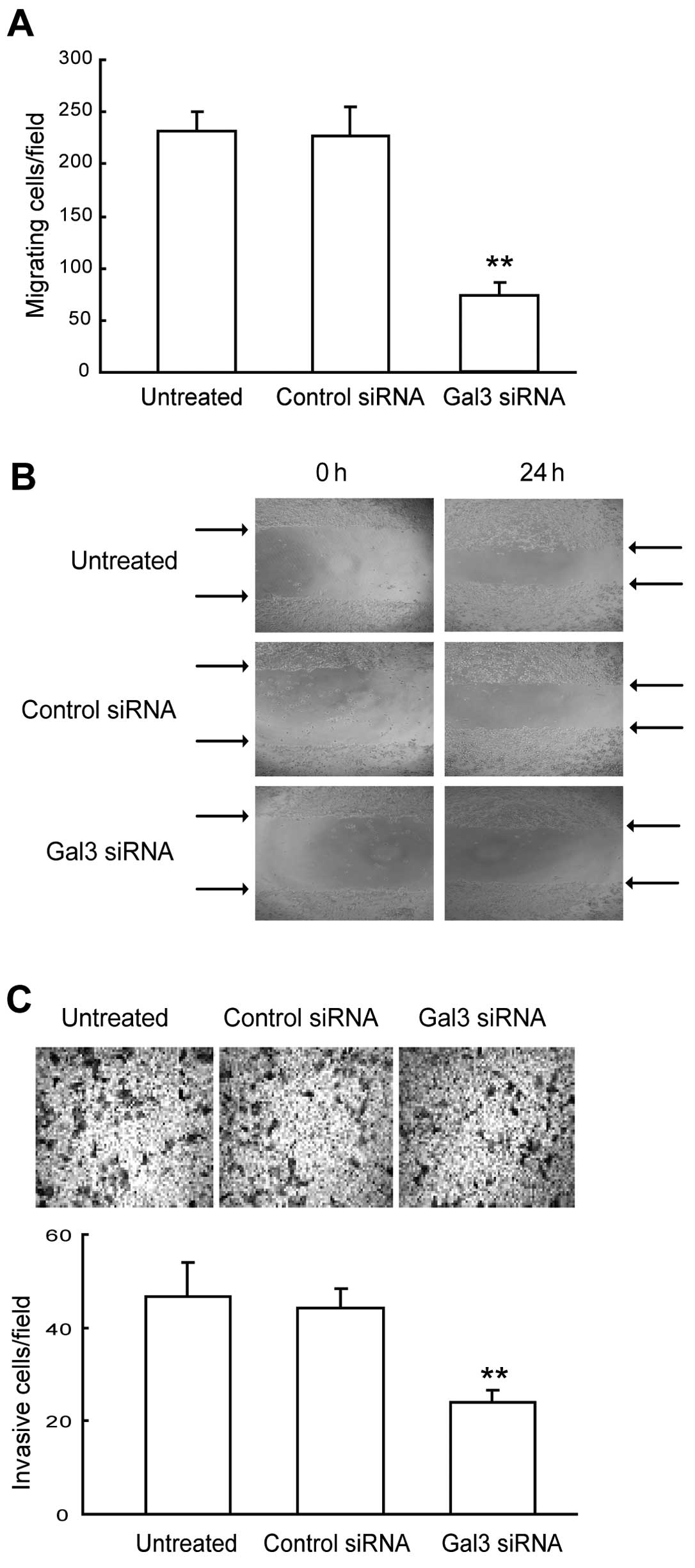
Downregulation of Gal-3 reduces cell
migration and invasion of HepG2 cells
To investigate the role of Gal-3 in HepG2 cell
migratory and invasive processes, we performed cell migration,
wound healing and invasion assays using HepG2 cells transfected
with control or Gal-3 siRNA. As shown in Fig. 3A, Gal-3 downregulation induced an
~2-fold decrease in the migration of HepG2 cells according to the
chamber-based assays (P<0.01). Moreover, wound-healing assays
confirmed the inhibitory effect of Gal-3 downregulation on cell
migration. We found that the time required for wound closure of the
Gal-3-knockdown HepG2 cells was significantly longer than the time
required for the corresponding control cells (Fig. 3B). In keeping with the migratory
patterns, Gal-3-knockdown cells displayed a significantly reduced
ability (P<0.01) to invade and migrate through a Matrigel
barrier relative to the control siRNA-treated cells (Fig. 3C). Collectively, these results
indicate that Gal-3 plays an important role in the migration and
invasion of HepG2 cells.
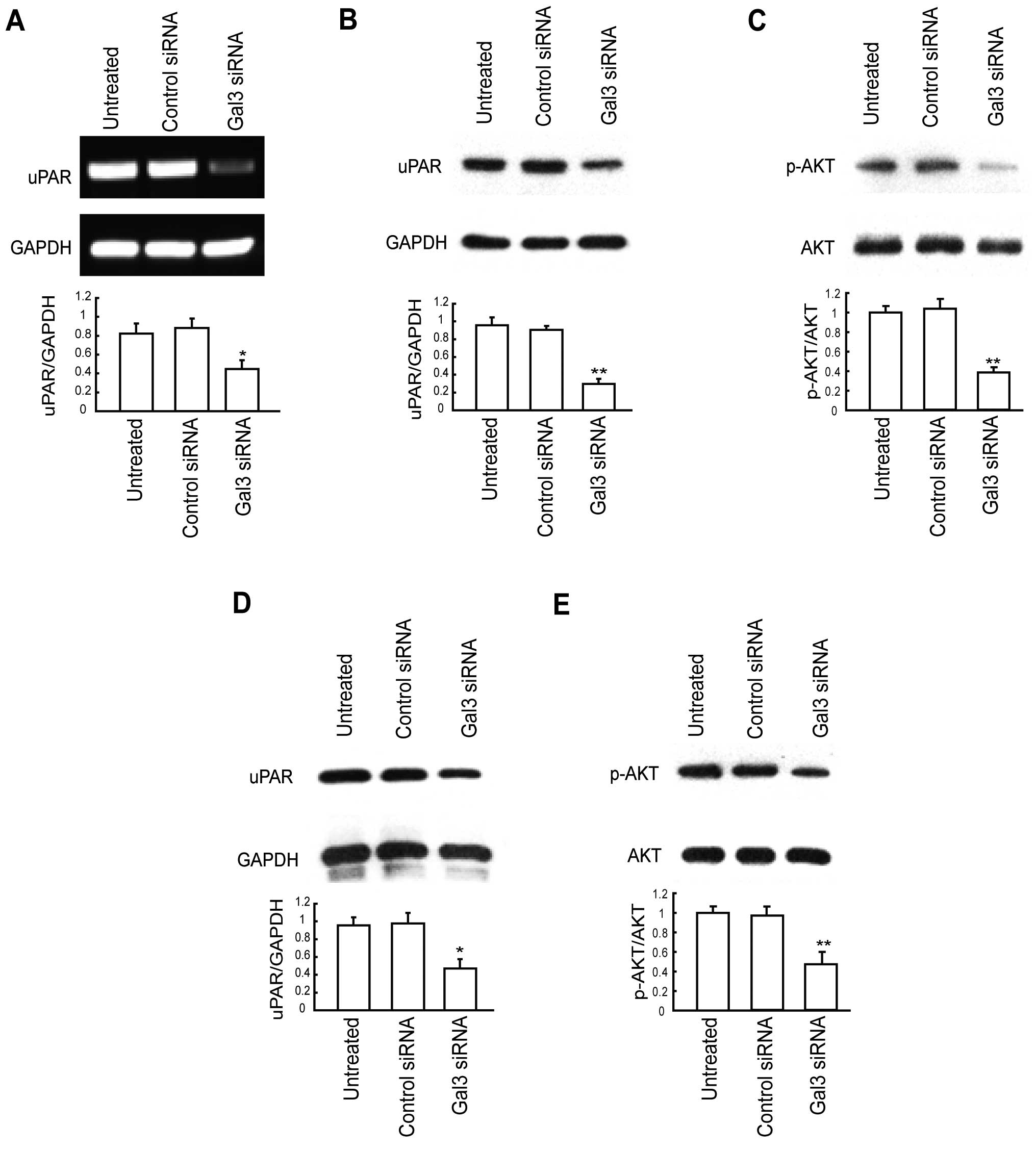
Downregulation of Gal-3 decreases uPAR
expression
To explore the underlying mechanism of reduced cell
proliferation, migration and invasion by Gal-3 downregulation,
expression of uPAR in the total cell lysate was examined by RT-PCR
and western blot analysis. As shown in Fig. 4A and B, mRNA and protein levels of
uPAR were significantly lower in the Gal-3 siRNA-treated HepG2
cells than levels in the corresponding controls. We also sought to
determine whether established downstream effectors of uPAR are
similarly disrupted. Fig. 4C shows
that the p-AKT levels were also greatly reduced in the
Gal-3-knockdown HepG2 cells, while total Akt levels remained
unchanged and comparable to those observed in the control cells.
Similar results were obtained in another HCC cell line, Huh7
(Fig. 4D and E). Taken together,
these results suggest that downregulation of Gal-3 decreases uPAR
expression and its downstream signaling.
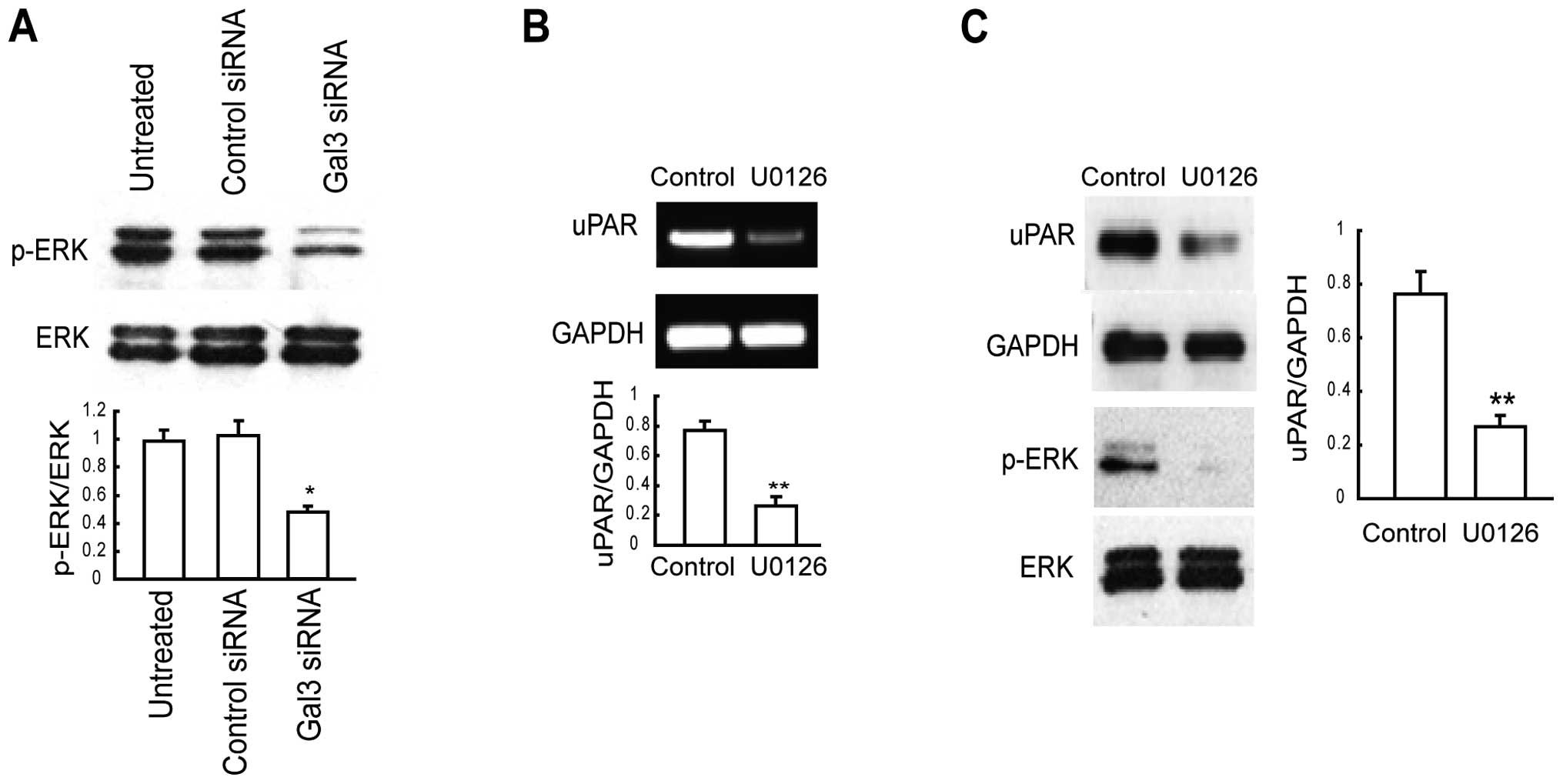
ERK is activated by Gal-3 and its
activation is correlated with uPAR expression
According to previous findings, Gal-3 regulates the
activation of ERK (32,33). In the present study, we determined
the activity of ERK in Gal-3-knockdown HepG2 cells. We found
markedly decreased phosphorylation of ERK in the Gal-3-knockdown
HepG2 cells (Fig. 5A). As ERK
activation is known to induce uPAR expression in a variety of
cancer cells (28–30), we determined whether uPAR expression
is positively regulated by MEK/ERK activity in HepG2 cells. We
treated HepG2 cells for 12 h with the synthetic MEK inhibitor U0126
(10 μM). RT-PCR data revealed decreased uPAR expression in
U0126-treated cells compared with that in the control cells
(Fig. 5B). uPAR expression was
reconfirmed at the protein level in the U0126-treated cells by
western blotting (Fig. 5C). Taken
together, these results indicate that downregulation of Gal-3 in
HepG2 cells reduced uPAR expression via the MEK/ERK pathway.
Discussion
Galectin-3 (Gal-3) is a member of the β-gal-binding
galectin family that exhibits pleiotropic biological functions.
Gal-3 is reported to be upregulated in many tumors and is involved
in several important processes, including cell-to-cell adhesion,
cell-to-extracellular matrix (ECM) interactions, cell growth,
differentiation, adhesion, migration, angiogenesis, malignant
transformation, apoptosis and cancer drug resistance (5,6,9,
10,12,35,36).
The expression of Gal-3 is detected in ~65% of HCC, and is highly
correlated with progression and prognosis of HCC (19). However, the actual biological
functions of Gal-3 in HCC have not yet been well described. The
present study was designed to address this issue using HepG2, a
hepatocellular liver carcinoma cell line that expresses high levels
of Gal-3.
In the present study, we knocked down expression of
Gal-3 in HCC cells with small interfering RNA (siRNA), and
demonstrated that siRNA targeting of Gal-3 in the HepG2 cell line
led to the efficient and specific inhibition of endogenous Gal-3
mRNA and protein in vitro. We found that downregulation of
Gal-3 in HepG2 cells contributed to reduced cell migration and cell
invasion. This suggests that Gal-3 is closely associated with the
metastatic events of HepG2 cells. Our data are consistent with
observations in the growth of many different types of human cancers
such as breast, colon, and brain tumors (32,37,38).
Previous research has implicated Gal-3 in the modulation of tumor
cell growth and tumorigenic phenotype of cancer cells (39,40).
We found that Gal-3-knockdown HepG2 cells displayed decreased cell
proliferation and colony formation efficiency. Coupled with the
observations in cell migration and cell invasion, we postulate that
Gal-3 expression is associated with migration, invasion,
proliferation and the tumorigenicity of HepG2 cells. Our finding is
different from a recent publication by Kobayashi et al
(41). They reported that transient
gene silencing of Gal-3 suppresses pancreatic cancer cell migration
and invasion, but failed to affect proliferation. We believe that
the observed discrepancy could be due to the different cell
systems.
Our findings also concern the key factors and
signaling pathways mediated by Gal-3, which are associated with HCC
progression. A number of recent studies have demonstrated a
correlation between Gal-3 and ERK in several cancer cell lines and
have implicated this association in cancer progression (33,42,43).
In the present study, we examined whether Gal-3 downregulation
affects ERK expression in HepG2 cells. We found that Gal-3
siRNA-transfected cells had markedly reduced phosphrylation of ERK
compared to their corresponding control cells, indicating that
Gal-3 regulates migration, invasion, proliferation and
tumorigenicity of HepG2 cells through the ERK pathway. Previous
research has shown that Gal-3 binds Ras and enhances Ras activity
and downstream signaling including phospshorylation of ERK, thereby
inducing cell proliferation and invasion in pancreatic cancer
(33). Therefore, based on our
study, we suggest that the decreased phosphorylation of ERK
observed in Gal-3 siRNA-transfected HepG2 cells may be due to
decreased Ras activity. Future experiments are needed to explore
this issue.
Recent studies have demonstrated that Gal-3
interacts with many signaling pathways, such as the Wnt/β-catenin
signaling pathway (9,33,44–47).
However, its interaction with the uPAR pathway has not yet been
reported. uPAR is a multifunctional protein which is involved in
several cellular processes such as cell proliferation, migration,
angiogenesis and invasion (48).
Studies have demonstrated that expression of uPAR is increased in
HCC, and is related to the invasiveness, metastasis and prognosis
of HCC (19,49). In this study, we found that Gal-3
silencing triggered the downregulation of uPAR in HepG2 and Huh7
cells. In addition, Gal-3 silencing significantly inhibited the
phosphorylation of AKT. The activation of PI3K/AKT signaling by
uPAR has been well documented in cancer research (23). It is known that the activated
PI3K/AKT pathway directly modulates cell growth and movement
behavior (23,50). Therefore, our results suggest that
Gal-3 modulates the PI3K/AKT pathway via uPAR, thereby affecting
cell proliferation, migration and invasion (23,50).
In addition, uPAR induction has been well documented
in several types of cancers by ERK (28–30). A
study by Bessard et al (31)
revealed that MEK/ERK-dependent uPAR expression is required for
motility in human hepatocarcinoma cells. We found that MEK
inhibitor significantly inhibited uPAR expression, indicating that
the induction of uPAR in HepG2 cells is MEK/ERK-dependent.
Therefore, we conclude that the decreased uPAR expression observed
in the present study might be due to decreased ERK activity induced
by Gal-3 downregulation.
In conclusion, our study revealed that Gal-3
regulates the level of uPAR in HCC cells. Gal-3 mediates cell
proliferation, migration and invasion by activating ERK, which
regulates uPAR expression. Understanding the underlying mechanisms
may provide new strategies for HCC treatment. RNA interference of
Gal-3 and uPAR expression could be considered as an effective
anti-HCC strategy.
Acknowledgements
This research was supported by grants from the
Health Department of Jiangsu (H200824), the Program for Advanced
Talents within Six Industries of Jiangsu to D.Z. (07-B-023), and
the Research Program funded by Nanjing Medical University to Z.H.
(2012NJMU088).
References
|
1
|
El-Serag HB, Mason AC and Key C: Trends in
survival of patients with hepatocellular carcinoma between 1977 and
1996 in the United States. Hepatology. 33:62–65. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar
|
|
4
|
Carr BI: Hepatocellular carcinoma: current
management and future trends. Gastroenterology. 127:S218–S224.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takenaka Y, Fukumori T and Raz A:
Galectin-3 and metastasis. Glycoconj J. 19:543–549. 2004.
View Article : Google Scholar
|
|
6
|
Radosavljevic G, Volarevic V, Jovanovic I,
et al: The roles of Galectin-3 in autoimmunity and tumor
progression. Immunol Res. 52:100–110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saussez S, Camby I, Toubeau G and Kiss R:
Galectins as modulators of tumor progression in head and neck
squamous cell carcinomas. Head Neck. 29:874–884. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McCubrey JA, Steelman LS, Abrams SL, et
al: Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in
malignant transformation and drug resistance. Adv Enzyme Regul.
46:249–279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wesley UV, Vemuganti R, Ayvaci ER and
Dempsey RJ: Galectin-3 enhances angiogenic and migratory potential
of microglial cells via modulation of integrin linked kinase
signaling. Brain Res. 1496:1–9. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Friedrichs J, Manninen A, Muller DJ and
Helenius J: Galectin-3 regulates integrin alpha2beta1-mediated
adhesion to collagen-I and -IV. J Biol Chem. 283:32264–32272. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Piccolo E, Tinari N, Semeraro D, et al:
LGALS3BP, lectin galactoside-binding soluble 3 binding protein,
induces vascular endothelial growth factor in human breast cancer
cells and promotes angiogenesis. J Mol Med. 91:83–94. 2013.
View Article : Google Scholar
|
|
12
|
Nangia-Makker P, Wang Y, Raz T, et al:
Cleavage of galectin-3 by matrix metalloproteases induces
angiogenesis in breast cancer. Int J Cancer. 127:2530–2541. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rossi ED, Straccia P, Palumbo M, et al:
Diagnostic and prognostic role of HBME-1, galectin-3, and
beta-catenin in poorly differentiated and anaplastic thyroid
carcinomas. Appl Immunohistochem Mol Morphol. 21:237–241.
2013.PubMed/NCBI
|
|
14
|
Yamaki S, Fujii T, Yajima R, et al:
Clinicopathological significance of decreased galectin-3 expression
and the long-term prognosis in patients with breast cancer. Surg
Today. 43:901–905. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cui W, Sang W, Zheng S, et al: Usefulness
of cytokeratin-19, galectin-3, and Hector Battifora mesothelial-1
in the diagnosis of benign and malignant thyroid nodules. Clin Lab.
58:673–680. 2012.PubMed/NCBI
|
|
16
|
Blanquart C, Gueugnon F, Nguyen JM, et al:
CCL2, galectin-3, and SMRP combination improves the diagnosis of
mesothelioma in pleural effusions. J Thorac Oncol. 7:883–889. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paunovic I, Isic T, Havelka M, et al:
Combined immunohistochemistry for thyroid peroxidase, galectin-3,
CK19 and HBME-1 in differential diagnosis of thyroid tumors. APMIS.
120:368–379. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chiu CG, Strugnell SS, Griffith OL, et al:
Diagnostic utility of galectin-3 in thyroid cancer. Am J Pathol.
176:2067–2081. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matsuda Y, Yamagiwa Y, Fukushima K, Ueno Y
and Shimosegawa T: Expression of galectin-3 involved in prognosis
of patients with hepatocellular carcinoma. Hepatol Res.
38:1098–1111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Baldini E, Sorrenti S, D’Armiento E, et
al: The urokinase plasminogen activating system in thyroid cancer:
clinical implications. G Chir. 33:305–310. 2012.PubMed/NCBI
|
|
21
|
Laerum OD, Ovrebo K, Skarstein A, et al:
Prognosis in adenocarcinomas of lower oesophagus,
gastro-oesophageal junction and cardia evaluated by
uPAR-immunohistochemistry. Int J Cancer. 131:558–569. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jacobsen B and Ploug M: The urokinase
receptor and its structural homologue C4.4A in human cancer:
expression, prognosis and pharmacological inhibition. Curr Med
Chem. 15:2559–2573. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smith HW and Marshall CJ: Regulation of
cell signalling by uPAR. Nat Rev Mol Cell Biol. 11:23–36. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dass K, Ahmad A, Azmi AS, Sarkar SH and
Sarkar FH: Evolving role of uPA/uPAR system in human cancers.
Cancer Treat Rev. 34:122–136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mani T, Wang F, Knabe WE, et al:
Small-molecule inhibition of the uPAR. uPA interaction: Synthesis,
biochemical, cellular, in vivo pharmacokinetics and efficacy
studies in breast cancer metastasis. Bioorg Med Chem. 21:2145–2155.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lebeau AM, Duriseti S, Murphy ST, et al:
Targeting uPAR with antagonistic recombinant human antibodies in
aggressive breast cancer. Cancer Res. 73:2070–2081. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soydinc HO, Duranyildiz D, Guney N, Derin
D and Yasasever V: Utility of serum and urine uPAR levels for
diagnosis of breast cancer. Asian Pac J Cancer Prev. 13:2887–2889.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park JS, Park JH, Khoi PN, Joo YE and Jung
YD: MSP-induced RON activation upregulates uPAR expression and cell
invasiveness via MAPK, AP-1 and NF-kappaB signals in gastric cancer
cells. Carcinogenesis. 32:175–181. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baek MK, Park JS, Park JH, et al:
Lithocholic acid upregulates uPAR and cell invasiveness via MAPK
and AP-1 signaling in colon cancer cells. Cancer Lett. 290:123–128.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoon SY, Lee YJ, Seo JH, et al: uPAR
expression under hypoxic conditions depends on iNOS modulated ERK
phosphorylation in the MDA-MB-231 breast carcinoma cell line. Cell
Res. 16:75–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bessard A, Fremin C, Ezan F, Coutant A and
Baffet G: MEK/ERK-dependent uPAR expression is required for
motility via phosphorylation of P70S6K in human hepatocarcinoma
cells. J Cell Physiol. 212:526–536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu KL, Huang EY, Jhu EW, et al:
Overexpression of galectin-3 enhances migration of colon cancer
cells related to activation of the K-Ras-Raf-Erk1/2 pathway. J
Gastroenterol. 48:350–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song S, Ji B, Ramachandran V, et al:
Overexpressed galectin-3 in pancreatic cancer induces cell
proliferation and invasion by binding Ras and activating Ras
signaling. PLoS One. 7:e426992012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Weng CJ, Tsai CM, Chen YC, et al:
Evaluation of the association of urokinase plasminogen activator
system gene polymorphisms with susceptibility and pathological
development of hepatocellular carcinoma. Ann Surg Oncol.
17:3394–3401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nangia-Makker P, Nakahara S, Hogan V and
Raz A: Galectin-3 in apoptosis, a novel therapeutic target. J
Bioenerg Biomembr. 39:79–84. 2007. View Article : Google Scholar
|
|
36
|
Fukumori T, Kanayama HO and Raz A: The
role of galectin-3 in cancer drug resistance. Drug Resist Updat.
10:101–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Song YK, Billiar TR and Lee YJ: Role of
galectin-3 in breast cancer metastasis: involvement of nitric
oxide. Am J Pathol. 160:1069–1075. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bresalier RS, Yan PS, Byrd JC, Lotan R and
Raz A: Expression of the endogenous galactose-binding protein
galectin-3 correlates with the malignant potential of tumors in the
central nervous system. Cancer. 80:776–787. 1997. View Article : Google Scholar
|
|
39
|
Cvejic DS, Savin SB, Petrovic IM, et al:
Galectin-3 expression in papillary thyroid carcinoma: relation to
histomorphologic growth pattern, lymph node metastasis,
extrathyroid invasion, and tumor size. Head Neck. 27:1049–1055.
2005. View Article : Google Scholar
|
|
40
|
John CM, Leffler H, Kahl-Knutsson B,
Svensson I and Jarvis GA: Truncated galectin-3 inhibits tumor
growth and metastasis in orthotopic nude mouse model of human
breast cancer. Clin Cancer Res. 9:2374–2383. 2003.PubMed/NCBI
|
|
41
|
Kobayashi T, Shimura T, Yajima T, et al:
Transient gene silencing of galectin-3 suppresses pancreatic cancer
cell migration and invasion through degradation of beta-catenin.
Int J Cancer. 129:2775–2786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Alge-Priglinger CS, Andre S, Schoeffl H,
et al: Negative regulation of RPE cell attachment by
carbohydrate-dependent cell surface binding of galectin-3 and
inhibition of the ERK-MAPK pathway. Biochimie. 93:477–488. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shalom-Feuerstein R, Cooks T, Raz A and
Kloog Y: Galectin-3 regulates a molecular switch from N-Ras to
K-Ras usage in human breast carcinoma cells. Cancer Res.
65:7292–7300. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Boscher C and Nabi IR: Galectin-3- and
phospho-caveolin-1-dependent outside-in integrin signaling mediates
the EGF motogenic response in mammary cancer cells. Mol Biol Cell.
24:2134–2145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Santiago-Gomez A, Barrasa JI, Olmo N, et
al: 4F2hc-silencing impairs tumorigenicity of HeLa cells via
modulation of galectin-3 and beta-catenin signaling, and MMP-2
expression. Biochim Biophys Acta. 1833:2045–2056. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sant’ana JM, Chammas R, Liu FT, et al:
Activation of the Wnt/beta-catenin signaling pathway during oral
carcinogenesis process is not influenced by the absence of
galectin-3 in mice. Anticancer Res. 31:2805–2811. 2011.PubMed/NCBI
|
|
47
|
Dakeng S, Duangmano S, Jiratchariyakul W,
et al: Inhibition of Wnt signaling by cucurbitacin B in breast
cancer cells: reduction of Wnt-associated proteins and reduced
translocation of galectin-3-mediated beta-catenin to the nucleus. J
Cell Biochem. 113:49–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kwaan HC and McMahon B: The role of
plasminogen-plasmin system in cancer. Cancer Treat Res. 148:43–66.
2009. View Article : Google Scholar
|
|
49
|
Zheng Q, Tang ZY, Xue Q, et al: Invasion
and metastasis of hepatocellular carcinoma in relation to
urokinase-type plasminogen activator, its receptor and inhibitor. J
Cancer Res Clin Oncol. 126:641–646. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Falasca M: PI3K/Akt signalling pathway
specific inhibitors: a novel strategy to sensitize cancer cells to
anti-cancer drugs. Curr Pharm Des. 16:1410–1416. 2010. View Article : Google Scholar : PubMed/NCBI
|