1. Introduction
Estrogens and their receptors (ERs) influence many
physiological processes in mammals and are also implicated in the
development or progression of numerous diseases, including various
types of cancer (breast, ovarian, colorectal, prostate and
endometrial), osteoporosis, neurodegenerative diseases,
cardiovascular disease, insulin resistance, lupus erythematosus,
endometriosis and obesity (1–3).
Breast cancer is the most frequently detected female neoplasm
worldwide. The ER is expressed by 60–70% of breast tumors (4,5) and
the mechanism has been studied; binding of estrogens to ER
stimulates proliferation of mammary cells, increasing the target
cell number within the tissue, and the increase in cell division
and DNA synthesis elevates the risk for replication errors, which
may result in the acquisition of detrimental mutations that disrupt
normal cellular processes such as apoptosis, cellular proliferation
or DNA repair. A second mechanism of estrogen metabolism leads to
the production of genotoxic by-products that could directly damage
DNA, again resulting in point mutations. There is evidence that
estrogen may act through both mechanisms to initiate and/or promote
mammary cancer (2). In the
treatment of breast cancer, tamoxifen is the most commonly used
anti-estrogen, but resistance remains an obstacle in the treatment
of hormone-dependent breast cancer. While up to one third of
patients are resistant to tamoxifen at the beginning of treatment,
the majority of patients who initially respond to tamoxifen will
later also become resistant. Some mechanisms may include changes in
the expression of ERα or ERβ, an alteration in
co-regulatory proteins and an alteration in transduction pathways,
altered expression of specific microRNA, and genetic polymorphisms
involved in tamoxifen metabolic activity. Due to the clinical
consequences of endocrine resistance, new treatment strategies are
arising to render the cells sensitive to tamoxifen. In the present
study, we reviewed the current knowledge on the mechanisms of
endocrine resistance in breast cancer cells.
2. Estrogen action and function
Estrogens perform their function by interacting with
ER. Two ER genes have been identified in mammals: ERα and ERβ,
which show similar DNA- and ligand-binding properties, but distinct
tissue distributions and functions (6–8). The
ER is a member of the nuclear receptor family of ligand-activated
transcription factors. After entering the cell, estrogen binds the
ER, which dissociates from heat shock proteins (HSPs) and undergoes
conformational changes, phosphorylation and dimerization before
binding to the estrogen response elements (ERE) upstream of
estrogen-dependent genes. This is referred to as the classical mode
of action. The majority of these genes in this model are involved
in cell proliferation and survival or in maintaining tissular
architecture (9,10). Ligand-bound ER can also modulate
gene expression through interaction of the receptor with Fos and
Jun at activator protein-1 (AP-1) binding sites (11) or with specificity protein 1 (SP-1)
sites in DNA, thereby functioning as a co-regulator (12). Co-regulators serve as a fine tuning
mechanism by increasing or reducing the receptor transcriptional
activity (13). Several
co-regulators have been implicated in cancer, most notably AIB1
(SRC-3), a gene that is amplified in a small percentage but
overexpressed in two thirds of all breast cancers. Overexpression
of this gene has been implicated in tamoxifen resistance (14,15).
In the ligand-independent mechanism, ER is
phosphorylated by membrane receptor tyrosine kinases including the
epidermal growth factor receptor (EGFR) (16), ErbB receptor 2 (ERBB2/HER2), and the
insulin-like growth factor receptor (IGF1-R) (17) or signaling molecules leading to
dimerization, DNA binding, and the activation of transcription
(18). Crosstalk between the growth
factor receptors (GFR) and ER pathways has been established through
several other mechanisms. Estrogen can increase the expression of
ligands such as transforming growth factor-α (TGFα) and IGF1
(12,19,20)
which can then activate the GFR pathway (17,20).
On the other hand, estrogen signaling downregulates the expression
of EGFR and ERBB2/HER2 while it induces the expression of IGF1-R
(21). Activation of the
phosphoinositide 3 kinase (PI3K)-kinase AKT (AKT) and the p42/44
mitogen-activated protein kinase (MAPK) pathways by these
receptors, in turn, downregulates the expression of ER and
progesterone receptors (PR) (22,23).
Thus, while receptor tyrosine kinases can activate the
transcriptional function of ER, they can also reduce estrogen
dependence by downregulating the expression of ER, perhaps
contributing to the relative resistance to endocrine therapies in
tumors amplified for ERBB2/HER2 (24). Studies also suggest that ER may work
by non-transcriptional mechanisms. Low levels of ER have been found
outside of the nucleus, in the membrane, cytoplasm or even in the
mitochondria, although the exact location for this receptor remains
controversial (25). Some of the
non-genomic action of estrogen appears to be too rapid for a
transcriptional effect to active GFR signaling, including the
PI3K/AKT and Ras/p42, 44 MAPK pathways. Thus, ER, through this
non-genomic activity, can alter the expression of genes normally
regulated by growth factors (25,26).
Finally, the stress kinase pathway via p38 and JNK can also
modulate ER function by phosphorylation of ER and its co-regulators
(27). The microenvironment and its
associated integrin signaling may exert similar activity (28). Thus, ER activity and signaling is
modulated by a variety of pathways, which could also contribute to
resistance to ER-targeted therapies, especially when the pathways
display aberrant activity in a cancer cell.
3. Mechanism of tamoxifen resistance
Several mechanisms of resistance to tamoxifen have
been studied and include the following: pharmacologic mechanisms;
loss or modification in ER expression; alterations in co-regulatory
proteins and regulation of different signaling pathways that
participate in the cellular process, such as survival,
proliferation, stress response, cell cycle, inhibition of apoptosis
regulated by the Bcl-2 family, autophagy, altered expression of
microRNAs and signaling pathways that regulate
epithelial-mesenchymal transition (EMT) in the tumor
microenvironment. Some proposed mechanisms responsible for
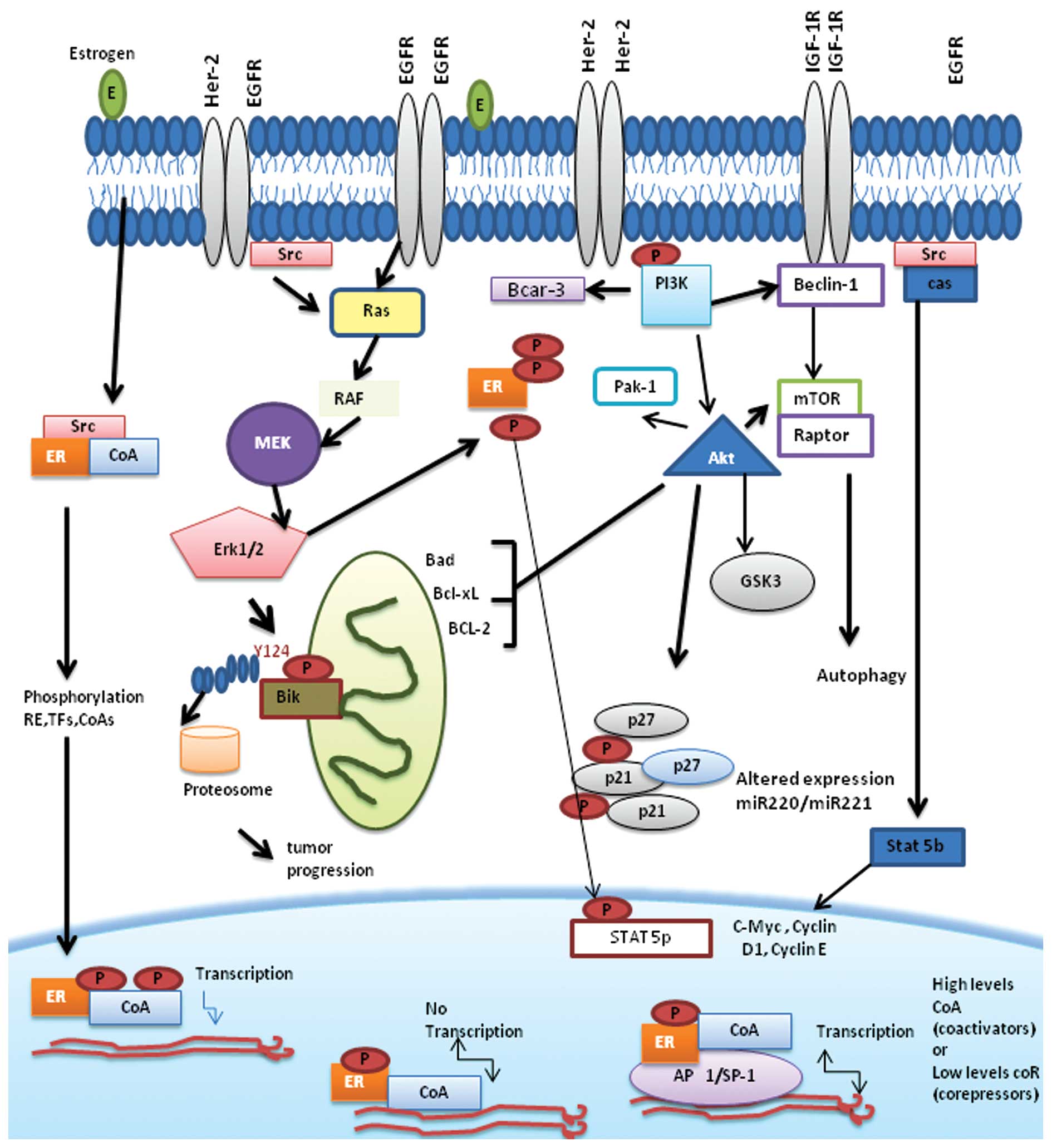
tamoxifen resistance are described below (Fig. 1).
Pharmacologic mechanisms
Cytochrome P450 2D6 (CYP2D6) is crucial in the
metabolism of tamoxifen to its active metabolite, endoxifen
(29). Retrospective clinical data
suggest that specific single nucleotide polymorphisms (SNPs) of
CYP2D6 can lead to null or reduced enzyme activity resulting in
poorer outcomes for patients with these when they are treated with
tamoxifen for hormone receptor (HR)-positive breast cancer
(30). Polymorphic CYP2D6 is the
key enzyme in this biotransformation, and recent mechanistic,
pharmacologic and clinical evidence suggests that genetic variants
and drug interaction with CYP2D6 inhibitors exert an influence on
the plasma concentrations of active tamoxifen metabolites and the
outcomes of tamoxifen-treated patients. In particular,
non-functional (poor metabolizer) and severely impaired
(intermediate metabolizer) CYP2D6 alleles are associated with
higher recurrence rates (31).
Loss or modification in ER
expression
Expression of ERα has long been considered a
determinant of a clinical response to endocrine or anti-estrogen
therapy. The ERα status of breast tumors provides prognostic
information and is the primary target for endocrine therapy.
Effective strategies to treat ER-positive breast cancer include
endocrine agents that compete with estrogen for binding to its
receptor, such as selective estrogen-receptor modulators (SERM) and
anti-estrogens or reducing the levels of circulating estrogens by
the administration of agents such as third-generation aromatase
inhibitors (32), which have been
shown to be more effective than tamoxifen in postmenopausal women
in neoadjuvant and adjuvant settings (33). Patients with tumors lacking ERα
generally do not benefit from tamoxifen therapy, although a
fraction of ERα-negative tumors appear to be sensitive to tamoxifen
(34). Several studies have
measured ERβ in breast cancer tumors and have sought to clarify the
relationship between ERβ and its role in response to endocrine
treatment; however, some of the results have been conflicting and
the majority have focused on ERβ as a resistance marker in
ERα-positive tumors (35,36). ERβ has been shown to bind tamoxifen
(37), and it has been suggested
that low levels of ERβ are associated with tamoxifen resistance
(35). Conversely, some studies
have shown that the expression of ERβ had a beneficial effect on
disease-free and overall survival in a group of 186
tamoxifen-treated tumors; however, the authors found no such
association in their set of 119 untreated patients, suggesting a
role for ERβ as a predictive marker for tamoxifen
sensitivity, but not as a prognostic marker (36).
Several mechanisms have been proposed to explain the
absence of ER expression. These mechanisms comprise epigenetic
changes, such as aberrant CpG island methylation of the ER promoter
and histone deacetylation, resulting in a compact nucleosome
structure that limits transcription (38–40).
Co-treatment with inhibitors of DNA methyltransferase-1 (DNMT-1),
such as 5-Aza-2-deoxycytidine (AZA), or inhibitors of histone
deacetylase (HDAC), like trichostatin A (TSA) and suberoylanilide
hydroxamic acid (SAHA), induce ER gene expression in ER(−) breast
cancer cells and restore sensitivity to anti-estrogen (38,41,42).
In ER(−) MDA-MB-231 cells, which overexpress EGFR, SAHA may not
only reactivate silenced ER, but it also simultaneously depletes
EGFR expression and abolishes EGF-initiated signaling pathways
including phosphorylated p21-activated kinase-1 (PAK1), p38MAPK and
AKT (43). In vitro and
in vivo studies showed that treatment with the histone
deacetylase inhibitor entinostat (ENT) increased the expression of
ERα and aromatase. Notably, ERα and aromatase
upregulation resulted in sensitization of breast cancer cells to
estrogen and letrozole (44).
Another mechanism that has been studied is mutations
in ER genes that may lead to a functionally negative ER phenotype
without the loss of ER expression as determined by protein-based
immunohistochemical assays. Using site-directed mutagenesis in the
AF-2 region of mouse ER, it is possible to reduce
estrogen-dependent transcriptional activation without significantly
affecting hormone and DNA binding (45). A mutation substituting aspartate for
tyrosine at position 351 has been identified in a
tamoxifen-stimulated cell line (46). More recently, substitution of
aspartate with glycine at amino acid 351 in the ER has been shown
in an experimental system to silence the agonist activity of
4-hydroxytamoxifen (47). However,
it has been reported that only 17–28% of patients with acquired
resistance to tamoxifen lose the expression of ERα (47).
Several kinase pathways have been associated with
tamoxifen resistance, including activation of the protein kinase A
(PKA) (17), MAPK (18) and PAK-1 signaling pathways (19). These kinases induce phosphorylation
of ERα or of its co-regulators. Some phospho-modification sites
have been studied in ERα that could contribute to an altered
response to tamoxifen and in which kinase pathways and upstream
activators are involved.
Serine residues S102, S104 and S106 at the
N-terminal AF-1 region of ERα are phosphorylated by glycogen
synthase kinase-3 (GSK-3) and by extracellular signal-regulated
kinases 1 and 2 (ERK1/2) and MEK1/2 pathways; these modifications
lead to ligand-independent transcription of ERα and to an agonistic
activity of tamoxifen (48,49). S102, a phosphorylation site
discovered by mass spectrometry, requires concurrent
phosphorylation of S104 (50). ERα
phosphorylation by GSK-3, which also targets S118, stabilizes ERα
without ligand and modulates ERα transcriptional activity upon
ligand binding. S104 and S106 can also be phosphorylated by the
CDK2/cyclin A complex (51). Cyclin
A has been reported as a predictive marker for tamoxifen resistance
in patients with breast cancer (52).
Serine 118 is one of the most reported
phosphorylation sites of ERα. It is targeted by a number of kinase
pathways: MAPK, GSK-3, IKKα, CDK7 and the mammalian target of
rapamycin (mTOR)-p70 S6 ribosomal kinase (p70S6K) and S118
phosphorylation by MAPK increases the binding of co-activator SRC3
and renders ERα hypersensitive to estradiol (53). Phosphorylated S118 decreases ERα
affinity for tamoxifen and reduces binding to DNA, when ERα is
tamoxifen bound (54). Upstream,
the RAS/MAPK pathway can be activated by IGF stimulation inducing
phosphorylation of ERα S118 and resulting in ERα activation and
enhanced response to estradiol (55).
Serine 282 resides in the hinge region and, similar
to S167, can be phosphorylated by CK2. Estradiol increases
phosphorylation of S282, stabilizes ER, and induces transcriptional
activity (50).
Serine 305 resides at the C-terminus of the hinge
region, which provides a center of rotation to the total
ERα. The region around Ser305 is a multifunctional domain
that binds to many co-regulatory proteins and is involved in the
regulation of the activity and stability of ERα (56,57).
Phosphorylation of Ser305 occurs by means of protein
kinase A (PKA) and is associated with resistance to tamoxifen in
patients.
Alterations in co-regulatory
proteins
The transcriptional regulatory activity of ER is
mainly mediated by the formation of complexes with co-activator or
co-repressor proteins. In general, co-activators bind the ER when
it is bound by estrogen, enhancing target gene transcription. When
an antagonist such as tamoxifen is bound to ER, co-repressors are
typically recruited, which results in repression of target gene
transcription. Under specific conditions, such as high ERBB2/HER2
activity, a tamoxifen-ER complex may also recruit co-activator
proteins, causing agonistic effects.
Altered expression of co-regulators may therefore
play a role in tamoxifen resistance (58,59).
It has been demonstrated in vitro that co-activator proteins
AIB1, PGC-1β and SRC1 enhance the agonistic activity of tamoxifen
(60,61). In patients receiving adjuvant
tamoxifen therapy, high levels of AIB1 alone or in combination with
high levels of ERBB2/HER2 are associated with shorter disease-free
survival in patients (62).
These findings support a role for the overexpression
of co-activators in tamoxifen resistance. However, high levels of
SRC1 were associated with favorable response to tamoxifen in
patients (61), which is not in
line with this hypothesis. The presence of other factors, such as
ERBB2/HER2, may play a role in this outcome (59). In addition, two studies showed that
low levels of the co-repressor protein NCOR1 predict poor response
to tamoxifen (63). These results
support the possibility that reductions in co-repressor activity
may also contribute to tamoxifen resistance.
Growth factor receptor signaling
pathways
Estrogen receptor may initiate rapid cellular
signaling via direct interaction with components of growth factor
signaling pathways. Several studies have found that overexpression
of EGFR or ERBB2/HER2 in ER-positive, anti-estrogen-sensitive
breast cancer confers resistance to this drug (3,64).
EGFR and HER2 are associated with reduced response to tamoxifen
that, in experimental systems, can actually stimulate their growth
(15,16).
Gefitinib, which inhibits ERBB2/HER2, improved the
antitumor effect of tamoxifen and delayed the acquisition of
resistance, but had no effect on estrogen-stimulated growth.
Phosphorylated levels of p42/44 and p38 MAPK (both downstream of
EGFR/HER2) were increased in tamoxifen-resistant tumors and were
suppressed by gefitinib. There was no apparent increase in
phosphorylated AKT, (also downstream of EGFR/HER in resistant
tumors, but it was nonetheless suppressed by gefitinib.
Phosphorylated IGF-1R, which can interact with both EGFR and
membrane ER, was elevated in the tamoxifen-resistant tumors
compared with the sensitive group. However, ER-regulated gene
products, including total IGF-IR itself and the PR, remained
suppressed even at the time of acquiring resistance. Tamoxifen’s
antagonism of classic ER genomic function was retained in these
resistant tumors and even in tumors that overexpress ERBB2/HER2
(MCF-7 HER2/18). In conclusion, EGFR/HER2 may mediate tamoxifen
resistance in ER-positive breast cancer despite continued
suppression of ER genomic function by tamoxifen. IGF-IR expression
remains dependent on ER but is activated in tamoxifen-resistant
tumors (65).
IGF-1R
The IGF-1R signaling pathway has important roles in
regulating energy metabolism, cellular proliferation and apoptosis.
IGF-1R is a receptor tyrosine kinase that exerts its biologic
effects through binding of the ligands IGF-I and IGF-II. Following
ligand binding and receptor activation, adaptor molecules are
recruited, leading to activation of downstream pathways, including
the RAS/MAPK and PI3K pathways (66,67).
The expression of ER is controlled by IGF in breast cancer cells
(68). Conversely, genomic and
non-genomic actions of ER can activate the mitogen-inducing signals
of the IGF pathway (69). Estrogen
signaling can also enhance IGF-1R signaling through transcriptional
upregulation of IGF1R, IRS-1 and IGF-II (70–72).
Reciprocally, IGF1R has been shown to phosphorylate and activate ER
in serine-167 through an S6-kinase mechanism (73). One mechanism by which IGF-1-treated
breast cancer cells may escape from tamoxifen-induced apoptosis is
via IGF-mediated activation of AKT and subsequent phosphorylation
of ER, leading to the ligand-independent activation of ER (74). Previous in vitro studies have
aimed to elucidate the role of IGF-1R in breast cancer resistance.
A gene that confers resistance to tamoxifen is for IGF binding
protein 5 (IGFBP5) (75). IGFBP5 is
a secreted protein that inhibits growth factor binding to IGF-1R
(76). In addition, IGFBP5 has
recently been shown to play a critical role in breast cancer
progression and metastasis (77).
Studies have demonstrated that knockdown of IGFBP5 resulted in
resistance to tamoxifen treatment in MCF-7 cells and in mice tumor
xenografts. IGFBP5 knockdown-induced resistance to tamoxifen
occurred potentially via altered IGF signaling and loss of ER
expression (55,68,78).
It has also been shown that inhibition of IGF-1R (by an anti-IGF-1R
antibody) (79) or by an IGF-1R
tyrosine kinase inhibitor (80)
reduces the growth of tamoxifen-resistant MCF-7 breast cancer
cells. Furthermore, it has been demonstrated in an in vivo
model that an IGF-1R monoclonal antibody enhances the antitumor
activity of tamoxifen in ER-receptor-positive breast cancer
xenografts (81).
In vitro study has shown that increased
IGF-IR signaling interferes with the action of the HER-2 antibody
trastuzumab in HER2 positive breast cancer cells (82). Unlike EGFR, several studies have
reported that tamoxifen continues to suppress the IGF-1R level,
even during the development of resistance (65).
Mechanism of resistance to tamoxifen
associated with signaling proteins p130Cas and c-Src
The focal adhesion adapter protein p130 (p130Cas)
also known as breast cancer anti-estrogen resistance 1, is involved
in different cellular process, including proliferation, survival,
cell adhesion and migration as an adapter or scaffolding protein
(83). High expression of p130Cas
is associated with resistance to tamoxifen and promotes the
proliferation and inhibition of apoptosis in MCF-7 cells and in
human tumors (84). Interactions
between c-Src and p130Cas result in activation of c-Src activity,
promotion of serum- and anchorage-independent growth and
enhancement of cellular migration; concomitantly Cas is
phosphorylated and activated by c-Src (83).
Two substrates of c-Src, tyrosine 845 (Tyr-845) on
the EGFR and signal transducer and activator of transcription 5b
(STAT 5b) (85), have been
implicated in p130Cas-dependent tamoxifen resistance (86). Pharmacologic inhibition of c-Src in
MCF-7 cells enhances the inhibitory effects of tamoxifen on cell
growth (86). It is clear that
p130Cas overexpression diminishes the antiproliferative and
pro-apoptotic effects of tamoxifen on breast cancer cells in
vitro, and data suggest that it accomplishes this via
activation of the p130Cas/c-Src/EGFR/STAT5 signaling pathway.
Whether these pathways are also clinically significant in
resistance to tamoxifen has not yet been established; however, a
large percentage of human breast tumors expresses high levels of
both c-Src and p130Cas, as well as elevated c-Src kinase activity,
and c-Src/Cas complexes can be isolated from a majority of breast
cancer cell lines (3). Taken
together, these data suggest that tamoxifen resistance may be
mediated in part through this mechanism involving c-Src and
p130Cas.
Breast cancer anti-estrogen resistance 3
(BCAR3) protein
BCAR3 is a member of the novel Src homology 2
(SH2)-containing protein family (87) that includes two other members,
Chat/SHEP1 and NSP1. These proteins share a common domain structure
consisting of an amino-terminal SH2 domain and a carboxyl-terminal
domain with sequence homology to the Cdc25-family of guanine
nucleotide exchange factors (GEF). Several studies have shown that
BCAR3 expression results in the activation of numerous small
GTPases, including Rap1, R-Ras, RalA, Cdc42 and Rac1 (88,89).
The carboxyl-terminal domain of BCAR3 has been shown to bind to the
carboxyl terminus of p130Cas, providing additional support for a
functional relationship between these proteins (88).
BCAR3 was discovered in a genetic screening for
genes associated with anti-estrogen resistance and cell motility
(90). It encodes a component of
intracellular signal transduction and interacts directly with
p130Cas. It has been demonstrated that breast cancer cells
transfected with BCAR3 to induce anti-estrogen resistance,
suggesting that upregulation of BCAR3 stimulates an alternative
growth path independent of hormone, both in the presence and
absence of anti-estrogens by activating PI3K, which induced Rac
activation (91).
PI3K and AKT pathway
The PI3K pathway is a key signal transduction system
that links oncogenes and multiple receptor classes to many
essential cellular functions, promotes anti-estrogen resistance and
is perhaps the most commonly activated signaling pathway in human
cancer (92–94). Studies have found that
overexpression of HER2 or FGF-1R, or loss of the inositol
polyphosphate phosphatase 4B (INPP4B), activate the PI3K pathway
and also confer anti-estrogen resistance in patients with
ER+ breast cancer (95).
PI3K is commonly activated in breast cancer cells by
growth factor receptor tyrosine kinases or G-protein-coupled
receptors. The signaling cascades triggered by PI3K, including
PDK1, AKT and SGK among others, promote cell growth and survival
(94). PI3K phosphorylates
phosphatidylinositol 4,5-bisphosphate (PIP2) to produce
phosphatidylinositol 3,4,5-trisphosphate (PIP3)
(96). In turn, PIP2
recruits several pleckstrin homology (PH) domain-containing
proteins to the plasma membrane such as the
serine/threonine-protein kinase PDK1 and AKT, which on activation
drive cell-cycle progression and survival. Negative regulation of
this pathway is conferred by the tumor suppressors phosphatase and
tensin homolog (PTEN) and INPP4B, which are lipid phosphatases and
dephosphorylate PIP3 and PIP2, respectively
(32,97).
AKT activates mTOR-containing complex 1 (TORC1),
which regulates protein synthesis. mTOR is also part of another
complex, TORC2, which lies upstream of AKT. In addition to its
pro-survival and growth-promoting roles, the PI3K pathway interacts
with ER directly and indirectly. ER phosphorylation at
Ser167 by AKT or p70S6K increases estrogen-induced,
tamoxifen-induced, and ligand-independent ER transcriptional
activity (74). Additionally, PI3K
and Ras promote c-Jun phosphorylation. c-Jun complexes with c-Fos
to form the AP-1 complex, which cooperates with ER transcription.
Other oncogenic kinase pathways (MAPK, protein kinase C) also
contribute to the modulation of ER and transcription cofactors
(98).
Stress-activated protein kinase/c-Jun
kinase pathway
Estrogens induce apoptosis by regulating the c-Jun
NH2-terminal kinases (JNKs). JNKs are stimulated by multiple
factors, including cytokines, DNA-damaging agents, and environment
stress, and are important in controlling apoptosis in the process
of cellular stress, increasing AP-1 transcriptional activity via
phosphorylation. AP-1 is a transcription factor heterodimer
composed of Jun and Fos family members and binds to DNA at AP-1
response elements. It has been reported that the development of
tamoxifen resistance in MCF-7 cells is accompanied by increased
AP-1 DNA binding (99).
These findings have been examined in a panel of 30
ER-positive primary human breast tumors with acquired tamoxifen
resistance, and these tumors also displayed a significant increase
in AP-1 DNA binding activity when compared with untreated control
tumor. In a tamoxifen-resistant xenograft model, increased
phosphorylated c-Jun and JNK levels accompanied the increase in
AP-1-dependent transcription following tamoxifen treatment, and the
conversion to a resistant phenotype was associated with an increase
in oxidative stress (OS) (100).
Cell cycle regulators associated with
resistance to tamoxifen
Cyclins, cyclin-dependent kinases (CDKs) and CDK
inhibitors are the major regulators of cell cycle progression
(3). Estrogen accelerates
progression from G1 to S phase, while tamoxifen inhibits cell cycle
progression by affecting these key cell cycle proteins (101). The expression and activity of
these proteins have been demonstrated to have the potential to
significantly impact tamoxifen sensitivity and resistance.
Cyclin D1 plays an important role in the regulation
of cell cycle, promoting progression through to G1-S phase. In
mammary epithelial cells, the expression of cyclin D1 is regulated
through the ER signaling (102).
Cyclin D1 is a direct transcriptional target of estrogen signaling;
thus, tamoxifen reduces cyclin D1 expression (103). In vitro sustained
expression of cyclin D1 is evident in breast cancer cells during
their acquisition of tamoxifen resistance, and downregulation of
cyclin D1 with siRNA restored the sensitivity of these cells to
tamoxifen (103). Cyclin D1 is one
co-regulator, known to interact with ERα and can potentiate its
transcriptional activity independently of estrogen and may not be
inhibited by tamoxifen (104).
Overexpression of cyclin D1 has been reported to result in a
conformational change in ERα that induces receptor activation in
the presence of tamoxifen, which in turn promotes growth of MCF-7
cells-indicating a change from antagonist to agonist (105,106). Patients with cyclin D1 negative
tumors show better relapse-free survival when they are treated with
tamoxifen (107), whereas multiple
clinical studies have demonstrated that overexpression of cyclin D1
is correlated with poor outcome on tamoxifen treatment (107,108).
Cyclin D1 inhibits Rb early in G1 phase, and the
transcription factor E2F strongly induces the expression of cyclin
E, which is associated with CDK2 to form an active complex that
promotes entry into S phase (108). Cyclin E2 expression has been
associated with poor outcome in ER-positive breast cancer (109), and cyclin E2 is included in genes
that predict disease progression in either tamoxifen-resistant
breast cancer or metastatic breast cancer, whereas cyclin E1 is
absent (110,111). Cyclin E1 overexpression can reduce
anti-estrogen sensitivity in vitro (112,113), but cyclin E2 has not been studied
in this context, although it is strongly estrogen regulated
(114). In addition, CDK2
activation is a possible mechanism of resistance to a CDK4
inhibitor that preferentially inhibits ER-positive breast cancer
cell lines and can overcome acquired resistance to tamoxifen
(115,116). We therefore considered cyclin E1
and E2 to be most important in anti-estrogen resistance in
ER-positive breast cancer and potential therapeutic targets in
endocrine-resistant disease.
Cell cycle progression upon ligand binding of ER has
been shown to be mediated by cyclin/cyclin-dependent kinase (CDK)
complexes and CDK inhibitors (117,118). p21 is a member of the Cip/Kip
family of CDK inhibitors and acts as a G1 checkpoint
protein, preventing cell cycle progression into S phase. p21
functions as a downstream effector of p53, and loss of p21
expression is seen in a high percentage of human breast cancers
(117–119). p21 and p27 are CDK inhibitors and
are negative regulators of cell cycle progression. These proteins
counteract the activities of cyclin D1 and E. Expression of the ER,
a good prognostic factor in breast cancer, is associated with
higher levels of both p21 and p27 proteins (120,121).
p27 induction in breast cancer cells by tamoxifen
induces quiescence and clinical data also support a role of these
CDK inhibitors in response to tamoxifen treatment. In premenopausal
women with early breast cancer, an increase in p27/KIP1 expression
was able to predict better relapse-free survival upon tamoxifen
combination treatment (122).
Studies using immortalized human breast epithelial
cells with somatic deletion of the p21 gene showed a growth
proliferative response to tamoxifen as absence of p21 enabled
cyclin-CDK complexes to aberrantly phosphorylate ER when bound to
tamoxifen, resulting in a growth-stimulatory phenotype. On the
other hand, p21 wild-type cells demonstrated growth inhibition upon
tamoxifen exposure (123).
Bcl-2 family of proteins and their effect
in resistance to tamoxifen
Deregulation of anti-apoptotic Bcl-2 family members,
including Bcl-2, Bcl-xL and MCL-l, has been implicated in the
progression of several different types of cancer. Studies have
shown that initial expression of Bcl-2 correlates with ER
expression, responsiveness to adjuvant hormonal therapy and
ultimately a favorable prognosis (124). Several studies have also suggested
that estrogen promotes resistance to chemotherapeutic drugs
tamoxifen and cisplatin by increasing the Bcl-2: Bax ratio
(196,214). Also, in vitro studies in MCF-7 cells have shown
that overexpression of HER-2 increases anti-apoptotic Bcl-2 and
Bcl-xL proteins, leading to suppression of tamoxifen-induced
apoptosis and ultimately tamoxifen resistance (50). Activation of the PI3K/Akt pathway
causes phosphorylation of Bad leading to modulation of cellular
apoptosis (125). Clinical studies
have shown that high levels of Bad expression, not phosho-Bad
levels, are associated with improved disease-free survival when
compared to tumors with low levels of Bad expression (125).
Another important member of the Bcl2 family is BIK,
which is inducible by estrogen-starvation and anti-estrogen
treatment and plays an important role in anti-estrogen-induced
apoptosis of breast cancer cells (126–128).
Our group has observed that Bik is a critical factor
for resistance to tamoxifen, and utilizing MCF-7 cells has
demonstrated that BIK mRNA and protein were strongly induced by
estrogen-starvation or anti-estrogen treatment (126,128). Conversely, knockdown of BIK by
siRNA significantly inhibited the apoptosis caused by tamoxifen
treatment, finding low expression of BAX, BAK and PUMA
pro-apoptotic proteins and high expression of some anti-apoptotic
proteins, such as BCL-2 and MCL-1 in BIK siRNA-transfected cells
after treatment with TAM (26,27).
These data demonstrated that Bik is an important factor in the
TAM-induced apoptosis process, which may regulate mitochondrial
integrity by modulation of pro- and anti-apoptotic proteins. Our
results showed that suppression of the BIK gene exhibited
anti-apoptotic effects in TAM-treated MCF-7 cells. These data may
be useful for future studies to establish the mechanisms of
regulation of TAM resistance in breast cancer. In women with this
neoplasm and with positive ER, it may be important to determine BIK
protein levels to define whether or not TAM is the appropriate
treatment (128). A possible
mechanism of resistance to apoptosis has been established and has
been described using mouse fibroblasts transformed with v-src as a
model. This group demonstrated that Src-dependent resistance to
cell death relies on Src ability to inhibit the mitochondrial
pathway of apoptosis by specifically increasing the degradation
rate of the BH3-only protein Bik by proteasome due to the
phosphorylation of Bik. This effect relies on the activation of the
Ras-Raf-Mek1/2-Erk1/2 pathway and on the phosphorylation of Bik on
Thr124, driving Bik ubiquitylation on Lys33 and subsequent
degradation by the proteasome. These results suggest that Bik could
be a rate-limiting factor for apoptosis induction of tumor cells
exhibiting deregulated Erk1/2 signaling, which may provide new
opportunities for cancer therapies (129).
Role for autophagy in anti-estrogen
resistance
Recent studies have demonstrated that autophagy
(also referred to as macroautophagy) is critical to the development
of anti-estrogen resistance (130). This pro-survival role for
autophagy in anti-estrogen-treated breast cancer cells is
consistent with one function of the autophagosomes, which is to
recycle metabolites from degraded cellular constituents to support
a basal level of cell maintenance under starvation conditions and
OS, thus rendering it essential for cellular viability.
The mammalian target of rapamycin (mTOR) kinase
represents the major negative regulator of autophagy in human
cells. Under physiological conditions, mTOR prevents autophagy by
maintaining the hyperphosphorylation of the proteins required for
the initiation of the autophagic cascade. Conversely, mTOR activity
is rapidly shut down under conditions of stress, which allows for
the rapid upregulation of autophagy (131). mTOR participates in multiple
signaling cascades that regulate cell growth, and especially in
those emanating from receptor tyrosine kinases (RTKs). In cancer
cells, constitutively active RTK and/or the production of autocrine
growth factors lead to the hyperphosphorylation of mTOR substrates.
Moreover, several tumors are characterized by activating mutations
of the key signal transducers connecting RTK to mTOR, including the
small GTPase Ras, phosphatidylinositol 3-kinase (PI3K), as well as
the Akt1 kinases and 3-phosphoinositide dependent protein kinase 1
(PDPK1) (132,133). The essential autophagy regulator
beclin 1 interacts with several cofactors (including Ambra1, Bif-1
and UVRAG) to activate the lipid kinase Vps34, which is required
for the initiation of the autophagic pathway (134).
Beclin 1 is a haploin-sufficient tumor suppressor
containing a BH3 domain that contributes to its interaction with
BCL-2, but the avidity of this interaction is quite weak compared
with the BH3 domains present in the BH3-only proteins typically
associated with apoptosis regulation (135). Moreover, BH3-containing Beclin 1
does not appear to antagonize the anti-apoptotic function of BCL-2
at either the ER or mitochondria (136). Thus, canonical BH3-only proteins
such as BAD have been shown to displace Beclin 1 from BCL-2
(137), leading to the proposal
that an analogous signaling cascade to that associated with the
release of BCL-2 antagonists by BH3-only proteins in the apoptosis
pathway also extends to autophagy (138). BIK is the founding member of the
BH3-only family proteins (139). A
number of reports have shown that ectopic overexpression of BIK
results in apoptotic cell death. However, BIK has also been
reported to cause non-apoptotic cell death in human malignant
glioma (140), melanoma cells
(141) and in samples of human
breast cancer (142), which could
be associated with a mechanism of autophagy.
Another important member in autophagy is Vps34,
which has a pro-survival function and increased Vps34 expression
levels, and tyrosine phosphorylation by pp60c-Scr contributes to
enhanced tumorigenic activity in breast cancer cells (143). In MCF-7 cells, knockdown of Vps34
by RNA interference reduced cytoprotective autophagy mediated by
BH3 domain and potentiated apoptosis induction (144). Lastly, Vps34 possesses a tumor
suppressor function in MCF-7 mouse xenograft tumors mediated
through distinct Beclin-1 binding and enhancement of
starvation-induced autophagy (145). These studies corroborate lines of
evidence demonstrating that the PI3K/AKT/mTOR pathway, implicated
in cell survival, contributes to anti-estrogen resistance (146). In contrast, our studies have
identified loss of Vps15 and Raptor as the candidate gene target
for mediating breast cancer tamoxifen resistance in vitro,
suggesting that Vps15- or Raptor mediated autophagy promotes cell
death induced by tamoxifen. In support of this suggestion,
inhibitors of PI3K, Akt and mTOR are in clinical trials and
inhibition of mTOR activity is thought to restore tamoxifen
sensitivity in breast cancer (147).
miRNA
Recent studies have also shown the critical role of
miRNAs in conferring drug resistance or responsiveness in cancer
(148,149). In breast cancer, the role of
miRNAs in tamoxifen resistance, through regulation of cell cycle
regulatory proteins, has been suggested. In particular, ectopic
expression of miR-101, miR-206 and miR-221/miR-222 was found to
render ER-positive MCF-7 cells resistant to tamoxifen (150–153). These miRNAs were also found to be
significantly increased in Her2-positive primary human breast
cancer tissues indicating an interrelationship between miR-221/222
expression and Her2 overexpression in primary breast tumors that
are generally resistant to tamoxifen therapy. In
tamoxifen-resistant breast cancer, it has been shown that
miRNA-221/222 play a critical role in the development of resistance
by targeting p27/kip1, a cell cycle inhibitor (153). When miRNA-221/222 downregulate
p27/kip1, p27/kip1 can no longer bind to the CDK2/cyclin-E complex,
allowing the cells to progress through cell cycle and facilitating
growth of cancerous cells, even when estrogen receptors are blocked
by tamoxifen (153).
miRNA-mediated targeting of ERα cofactors, which
influence agonistic and antagonistic effects of ligand binding, has
also been linked to impaired chemotherapeutic response. Indeed, one
of the major co-activators for ERα, AIB1, is a target for miR-17
and miR-106, which are found to be dysregulated in breast cancer
cells exhibiting drug resistance (154,155). The transcriptional co-repressor,
receptor interacting protein 140 (RIP140), was found to be targeted
by miR-346, which is downregulated in endocrine-resistant cell
lines (156), indicating a role
for miRNAs in regulating estrogen-responsive genes.
4. Signaling pathways that regulate EMT in
tumor microenvironment
The tubular EMT (TEMT)-related signaling associated
with endocrine resistance are WNT, nuclear factor-κβ (NFκβ), Notch,
keratinocyte growth factor (KGF), the platelet-derived growth
factor (PDGF)/Abl signaling pathway and enolase-1.
Wnt signaling pathway is an important developmental
pathway that is frequently dysregulated in human cancers including
breast cancer (157–160). Wnt signaling is important for cell
migration, invasion, adhesion and survival. Wnt ligands primarily
signal via membrane bound Frizzled receptors through a number of
different but interconnected signaling pathways, including the
β-catenin, Wnt/Ca2+, and planar-cell polarity pathways
(1,161). Wnt signaling is altered in nearly
one half of all breast cancers. Both upregulation of Wnt pathway
activators and downregulation of pathway inhibitors have been
identified in breast cancer. Previous studies have reported that
the activity of β-catenin is altered in acquired
tamoxifen-resistant (TamR) breast cancer cells compared to
endocrine-sensitive parental MCF7 cells, with an increase in
β-catenin-mediated gene transcription. These observations suggest
that deregulated Wnt signaling may play a role in acquired
tamoxifen resistance in breast cancer where it may act to promote
growth and the development of a more aggressive phenotype.
Keratinocyte growth factor/fibroblast growth
factor-7 (KGF/FGF-7) is a member of the FGF family, which interacts
with fibroblast growth factor receptors (FGFRs). KGF has a stromal
origin and appears to act specifically on epithelial cells in human
tissue and is therefore an exclusively paracrine growth factor in
human tissues (162,163). KGF stimulates normal human breast
and human breast cancer epithelial cell proliferation in a
dose-dependent manner. KGF may increase endocrine resistance via
decreasing ER, progesterone receptor (PR) and protein tyrosine
phosphatases γ (PTPγ). Results suggested that the presence of ER
and PR is a good prognostic factor and indicator of benefit from
endocrine therapy. Low levels of ER and PR in human breast cancers
have been associated with resistance to tamoxifen and increased
risk of breast cancer. The signal transduction of KGF/KGFR can
proceed via the Ras/MAPK and PI3K/Akt pathways (164,165). The findings on KGF stimulation of
cell proliferation and motility via Ras/MAPK suggest that KGF/KGFR
could potentially influence the development and progression of
breast cancer (166).
The Notch pathway has been reported to be involved
in drug resistance. Studies have demonstrated that Notch regulates
the formation of cancer stem cells (CSCs) and contributes to EMT
leading to the acquisition of the invasive phenotype, which are
associated with drug resistance (167,168).
The Notch pathway is implicated in both cell fate
in normal human mammary gland and regulation of CSCs in both ductal
carcinoma in situ and invasive carcinoma of the breast
(169–171). Binding of Notch ligand (Jagged or
Δ) to the Notch receptor cleaves its intracellular domain (NICD),
which translocates to the nucleus where it binds with co-activators
to induce transcription of its target genes and regulates migration
and invasion of breast cancer cells. Estradiol inhibits Notch
activity and affects Notch receptor cellular distribution.
Tamoxifen and raloxifene block this effect, reactivating the Notch
pathway. Pharmacologic inhibition of Notch activation with
γ-secretase inhibitors (GSI) was more effective in combination with
tamoxifen than tamoxifen alone (172). These data indicate that GSIs block
the proliferative effect of tamoxifen through the Notch pathway,
and at the same time allow tamoxifen to exert its antagonistic
effect. In a previous study, Rizzo et al found that
downregulation of Notch-1 by siRNA or GSI potentiated the effects
of tamoxifen in breast cancer cells. Moreover, GSI in combination
with tamoxifen caused regression of breast cancer cell growth in
mice. These data indicate that the combinations of tamoxifen and
Notch inhibitors may be effective in ERα (+) breast cancer, and
such a combination treatment could eliminate the emergence of
tamoxifen-resistance (172,173).
PDGF/Ableson (Abl) canonical signaling pathway has
also been associated with resistance (174). PDGF receptor (PDGFR) is classified
as a TRK whose activation is dependent on the binding of PDGF
resulting in stimulation of several intracellular pathways. PDGF
can promote tumor growth via autocrine stimulation of malignant
cells, overexpression or overactivation of PDGFRs, or by
stimulating tumor angiogenesis. Abl is an Src-like non-receptor
protein kinase that acts downstream of the PDGFR (175). Its corresponding gene c-ABL
is a proto-oncogene with multiple functions; it regulates a variety
of cellular activities, including cell migration, response to
oxidative stress and DNA damage, cell proliferation and survival.
Data revealed the PDGF/Abl canonical pathway as significantly
upregulated as early as one-week post-estrogen deprivation and
revealed that this could be the top adaptive pathway at the point
of full resistance. In studies of molecular changes occurring in
tumors in a cohort of patients treated with an aromatase inhibitor
in the neoadjuvant setting, it was found that PDGFR β expression
was significantly associated with poor antiproliferative response
to therapy (176).
α-enolase (ENO1) is a glycolytic enzyme that
converts 2-phosphoglycerate into phosphoenolpyruvate in glycolysis
and a multifunctional protein that plays a crucial role in a
variety of biological and pathophysiological processes (177). ENO1 may act as a stress protein
that promotes hypoxic tolerance in tumor cells by increasing
anaerobic metabolism (178). ENO1
may also function as a plasminogen receptor on the surface of a
variety of hematopoetic, epithelial, endothelial and cancerous
cells (179,180). Previously, several lines of
evidence suggested that ENO1 may contribute to tumor malignancy
(180). Upregulation of the
ENO1 gene has been observed in several highly tumorigenic or
metastatic cell lines (181,182) and its enzymatic activity in breast
cancer suggests a role of ENO1 in tumor progression. Increased
expression of enolase α in human breast cancer confers tamoxifen
resistance in humans. The treatment with 4-OH tamoxifen-induced
ENO-1 overexpression, which results in endocrine resistance in
human breast cancer (MCF-7) cells. ENO-1 was induced by 4-OH
tamoxifen treatment through transcriptional upregulation of ERα and
NFκB. The enhanced expression of ENO-1 exerts a negative regulatory
effect on c-Myc transcription that counteracts the 4-OH
tamoxifen-induced apoptosis of breast cancer cells. In contrast,
inhibition of ENO-1 transcriptional regulation, either by blocking
the NFκB signal pathway or by siRNA knockdown, significantly
sensitized human breast cancer cells to 4-OH tamoxifen-induced
cytotoxicity (183). These results
suggest that ENO-1 may facilitate resistance to 4-OH
tamoxifen-induced cell death and that the combined use of 4-OH
tamoxifen and an NFκB pathway inhibitor (PDTC) may provide a novel
approach to overcome tamoxifen resistance in breast cancer
(184).
5. Conclusion
Endocrine treatment of ER-positive breast cancer
with tamoxifen, and later with aromatase inhibitors and the
estrogen receptor antagonist fulvestrant, are the first
target-based therapeutic strategies in oncology. However, a
substantial proportion of patients, despite being ER and/or
PR-positive, are either primarily resistant or will develop
resistance during the course of their disease. Delineation of the
molecular mechanisms underlying the development of resistance will
allow the development of therapeutic strategies to improve the
efficiency of treatment and to prevent or overcome the drug
resistance.
Acknowledgements
This study was performed in partial fulfillment of
the requirements for the PhD degree in Biomedical Sciences of Rubí
Viedma-Rodríguez at the Universidad Nacional Autónoma de México,
with a doctoral fellowship provided by CONACyT-México (grant no.
207148). Supported, in part, by PAPIIT-DGAPA-UNAM, grants IN 230611
and IN 223014 to Luis Baiza-Gutman.
References
|
1
|
Kohn AD and Moon RT: Wnt and calcium
signaling: β-catenin-independent pathways. Cell Calcium.
38:439–446. 2005.
|
|
2
|
Deroo BJ and Korach KS: Estrogen receptors
and human disease. J Clin Invest. 116:561–570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Riggins RB, Schrecengost RS, Guerrero MS
and Bouton AH: Pathways to tamoxifen resistance. Cancer Lett.
256:1–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mueller SO and Korach KS: Estrogen
receptors and endocrine diseases: lessons from estrogen receptor
knockout mice. Curr Opin Pharmacol. 1:613–619. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Henderson BE and Feigelson HS: Hormonal
carcinogenesis. Carcinogenesis. 21:427–433. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuiper GG, Enmark E, Pelto-Huikko M,
Nilsson S and Gustafsson JA: Cloning of a novel receptor expressed
in rat prostate and ovary. Proc Natl Acad Sci USA. 93:5925–5930.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mosselman S, Polman J and Dijkema R: ERβ:
identification and characterization of a novel human estrogen
receptor. FEBS Lett. 392:49–53. 1996.
|
|
8
|
Dixon D, Couse JF and Korach KS:
Disruption of the estrogen receptor gene in mice. Toxicol Pathol.
25:518–520. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elliston JF, Fawell SE, Klein-Hitpass L,
et al: Mechanism of estrogen receptor-dependent transcription in a
cell-free system. Mol Cell Biol. 10:6607–6612. 1990.PubMed/NCBI
|
|
10
|
Frasor J, Danes JM, Komm B, Chang KC,
Lyttle CR and Katzenellenbogen BS: Profiling of estrogen up- and
down-regulated gene expression in human breast cancer cells:
insights into gene networks and pathways underlying estrogenic
control of proliferation and cell phenotype. Endocrinology.
144:4562–4574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Webb P, Lopez GN, Uht RM and Kushner PJ:
Tamoxifen activation of the estrogen receptor/AP-1 pathway:
potential origin for the cell-specific estrogen-like effects of
antiestrogens. Mol Endocrinol. 9:443–456. 1995.PubMed/NCBI
|
|
12
|
Kushner PJ, Agard DA, Greene GL, et al:
Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol.
74:311–317. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smith CL and O’Malley BW: Coregulator
function: a key to understanding tissue specificity of selective
receptor modulators. Endocr Rev. 25:45–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Osborne CK, Bardou V, Hopp TA, et al: Role
of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in
tamoxifen resistance in breast cancer. J Natl Cancer Inst.
95:353–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saville B, Wormke M, Wang F, et al:
Ligand-, cell-, and estrogen receptor subtype (α/β)-dependent
activation at GC-rich (Sp1) promoter elements. J Biol Chem.
275:5379–5387. 2000.
|
|
16
|
Kelloff GJ, Lippman SM, Dannenberg AJ, et
al: Progress in chemoprevention drug development: the promise of
molecular biomarkers for prevention of intraepithelial neoplasia
and cancer - a plan to move forward. Clin Cancer Res. 12:3661–3697.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schiff R, Massarweh SA, Shou J, Bharwani
L, Mohsin SK and Osborne CK: Cross-talk between estrogen receptor
and growth factor pathways as a molecular target for overcoming
endocrine resistance. Clin Cancer Res. 10:331S–336S. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Le Goff P, Montano MM, Schodin DJ and
Katzenellenbogen BS: Phosphorylation of the human estrogen
receptor. Identification of hormone-regulated sites and examination
of their influence on transcriptional activity. J Biol Chem.
269:4458–4466. 1994.PubMed/NCBI
|
|
19
|
Vyhlidal C, Samudio I, Kladde MP and Safe
S: Transcriptional activation of transforming growth factor α by
estradiol: requirement for both a GC-rich site and an estrogen
response element half-site. J Mol Endocrinol. 24:329–338. 2000.
|
|
20
|
Lee AV, Cui X and Oesterreich S:
Cross-talk among estrogen receptor, epidermal growth factor, and
insulin-like growth factor signaling in breast cancer. Clin Cancer
Res. 7(Suppl 12): S4429–S4435. 2001.PubMed/NCBI
|
|
21
|
Yarden RI, Wilson MA and Chrysogelos SA:
Estrogen suppression of EGFR expression in breast cancer cells: a
possible mechanism to modulate growth. J Cell Biochem. (Suppl 36):
232–246. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bayliss J, Hilger A, Vishnu P, Diehl K and
El-Ashry D: Reversal of the estrogen receptor negative phenotype in
breast cancer and restoration of antiestrogen response. Clin Cancer
Res. 13:7029–7036. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cui X, Zhang P, Deng W, et al:
Insulin-like growth factor-I inhibits progesterone receptor
expression in breast cancer cells via the phosphatidylinositol
3–kinase/Akt/mammalian target of rapamycin pathway: progesterone
receptor as a potential indicator of growth factor activity in
breast cancer. Mol Endocrinol. 17:575–588. 2003.PubMed/NCBI
|
|
24
|
Brinkman JA and El-Ashry D: ER
re-expression and re-sensitization to endocrine therapies in
ER-negative breast cancers. J Mammary Gland Biol Neoplasia.
14:67–78. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Levin ER and Pietras RJ: Estrogen
receptors outside the nucleus in breast cancer. Breast Cancer Res
Treat. 108:351–361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pedram A, Razandi M, Lubahn D, Liu J,
Vannan M and Levin ER: Estrogen inhibits cardiac hypertrophy: role
of estrogen receptor-β to inhibit calcineurin. Endocrinology.
149:3361–3369. 2008.PubMed/NCBI
|
|
27
|
Wu RC, Qin J, Yi P, et al: Selective
phosphorylations of the SRC-3/AIB1 coactivator integrate genomic
reponses to multiple cellular signaling pathways. Mol Cell.
15:937–949. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pontiggia O, Rodriguez V, Fabris V, et al:
Establishment of an in vitro estrogen-dependent mouse mammary tumor
model: a new tool to understand estrogen responsiveness and
development of tamoxifen resistance in the context of
stromal-epithelial interactions. Breast Cancer Res Treat.
116:247–255. 2009. View Article : Google Scholar
|
|
29
|
Machuca TN, Hsin MK, Ott HC, et al:
Injury-specific ex vivo treatment of the donor lung: pulmonary
thrombolysis followed by successful lung transplantation. Am J
Respir Crit Care Med. 188:878–880. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Goetz MP, Rae JM, Suman VJ, et al:
Pharmacogenetics of tamoxifen biotransformation is associated with
clinical outcomes of efficacy and hot flashes. J Clin Oncol.
23:9312–9318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stearns V and Rae JM: Pharmacogenetics and
breast cancer endocrine therapy: CYP2D6 as a predictive factor for
tamoxifen metabolism and drug response? Expert Rev Mol Med.
10:e342008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jaiswal BS, Janakiraman V, Kljavin NM, et
al: Somatic mutations in p85α promote tumorigenesis through class
IA PI3K activation. Cancer Cell. 16:463–474. 2009.
|
|
33
|
Madeira M, Mattar A, Logullo AF, Soares FA
and Gebrim LH: Estrogen receptor alpha/beta ratio and estrogen
receptor beta as predictors of endocrine therapy responsiveness-a
randomized neoadjuvant trial comparison between anastrozole and
tamoxifen for the treatment of postmenopausal breast cancer. BMC
Cancer. 13:4252013. View Article : Google Scholar
|
|
34
|
McGuire WL: Current status of estrogen
receptors in human breast cancer. Cancer. 36:638–644. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Esslimani-Sahla M, Simony-Lafontaine J,
Kramar A, et al: Estrogen receptor β (ERβ) level but not its ERβcx
variant helps to predict tamoxifen resistance in breast cancer.
Clin Cancer Res. 10:5769–5776. 2004.
|
|
36
|
Hopp TA, Weiss HL, Parra IS, Cui Y,
Osborne CK and Fuqua SA: Low levels of estrogen receptor β protein
predict resistance to tamoxifen therapy in breast cancer. Clin
Cancer Res. 10:7490–7499. 2004.
|
|
37
|
Kuiper GG, Lemmen JG, Carlsson B, et al:
Interaction of estrogenic chemicals and phytoestrogens with
estrogen receptor β. Endocrinology. 139:4252–4263. 1998.
|
|
38
|
Yang X, Phillips DL, Ferguson AT, Nelson
WG, Herman JG and Davidson NE: Synergistic activation of functional
estrogen receptor (ER)-α by DNA methyltransferase and histone
deacetylase inhibition in human ER-α-negative breast cancer cells.
Cancer Res. 61:7025–7029. 2001.
|
|
39
|
Parl FF: Multiple mechanisms of estrogen
receptor gene repression contribute to ER-negative breast cancer.
Pharmacogenomics J. 3:251–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ottaviano YL, Issa JP, Parl FF, Smith HS,
Baylin SB and Davidson NE: Methylation of the estrogen receptor
gene CpG island marks loss of estrogen receptor expression in human
breast cancer cells. Cancer Res. 54:2552–2555. 1994.PubMed/NCBI
|
|
41
|
Robertson KD, Ait-Si-Ali S, Yokochi T,
Wade PA, Jones PL and Wolffe AP: DNMT1 forms a complex with Rb,
E2F1 and HDAC1 and represses transcription from E2F-responsive
promoters. Nat Genet. 25:338–342. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fan J, Yin WJ, Lu JS, et al: ERα negative
breast cancer cells restore response to endocrine therapy by
combination treatment with both HDAC inhibitor and DNMT inhibitor.
J Cancer Res Clin Oncol. 134:883–890. 2008.
|
|
43
|
Zhou Q, Shaw PG and Davidson NE:
Inhibition of histone deacetylase suppresses EGF signaling pathways
by destabilizing EGFR mRNA in ER-negative human breast cancer
cells. Breast Cancer Res Treat. 117:443–451. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sabnis GJ, Goloubeva O, Chumsri S, Nguyen
N, Sukumar S and Brodie AM: Functional activation of the estrogen
receptor-α and aromatase by the HDAC inhibitor entinostat
sensitizes ER-negative tumors to letrozole. Cancer Res.
71:1893–1903. 2011.
|
|
45
|
Mahfoudi A, Roulet E, Dauvois S, Parker MG
and Wahli W: Specific mutations in the estrogen receptor change the
properties of antiestrogens to full agonists. Proc Natl Acad Sci
USA. 92:4206–4210. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wolf DM and Jordan VC: The estrogen
receptor from a tamoxifen stimulated MCF-7 tumor variant contains a
point mutation in the ligand binding domain. Breast Cancer Res
Treat. 31:129–138. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
MacGregor Schafer J, Liu H, Bentrem DJ,
Zapf JW and Jordan VC: Allosteric silencing of activating function
1 in the 4-hydroxytamoxifen estrogen receptor complex is induced by
substituting glycine for aspartate at amino acid 351. Cancer Res.
60:5097–5105. 2000.
|
|
48
|
Thomas RS, Sarwar N, Phoenix F, Coombes RC
and Ali S: Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is
important for estrogen receptor-α activity. J Mol Endocrinol.
40:173–184. 2008.PubMed/NCBI
|
|
49
|
Chen D, Washbrook E, Sarwar N, et al:
Phosphorylation of human estrogen receptor α at serine 118 by two
distinct signal transduction pathways revealed by
phosphorylation-specific antisera. Oncogene. 21:4921–4931.
2002.
|
|
50
|
Williams CC, Basu A, El-Gharbawy A,
Carrier LM, Smith CL and Rowan BG: Identification of four novel
phosphorylation sites in estrogen receptor α: impact on
receptor-dependent gene expression and phosphorylation by protein
kinase CK2. BMC Biochem. 10:362009.PubMed/NCBI
|
|
51
|
Rogatsky I, Trowbridge JM and Garabedian
MJ: Potentiation of human estrogen receptor α transcriptional
activation through phosphorylation of serines 104 and 106 by the
cyclin A-CDK2 complex. J Biol Chem. 274:22296–22302. 1999.
|
|
52
|
Michalides R, van Tinteren H, Balkenende
A, et al: Cyclin A is a prognostic indicator in early stage breast
cancer with and without tamoxifen treatment. Br J Cancer.
86:402–408. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vendrell JA, Bieche I, Desmetz C, et al:
Molecular changes associated with the agonist activity of
hydroxy-tamoxifen and the hyper-response to estradiol in
hydroxy-tamoxifen-resistant breast cancer cell lines. Endocr Relat
Cancer. 12:75–92. 2005. View Article : Google Scholar
|
|
54
|
Likhite VS, Stossi F, Kim K,
Katzenellenbogen BS and Katzenellenbogen JA: Kinase-specific
phosphorylation of the estrogen receptor changes receptor
interactions with ligand, deoxyribonucleic acid, and coregulators
associated with alterations in estrogen and tamoxifen activity. Mol
Endocrinol. 20:3120–3132. 2006. View Article : Google Scholar
|
|
55
|
Kato S, Endoh H, Masuhiro Y, et al:
Activation of the estrogen receptor through phosphorylation by
mitogen-activated protein kinase. Science. 270:1491–1494. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Barone I, Brusco L and Fuqua SA: Estrogen
receptor mutations and changes in downstream gene expression and
signaling. Clin Cancer Res. 16:2702–2708. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
de Leeuw R, Neefjes J and Michalides R: A
role for estrogen receptor phosphorylation in the resistance to
tamoxifen. Int J Breast Cancer. 2011:2324352011.PubMed/NCBI
|
|
58
|
Osborne CK and Schiff R: Estrogen-receptor
biology: continuing progress and therapeutic implications. J Clin
Oncol. 23:1616–1622. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Girault I, Bièche I and Lidereau R: Role
of estrogen receptor α transcriptional coregulators in tamoxifen
resistance in breast cancer. Maturitas. 54:342–351. 2006.
|
|
60
|
Webb P, Nguyen P, Shinsako J, et al:
Estrogen receptor activation function 1 works by binding p160
coactivator proteins. Mol Endocrinol. 12:1605–1618. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kressler D, Hock MB and Kralli A:
Coactivators PGC-1β and SRC-1 interact functionally to promote the
agonist activity of the selective estrogen receptor modulator
tamoxifen. J Biol Chem. 282:26897–26907. 2007.
|
|
62
|
Fuqua SA, Schiff R, Parra I, et al:
Estrogen receptor β protein in human breast cancer: correlation
with clinical tumor parameters. Cancer Res. 63:2434–2439. 2003.
|
|
63
|
Lavinsky RM, Jepsen K, Heinzel T, et al:
Diverse signaling pathways modulate nuclear receptor recruitment of
N-CoR and SMRT complexes. Proc Natl Acad Sci USA. 95:2920–2925.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kurokawa H, Lenferink AE, Simpson JF, et
al: Inhibition of HER2/neu (erbB-2) and
mitogen-activated protein kinases enhances tamoxifen action against
HER2-overexpressing, tamoxifen-resistant breast cancer cells.
Cancer Res. 60:5887–5894. 2000.
|
|
65
|
Massarweh S, Osborne CK, Creighton CJ, et
al: Tamoxifen resistance in breast tumors is driven by growth
factor receptor signaling with repression of classic estrogen
receptor genomic function. Cancer Res. 68:826–833. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fagan DH, Uselman RR, Sachdev D and Yee D:
Acquired resistance to tamoxifen is associated with loss of the
type I insulin-like growth factor receptor: implications for breast
cancer treatment. Cancer Res. 72:3372–3380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ciampolillo A, De Tullio C and Giorgino F:
The IGF-I/IGF-I receptor pathway: implications in the
pathophysiology of thyroid cancer. Curr Med Chem. 12:2881–2891.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lee AV, Weng CN, Jackson JG and Yee D:
Activation of estrogen receptor-mediated gene transcription by
IGF-I in human breast cancer cells. J Endocrinol. 152:39–47. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fagan DH and Yee D: Crosstalk between
IGF1R and estrogen receptor signaling in breast cancer. J Mammary
Gland Biol Neoplasia. 13:423–429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lee AV, Darbre P and King RJ: Processing
of insulin-like growth factor-II (IGF-II) by human breast cancer
cells. Mol Cell Endocrinol. 99:211–220. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Umayahara Y, Kawamori R, Watada H, et al:
Estrogen regulation of the insulin-like growth factor I gene
transcription involves an AP-1 enhancer. J Biol Chem.
269:16433–16442. 1994.PubMed/NCBI
|
|
72
|
Salerno M, Sisci D, Mauro L, Guvakova MA,
Ando S and Surmacz E: Insulin receptor substrate 1 is a target for
the pure antiestrogen ICI 182,780 in breast cancer cells. Int J
Cancer. 81:299–304. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Becker MA, Ibrahim YH, Cui X, Lee AV and
Yee D: The IGF pathway regulates ERα through a S6K1-dependent
mechanism in breast cancer cells. Mol Endocrinol. 25:516–528.
2011.
|
|
74
|
Campbell RA, Bhat-Nakshatri P, Patel NM,
Constantinidou D, Ali S and Nakshatri H: Phosphatidylinositol
3-kinase/AKT- mediated activation of estrogen receptor α: a new
model for anti-estrogen resistance. J Biol Chem. 276:9817–9824.
2001.
|
|
75
|
Ahn BY, Elwi AN, Lee B, et al: Genetic
screen identifies insulin-like growth factor binding protein 5 as a
modulator of tamoxifen resistance in breast cancer. Cancer Res.
70:3013–3019. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Beattie J, Allan GJ, Lochrie JD and Flint
DJ: Insulin-like growth factor-binding protein-5 (IGFBP-5): a
critical member of the IGF axis. Biochem J. 395:1–19. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Akkiprik M, Feng Y, Wang H, et al:
Multifunctional roles of insulin-like growth factor binding protein
5 in breast cancer. Breast Cancer Res. 10:2122008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bunone G, Briand PA, Miksicek RJ and
Picard D: Activation of the unliganded estrogen receptor by EGF
involves the MAP kinase pathway and direct phosphorylation. EMBO J.
15:2174–2183. 1996.PubMed/NCBI
|
|
79
|
Parisot JP, Hu XF, DeLuise M and Zalcberg
JR: Altered expression of the IGF-1 receptor in a
tamoxifen-resistant human breast cancer cell line. Br J Cancer.
79:693–700. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Knowlden JM, Hutcheson IR, Barrow D, Gee
JM and Nicholson RI: Insulin-like growth factor-I receptor
signaling in tamoxifen-resistant breast cancer: a supporting role
to the epidermal growth factor receptor. Endocrinology.
146:4609–4618. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Cohen BD, Baker DA, Soderstrom C, et al:
Combination therapy enhances the inhibition of tumor growth with
the fully human anti-type 1 insulin-like growth factor receptor
monoclonal antibody CP-751,871. Clin Cancer Res. 11:2063–2073.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lu Y, Zi X, Zhao Y, Mascarenhas D and
Pollak M: Insulin-like growth factor-I receptor signaling and
resistance to trastuzumab (Herceptin). J Natl Cancer Inst.
93:1852–1857. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Bouton AH, Riggins RB and Bruce-Staskal
PJ: Functions of the adapter protein Cas: signal convergence and
the determination of cellular responses. Oncogene. 20:6448–6458.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Defilippi P, Di Stefano P and Cabodi S:
p130Cas: a versatile scaffold in signaling networks. Trends Cell
Biol. 16:257–263. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lu Y, Mani S, Kandimalla ER, et al: The
Cockayne syndrome group B DNA repair protein as an anti-cancer
target. Int J Oncol. 19:1089–1097. 2001.PubMed/NCBI
|
|
86
|
Planas-Silva MD and Hamilton KN: Targeting
c-Src kinase enhances tamoxifen’s inhibitory effect on cell growth
by modulating expression of cell cycle and survival proteins.
Cancer Chemother Pharmacol. 60:535–543. 2007.PubMed/NCBI
|
|
87
|
Schuh NR, Guerrero MS, Schrecengost RS and
Bouton AH: BCAR3 regulates Src/p130 Cas association, Src kinase
activity, and breast cancer adhesion signaling. J Biol Chem.
285:2309–2317. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Gotoh T, Cai D, Tian X, Feig LA and Lerner
A: p130Cas regulates the activity of AND-34, a novel
Ral, Rap1, and R-Ras guanine nucleotide exchange factor. J Biol
Chem. 275:30118–30123. 2000.
|
|
89
|
Cai D, Iyer A, Felekkis KN, et al:
AND-34/BCAR3, a GDP exchange factor whose overexpression confers
antiestrogen resistance, activates Rac, PAK1, and the cyclin D1
promoter. Cancer Res. 63:6802–6808. 2003.PubMed/NCBI
|
|
90
|
van Agthoven T, van Agthoven TL, Dekker A,
van der Spek PJ, Vreede L and Dorssers LC: Identification of BCAR3
by a random search for genes involved in antiestrogen resistance of
human breast cancer cells. EMBO J. 17:2799–2808. 1998.PubMed/NCBI
|
|
91
|
Felekkis KN, Narsimhan RP, Near R, et al:
AND-34 activates phosphatidylinositol 3-kinase and induces
anti-estrogen resistance in a SH2 and GDP exchange factor-like
domain-dependent manner. Mol Cancer Res. 3:32–41. 2005.PubMed/NCBI
|
|
92
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Miller TW, Balko JM and Arteaga CL:
Phosphatidylinositol 3-kinase and antiestrogen resistance in breast
cancer. J Clin Oncol. 29:4452–4461. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Fox EM, Arteaga CL and Miller TW:
Abrogating endocrine resistance by targeting ERα and PI3K in breast
cancer. Front Oncol. 2:1452012.
|
|
95
|
Turner N, Pearson A, Sharpe R, et al:
FGFR1 amplification drives endocrine therapy resistance and
is a therapeutic target in breast cancer. Cancer Res. 70:2085–2094.
2010. View Article : Google Scholar
|
|
96
|
van der Kaay J, Cullen PJ and Downes CP:
Phosphatidylinositol(3,4,5)trisphosphate
(Ptdins(3,4,5)P3) mass measurement using a radioligand
displacement assay. Methods Mol Biol. 105:109–125. 1998.
|
|
97
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Massarweh S and Schiff R: Unraveling the
mechanisms of endocrine resistance in breast cancer: new
therapeutic opportunities. Clin Cancer Res. 13:1950–1954. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Lewis-Wambi JS and Jordan VC: Estrogen
regulation of apoptosis: how can one hormone stimulate and inhibit?
Breast Cancer Res. 11:2062009. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Schiff R, Reddy P, Ahotupa M, et al:
Oxidative stress and AP-1 activity in tamoxifen-resistant breast
tumors in vivo. J Natl Cancer Inst. 92:1926–1934. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Nair BC and Vadlamudi RK: Regulation of
hormonal therapy resistance by cell cycle machinery. Gene Ther Mol
Biol. 12:3952008.PubMed/NCBI
|
|
102
|
Butt AJ, McNeil CM, Musgrove EA and
Sutherland RL: Downstream targets of growth factor and oestrogen
signalling and endocrine resistance: the potential roles of c-Myc,
cyclin D1 and cyclin E. Endocr Relat Cancer. 12(Suppl 10): S47–S59.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Kilker RL, Hartl MW, Rutherford TM and
Planas-Silva MD: Cyclin D1 expression is dependent on estrogen
receptor function in tamoxifen-resistant breast cancer cells. J
Steroid Biochem Mol Biol. 92:63–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zwijsen RM, Wientjens E, Klompmaker R, van
der Sman J, Bernards R and Michalides RJ: CDK-independent
activation of estrogen receptor by cyclin D1. Cell. 88:405–415.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wilcken NR, Prall OW, Musgrove EA and
Sutherland RL: Inducible overexpression of cyclin D1 in breast
cancer cells reverses the growth-inhibitory effects of
antiestrogens. Clin Cancer Res. 3:849–854. 1997.PubMed/NCBI
|
|
106
|
Osborne CK and Schiff R: Growth factor
receptor cross-talk with estrogen receptor as a mechanism for
tamoxifen resistance in breast cancer. Breast. 12:362–367. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Rudas M, Lehnert M, Huynh A, et al: Cyclin
D1 expression in breast cancer patients receiving adjuvant
tamoxifen-based therapy. Clin Cancer Res. 14:1767–1774. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Stendahl M, Kronblad A, Rydén L, Emdin S,
Bengtsson NO and Landberg G: Cyclin D1 overexpression is a negative
predictive factor for tamoxifen response in postmenopausal breast
cancer patients. Br J Cancer. 90:1942–1948. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sieuwerts AM, Look MP, Meijer-van Gelder
ME, et al: Which cyclin E prevails as prognostic marker for breast
cancer? Results from a retrospective study involving 635 lymph
node-negative breast cancer patients. Clin Cancer Res.
12:3319–3328. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bosco EE, Wang Y, Xu H, et al: The
retinoblastoma tumor suppressor modifies the therapeutic response
of breast cancer. J Clin Invest. 117:218–228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Musgrove EA, Sergio CM, Anderson LR, et
al: Identification of downstream targets of estrogen and c-myc in
breast cancer cells. Adv Exp Med Biol. 617:445–451. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Dhillon NK and Mudryj M: Ectopic
expression of cyclin E in estrogen responsive cells abrogates
antiestrogen mediated growth arrest. Oncogene. 21:4626–4634. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Hui R, Finney GL, Carroll JS, Lee CS,
Musgrove EA and Sutherland RL: Constitutive overexpression of
cyclin D1 but not cyclin E confers acute resistance to
antiestrogens in T-47D breast cancer cells. Cancer Res.
62:6916–6923. 2002.PubMed/NCBI
|
|
114
|
Caldon CE, Sergio CM, Schütte J, et al:
Estrogen regulation of cyclin E2 requires cyclin D1 but not c-Myc.
Mol Cell Biol. 29:4623–4639. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Finn RS, Dering J, Conklin D, et al: PD
0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially
inhibits proliferation of luminal estrogen receptor-positive human
breast cancer cell lines in vitro. Breast Cancer Res. 11:R772009.
View Article : Google Scholar
|
|
116
|
Wang L, Wang J, Blaser BW, et al:
Pharmacologic inhibition of CDK4/6: mechanistic evidence for
selective activity or acquired resistance in acute myeloid
leukemia. Blood. 110:2075–2083. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Planas-Silva MD and Weinberg RA:
Estrogen-dependent cyclin E-cdk2 activation through p21
redistribution. Mol Cell Biol. 17:4059–4069. 1997.PubMed/NCBI
|
|
118
|
Cariou S, Donovan JC, Flanagan WM, Milic
A, Bhattacharya N and Slingerland JM: Down-regulation of
p21WAF1/CIP1 or p27Kip1 abrogates
antiestrogen-mediated cell cycle arrest in human breast cancer
cells. Proc Natl Acad Sci USA. 97:9042–9046. 2000.PubMed/NCBI
|
|
119
|
Bachman KE, Blair BG, Brenner K, et al:
p21WAF1/CIP1 mediates the growth response to TGF-β in
human epithelial cells. Cancer Biol Ther. 3:221–225. 2004.
|
|
120
|
Mokbel K: The evolving role of aromatase
inhibitors in breast cancer. Int J Clin Oncol. 7:279–283.
2002.PubMed/NCBI
|
|
121
|
Jordan VC: Tamoxifen (ICI46,474) as a
targeted therapy to treat and prevent breast cancer. Br J
Pharmacol. 147(Suppl 1): S269–S276. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Pohl G, Rudas M, Dietze O, et al: High
p27Kip1 expression predicts superior relapse-free and
overall survival for premenopausal women with early-stage breast
cancer receiving adjuvant treatment with tamoxifen plus goserelin.
J Clin Oncol. 21:3594–3600. 2003.
|
|
123
|
Abukhdeir AM and Park BH: P21 and p27:
roles in carcinogenesis and drug resistance. Expert Rev Mol Med.
10:e192008. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ellis PA, Smith IE, Detre S, et al:
Reduced apoptosis and proliferation and increased Bcl-2 in residual
breast cancer following preoperative chemotherapy. Breast Cancer
Res Treat. 48:107–116. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cannings E, Kirkegaard T, Tovey SM, Dunne
B, Cooke TG and Bartlett JM: Bad expression predicts outcome in
patients treated with tamoxifen. Breast Cancer Res Treat.
102:173–179. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Hur J, Chesnes J, Coser KR, et al: The Bik
BH3-only protein is induced in estrogen-starved and
antiestrogen-exposed breast cancer cells and provokes apoptosis.
Proc Natl Acad Sci USA. 101:2351–2356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Fu Y, Li J and Lee AS: GRP78/BiP inhibits
endoplasmic reticulum BIK and protects human breast cancer cells
against estrogen starvation-induced apoptosis. Cancer Res.
67:3734–3740. 2007. View Article : Google Scholar
|
|
128
|
Viedma-Rodriguez R, Baiza-Gutman LA,
García-Carrancá A, Moreno-Fierros L, Salamanca-Gómez F and
Arenas-Aranda D: Suppression of the death gene BIK is a critical
factor for resistance to tamoxifen in MCF-7 breast cancer cells.
Int J Oncol. 43:1777–1786. 2013.PubMed/NCBI
|
|
129
|
Lopez J, Hesling C, Prudent J, et al: Src
tyrosine kinase inhibits apoptosis through the Erk1/2-dependent
degradation of the death accelerator Bik. Cell Death Differ.
19:1459–1469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Schoenlein PV, Periyasamy-Thandavan S,
Samaddar JS, Jackson WH and Barrett JT: Autophagy facilitates the
progression of ERα-positive breast cancer cells to antiestrogen
resistance. Autophagy. 5:400–403. 2009.
|
|
131
|
Galluzzi L, Vicencio JM, Kepp O, Tasdemir
E, Maiuri MC and Kroemer G: To die or not to die: that is the
autophagic question. Curr Mol Med. 8:78–91. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Cully M, You H, Levine AJ and Mak TW:
Beyond PTEN mutations: the PI3K pathway as an integrator of
multiple inputs during tumorigenesis. Nat Rev Cancer. 6:184–192.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Degenhardt K, Mathew R, Beaudoin B, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Feng W, Huang S, Wu H and Zhang M:
Molecular basis of Bcl-xL’s target recognition versatility revealed
by the structure of Bcl-xL in complex with the BH3 domain of
Beclin-1. J Mol Biol. 372:223–235. 2007.
|
|
136
|
Ciechomska IA, Goemans GC, Skepper JN and
Tolkovsky AM: Bcl-2 complexed with Beclin-1 maintains full
anti-apoptotic function. Oncogene. 28:2128–2141. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Maiuri MC, Le Toumelin G, Criollo A, et
al: Functional and physical interaction between Bcl-XL
and a BH3-like domain in Beclin-1. EMBO J. 26:2527–2539. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Boyd JM, Gallo GJ, Elangovan B, et al:
Bik, a novel death-inducing protein shares a distinct sequence
motif with Bcl-2 family proteins and interacts with viral and
cellular survival-promoting proteins. Oncogene. 11:1921–1928.
1995.PubMed/NCBI
|
|
140
|
Naumann U, Bähr O, Wolburg H, et al:
Adenoviral expression of XIAP antisense RNA induces apoptosis in
glioma cells and suppresses the growth of xenografts in nude mice.
Gene Ther. 14:147–161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Oppermann M, Geilen CC, Fecker LF,
Gillissen B, Daniel PT and Eberle J: Caspase-independent induction
of apoptosis in human melanoma cells by the proapoptotic
Bcl-2-related protein Nbk/Bik. Oncogene. 24:7369–7380. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Garcia N, Salamanca F, Astudillo-de la
Vega H, et al: A molecular analysis by gene expression profiling
reveals Bik/NBK overexpression in sporadic breast tumor
samples of Mexican females. BMC Cancer. 5:932005. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Hirsch DS, Shen Y, Dokmanovic M and Wu WJ:
pp60c-Src phosphorylates and activates vacuolar protein
sorting 34 to mediate cellular transformation. Cancer Res.
70:5974–5983. 2010.
|
|
144
|
Gao P, Bauvy C, Souquère S, et al: The
Bcl-2 homology domain 3 mimetic gossypol induces both Beclin
1-dependent and Beclin 1-independent cytoprotective autophagy in
cancer cells. J Biol Chem. 285:25570–25581. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Furuya N, Yu J, Byfield M, Pattingre S and
Levine B: The evolutionarily conserved domain of Beclin 1 is
required for Vps34 binding, autophagy and tumor suppressor
function. Autophagy. 1:46–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Dancey J: mTOR signaling and drug
development in cancer. Nat Rev Clin Oncol. 7:209–219. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Ciafrè SA, Galardi S, Mangiola A, et al:
Extensive modulation of a set of microRNAs in primary glioblastoma.
Biochem Biophys Res Commun. 334:1351–1358. 2005.PubMed/NCBI
|
|
149
|
Pallante P, Visone R, Ferracin M, et al:
MicroRNA deregulation in human thyroid papillary carcinomas. Endocr
Relat Cancer. 13:497–508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Kondo N, Toyama T, Sugiura H, Fujii Y and
Yamashita H: miR-206 expression is down-regulated in estrogen
receptor α-positive human breast cancer. Cancer Res. 68:5004–5008.
2008.
|
|
151
|
Rao X, Di Leva G, Li M, et al:
MicroRNA-221/222 confers breast cancer fulvestrant resistance by
regulating multiple signaling pathways. Oncogene. 30:1082–1097.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Sachdeva M, Wu H, Ru P, Hwang L, Trieu V
and Mo YY: MicroRNA-101-mediated Akt activation and
estrogen-independent growth. Oncogene. 30:822–831. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Miller TE, Ghoshal K, Ramaswamy B, et al:
MicroRNA-221/222 confers tamoxifen resistance in breast cancer by
targeting p27Kip1. J Biol Chem. 283:29897–29903. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Kovalchuk O, Filkowski J, Meservy J, et
al: Involvement of microRNA-451 in resistance of the MCF-7 breast
cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther.
7:2152–2159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Liang Z, Wu H, Xia J, et al: Involvement
of miR-326 in chemotherapy resistance of breast cancer through
modulating expression of multidrug resistance-associated protein 1.
Biochem Pharmacol. 79:817–824. 2010. View Article : Google Scholar
|
|
156
|
Xin F, Li M, Balch C, et al: Computational
analysis of microRNA profiles and their target genes suggests
significant involvement in breast cancer antiestrogen resistance.
Bioinformatics. 25:430–434. 2009. View Article : Google Scholar
|
|
157
|
Guardavaccaro D and Clevers H:
Wnt/β-catenin and MAPK signaling: allies and enemies in different
battlefields. Sci Signal. 5:pe152012.
|
|
158
|
Gabrovska PN, Smith RA, Tiang T, Weinstein
SR, Haupt LM and Griffiths LR: Development of an eight gene
expression profile implicating human breast tumours of all grade.
Mol Biol Rep. 39:3879–3892. 2012. View Article : Google Scholar
|
|
159
|
Khramtsov AI, Khramtsova GF, Tretiakova M,
Huo D, Olopade OI and Goss KH: Wnt/β-catenin pathway activation is
enriched in basal-like breast cancers and predicts poor outcome. Am
J Pathol. 176:2911–2920. 2010.
|
|
160
|
Loh YN, Hedditch EL, Baker LA, Jary E,
Ward RL and Ford CE: The Wnt signalling pathway is upregulated in
an in vitro model of acquired tamoxifen resistant breast cancer.
BMC Cancer. 13:1742013. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Katoh M: Comparative genomics on
Wnt3-Wnt9b gene cluster. Int J Mol Med. 15:743–747. 2005.PubMed/NCBI
|
|
162
|
Rubin JS, Bottaro DP, Chedid M, et al:
Keratinocyte growth factor. Cell Biol Int. 19:399–411. 1995.
View Article : Google Scholar
|
|
163
|
Rubin JS, Bottaro DP, Chedid M, et al:
Keratinocyte growth factor as a cytokine that mediates
mesenchymal-epithelial interaction. EXS. 74:191–214.
1995.PubMed/NCBI
|
|
164
|
Kumar V, Green S, Staub A and Chambon P:
Localisation of the oestradiol-binding and putative DNA-binding
domains of the human oestrogen receptor. EMBO J. 5:2231–2236.
1986.PubMed/NCBI
|
|
165
|
Berry M, Metzger D and Chambon P: Role of
the two activating domains of the oestrogen receptor in the
cell-type and promoter-context dependent agonistic activity of the
anti-oestrogen 4-hydroxytamoxifen. EMBO J. 9:2811–2818. 1990.
|
|
166
|
Mader S, Chambon P and White JH: Defining
a minimal estrogen receptor DNA binding domain. Nucleic Acids Res.
21:1125–1132. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Wang Z, Li Y, Banerjee S and Sarkar FH:
Emerging role of Notch in stem cells and cancer. Cancer Lett.
279:8–12. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Shi TP, Xu H, Wei JF, et al: Association
of low expression of notch-1 and jagged-1 in human papillary
bladder cancer and shorter survival. J Urol. 180:361–366. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Al Saleh S, Sharaf LH and Luqmani YA:
Signalling pathways involved in endocrine resistance in breast
cancer and associations with epithelial to mesenchymal transition
(Review). Int J Oncol. 38:1197–1217. 2011.PubMed/NCBI
|
|
170
|
Dontu G, Jackson KW, McNicholas E,
Kawamura MJ, Abdallah WM and Wicha MS: Role of Notch signaling in
cell-fate determination of human mammary stem/progenitor cells.
Breast Cancer Res. 6:R605–R615. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
171
|
Stylianou S, Clarke RB and Brennan K:
Aberrant activation of notch signaling in human breast cancer.
Cancer Res. 66:1517–1525. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Rizzo P, Miao H, D’Souza G, et al:
Cross-talk between notch and the estrogen receptor in breast cancer
suggests novel therapeutic approaches. Cancer Res. 68:5226–5235.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Wang Z, Li Y, Ahmad A, et al: Targeting
Notch signaling pathway to overcome drug resistance for cancer
therapy. Biochim Biophys Acta. 1806:258–267. 2010.PubMed/NCBI
|
|
174
|
Hurvitz SA and Pietras RJ: Rational
management of endocrine resistance in breast cancer: a
comprehensive review of estrogen receptor biology, treatment
options, and future directions. Cancer. 113:2385–2397. 2008.
View Article : Google Scholar
|
|
175
|
Dibb NJ, Dilworth SM and Mol CD: Switching
on kinases: oncogenic activation of BRAF and the PDGFR family. Nat
Rev Cancer. 4:718–727. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Weigel MT, Ghazoui Z, Dunbier A, Pancholi
S, Dowsett M and Martin LA: Preclinical and clinical studies of
estrogen deprivation support the PDGF/Abl pathway as a novel
therapeutic target for overcoming endocrine resistance in breast
cancer. Breast Cancer Res. 14:R782012. View Article : Google Scholar
|
|
177
|
Pancholi V: Multifunctional α-enolase: its
role in diseases. Cell Mol Life Sci. 58:902–920. 2001.
|
|
178
|
Jiang BH, Agani F, Passaniti A and Semenza
GL: V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1)
and transcription of genes encoding vascular endothelial growth
factor and enolase 1: involvement of HIF-1 in tumor progression.
Cancer Res. 57:5328–5335. 1997.
|
|
179
|
Wygrecka M, Marsh LM, Morty RE, et al:
Enolase-1 promotes plasminogen-mediated recruitment of monocytes to
the acutely inflamed lung. Blood. 113:5588–5598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Dudani AK, Cummings C, Hashemi S and Ganz
PR: Isolation of a novel 45 kDa plasminogen receptor from human
endothelial cells. Thromb Res. 69:185–196. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Peebles KA, Duncan MW, Ruch RJ and
Malkinson AM: Proteomic analysis of a neoplastic mouse lung
epithelial cell line whose tumorigenicity has been abrogated by
transfection with the gap junction structural gene for connexin 43,
Gja1. Carcinogenesis. 24:651–657. 2003. View Article : Google Scholar
|
|
182
|
Wu W, Tang X, Hu W, Lotan R, Hong WK and
Mao L: Identification and validation of metastasis-associated
proteins in head and neck cancer cell lines by two-dimensional
electrophoresis and mass spectrometry. Clin Exp Metastasis.
19:319–326. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Ray R and Miller DM: Cloning and
characterization of a human c-myc promoter-binding protein. Mol
Cell Biol. 11:2154–2161. 1991.PubMed/NCBI
|
|
184
|
Tu SH, Chang CC, Chen CS, et al: Increased
expression of enolase α in human breast cancer confers tamoxifen
resistance in human breast cancer cells. Breast Cancer Res Treat.
121:539–553. 2010.
|