Introduction
Lung cancer is one of the main causes of
cancer-related mortality. Approximately 85% of lung cancers are
diagnosed as non-small cell lung cancer (NSCLC), and the overall
survival (OS) rate for advanced NSCLC is poor. The 5-year survival
rate is 5% for stage IIIb NSCLC and <1% for stage IV NSCLC
(1). Treatment for NCSLC is
determined by the patient’s clinical and tumor characteristics,
performance status (PS), the histological subtype and tumor
genotype/phenotype.
Recently, there have been many studies concerning
agents that target molecular changes, such as mutations in the
epidermal growth factor receptor (EGFR) and the fusion oncogene
EML4-ALK, in which the echinoderm microtubule-associated
protein-like 4 (EML4) is fused with the intracellular domain of
anaplastic kinase (ALK) (2–4). Although significant advances have been
made in the treatment of NSCLC using molecular targeted therapies
such as erlotinib and crizotinib, the median OS for patients with
advanced NSCLC remains low (5,6), and
acquired resistance to target agents is a major clinical problem.
Therefore, the development of novel therapies is needed (7).
Immunotherapy manipulates the immune system to
control and eradicate cancer. Many recent studies provide evidence
suggesting that immunotherapeutic manipulations are viable in many
tumor types, including lung cancer. Numerous trials of peptide
vaccines, autologous cellular therapy, T cell-directed antibody
therapy and monoclonal antibody therapy for lung cancer have been
carried out around the world (8–10) and
some of them have shown favorable results (11–13).
The EML4-ALK fusion gene was identified in NSCLC
patients by a team led by Professor H. Mano. This fusion gene was
formed as the result of a small inversion within the short arm of
chromosome 2 that joins differing portions of the EML4 gene with a
portion of the ALK gene (14,15).
As a result of this fusion, constant dimerization of the kinase
domain of ALK is induced and its catalytic activity increases
consequently.
The EML4-ALK fusion gene is mainly identified in
young, never/former light smokers with NSCLC (16). It is estimated that approximately 5%
of all NSCLC cases have this fusion gene. A few reports have also
identified EML4-ALK in other cancers, namely breast cancer and
colorectal cancer (17,18). For the most part, the EML4-ALK
fusion gene and other mutations, such as those in EGFR and KRAS,
are mutually exclusive (19).
The chromosomal inversion does not always occur in
the same location, and multiple EML4-ALK variants have been
identified (19). At least 11
variants have been reported. The most common variants are E13;A20
(variant 1) and E6a/b;A20 (variant 3a/b), which have been detected
in 33% and 29% of NSCLC patients, respectively (14).
PF-02341066 (crizotinib) is an ALK inhibitor
currently under clinical development. Kwak et al conducted
an open-label, multi-center, two-part phase I trial and found a
remarkable 57% overall response rate and a 72% 6-month
progression-free survival rate (20).
In spite of the marked antitumor activity of
crizotinib, ALK-positive cancers invariably gain resistance to
crizotinib. In the case of ALK-positive cancers, as well as
EGFR-mutant lung cancer, resistance develops on average within the
first 2 years of therapy (21). The
main resistance mutations are L1196M, a gatekeeper mutation and
C1156M. In addition to ALK mutations, other known mechanisms for
acquired resistance include ALK amplification (21,22)
and EGFR activation (23,24). To overcome resistance, new ALK
inhibitors are currently in early phase studies (25). Novel combinatorial strategies to
overcome crizotinib resistance and further improve the clinical
outcome are needed.
We focused on this new fusion array as a novel
target of immunotherapy. There are several methods to detect
EML4-ALK NSCLC, including polymerase chain reaction (PCR),
immunohistochemistry (IHC) and fluorescence in situ hybridization
(FISH) (19). These methods detect
high-level EML4-ALK fusion gene expression. Passoni et al
identified two HLA-A*02:01-restricted ALK-derived
peptides that induce peptide-specific CTL lines (26).
We focused on the EML4 array as a novel epitope of
immunotherapy. We identified a candidate 9- or 10-amino acid array
of novel epitopes using the Bioinformatics and Molecular Analysis
Section (BIMAS) software and analyzed its potential as a new
immunotherapy epitope, with respect to its ability to induce
anticancer activity. We then induced and generated a
peptide-specific CTL clone from peripheral blood lymphocytes of
HLA-A*02:01-positive healthy donors. We report here that
an EML4-ALK-derived peptide-specific human CTL clone recognized
peptide-pulsed T2 cells and HLA-A*02:01-positive and
EML4-ALK-positive tumor cells pretreated with IFN-γ. Furthermore,
we showed that immunotherapy with this novel epitope peptide has
potential for treatment of EML4-ALK-positive NSCLC.
Materials and methods
Peptides
Human EML4-ALK-derived peptides carrying binding
motifs for HLA-A*02:01-/HLA-A*24:02-encoded
molecules were identified by HLA-peptide binding predictions using
the BIMAS program (http://bimas.dcrt.nih.gov/molbio/hla_bind/index.html).
We purchased a total of seven EML4-ALK-derived peptides carrying
HLA-A*02:01 binding motifs and two peptides carrying
HLA-A*24:02 binding motifs from Geneworld (Tokyo,
Japan).
Cell lines
The H2228 human lung adenocarcinoma cell line and
EML4-ALK fusion protein variant 3 (E6; A20) were kindly provided by
Professor S. Yano (Kanazawa University).
T2 is a lymphoblastoid cell line that lacks TAP
function and has HLA-A*02:01 molecules that can easily
be loaded with exogenous peptides. T2A24 is the same cell line but
with HLA-A*24:02 instead. T2 and T2A24 cells were
cultured in RPMI medium supplemented with 10% heat-inactivated
FBS.
HLA-A*02:01/HLA-A*24:02 binding assay
In order to determine the binding ability of the
predicted peptides to HLA-A*02:01/HLA-A*24:02
molecules, an in vitro cellular binding assay was performed
as reported previously (27).
Briefly, after incubation of the T2/T2A24 cells in
culture medium at 26°C for 18 h, cells were washed with PBS and
suspended in 1 ml Opti-MEM (Invitrogen, Carlsbad, CA, USA) with or
without 100 μg peptide and then incubated at 26°C for 3 h and at
37°C for 3 h. After washing with PBS,
HLA-A*02:01/HLA-A*24:02 expression was
measured by flow cytometry using a FITC-conjugated and
HLA-A*02:01-/HLA-A*24:02-specific monoclonal
antibody (mAb) and the mean fluorescence intensity was
recorded.
Generation of dendritic cells
CD14+ cells were isolated from human
peripheral blood mononuclear cells (PBMCs) using human CD14
microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Immature
dendritic cells (DCs) were generated from CD14+ cells
using interleukin (IL)-4 (10 ng/ml; PeproTech Inc., Rocky Hill, NJ,
USA) and granulocyte-macrophage colony-stimulating factor (GM-CSF;
10 ng/ml; PeproTech) in RPMI-1640 medium supplemented with 10% FBS.
Maturation of DCs was induced by prostaglandin E2 (PGE2; 1 μg/ml;
Sigma, St. Louis, MO, USA) and tumor necrosis factor (TNF-)-α (10
ng/ml; PeproTech).
Induction of EML4-ALK-derived
peptide-specific CTLs from PBMCs
CD8+ cells were isolated from PBMCs using
human CD8 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany).
CD8+ cells (2×106) were stimulated by
peptide-pulsed irradiated autologous mature DCs (1×105).
Autologous DCs were prepared from a limited supply; artificial
antigen presenting cells (aAPCs) (K562/A2 or A24/CD80/CD83) were
alternatively used for further examination. After 1 week, these
cells were stimulated twice per week by peptide-pulsed irradiated
artificial APC-A2 or artificial APC-A24 cells (1×105).
Supplementation with 10 IU/ml IL-2 (Proleukin; Novartis
Pharmaceuticals, Basel, Switzerland) and 10 ng/ml IL-15 (PeproTech)
was performed every 3 to 4 days between stimulations (28).
IFN-γ ELISPOT assay
Specific secretion of IFN-γ from human CTLs in
response to stimulator cells was assayed using the IFN-γ ELISPOT
kit (BD Biosciences), according to the manufacturer’s instructions.
Stimulator cells were pulsed with peptide for 2 h at room
temperature and then washed. Responder cells were incubated with
stimulator cells for 20 h. The resulting spots were counted using
an ELIPHOTO counter (Minerva Tech, Tokyo, Japan). HIV-gag (77–85)
(SLYNTYATL) was used as an irrelevant peptide in the CTL assay.
Generation of CTL clones
Cultured cells were incubated with peptide-pulsed
T2/T2A24 cells at a ratio of 2:1 for 3.5 h at 37°C. CD107a-specific
antibodies (BioLegend, San Diego, CA, USA) were included in the
mixture during the incubation period.
CD8+CD107a+ cells were sorted using a
FACSAria II cell sorter (BD Biosciences). Sorted CTLs were
stimulated and the CTL clones were established as described
previously (29).
Flow cytometry
H2228 cells with or without pretreatment with 100
U/ml IFN-γ (PeproTech) for 48 h were harvested and stained with
anti-HLA-A2 Ab-FITC (MBL, Japan) and analyzed using a FACSCanto II
flow cytometer (BD Biosciences). Flow cytometry data were analyzed
using FlowJo software.
Cytotoxicity assay
The cytotoxic capacity was analyzed using the
Terascan VPC system (Minerva Tech, Tokyo). The CTL clone was used
as the effector cell type. Target cells treated with 100 U/ml IFN-γ
(PeproTech) 42 h previously were labeled through incubation in
calcein-AM solution for 30 min at 37°C. The labeled cells
(1×104) were then co-cultured with the effector cells
for 4–6 h. Fluorescence intensity was measured before and after the
culture period, and specific cytotoxic activity was calculated as
described previously (29).
HLA-A*02:01 blocking of T-cell activity
was tested by pre-incubating the target cells with anti-HLA-A, -B,
-C mAb (W6/32) or an isotype control mAb (mIgG2a,κ; BioLegend San
Diego, CA, USA).
Results
Identification of
HLA-A*02:01-/HLA-A*24:02-restricted
EML4-ALK-derived peptides
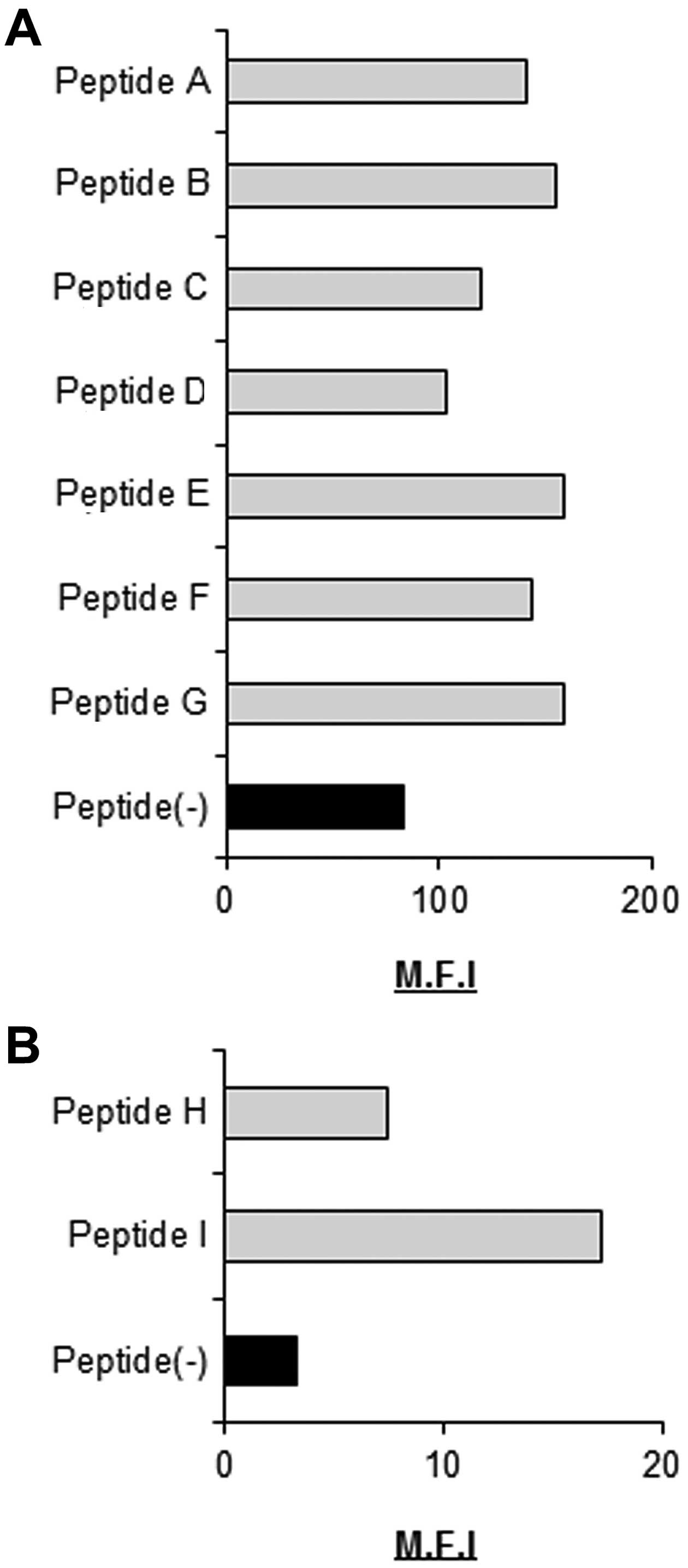
As candidate EML4-ALK- derived and
HLA-A*02:01-/HLA-A*24:02-restricted CTL
epitopes, we selected nine peptides with highly predicted scores
for HLA-A*02:01/HLA-A*24:02 binding
calculated using BIMAS software (Tables
I and II) and evaluated their
ability to bind to HLA-A*02:01/HLA-A*24:02
molecules. All nine peptides were able to bind
HLA-A*02:01/HLA-A*24:02 molecules (Fig. 1).
 | Table IHLA-A2 peptide binding predictions of
the BIMAS program. |
Table I
HLA-A2 peptide binding predictions of
the BIMAS program.
| Peptide name | Peptide
sequence | Binding
scorea |
|---|
| A | RLSALESRV | 69.552 |
| B | AISEDHVASV | 90.183 |
| C | TVLKAALADV | 51.79 |
| D | KLIPKVTKT | 59.989 |
| E | YLLPTGEIV | 237.82 |
| F | MLIWSKTTV | 118.238 |
| G | VMLIWSKTTV | 315.95 |
| Table IIHLA-A24 peptide binding predictions
of the BIMAS program. |
Table II
HLA-A24 peptide binding predictions
of the BIMAS program.
| Peptide name | Peptide
sequence | Binding
scorea |
|---|
| H | NYDDIRTEL | 369.6 |
| I | VYFIASVVVL | 200 |
Generation of an EML4-ALK-derived
peptide-specific CTL clone from human PBMCs
We next assessed the capacity of EML4-ALK-derived
peptides to generate peptide-specific CTLs in vitro from
human PBMCs of HLA-A*02:01/HLA-A*24:02
healthy donors. CTLs were induced by three stimulations with DCs or
artificial APCs loaded with the EML4-ALK-derived peptides. CTLs
were tested for specificity for each peptide using the IFN-γ
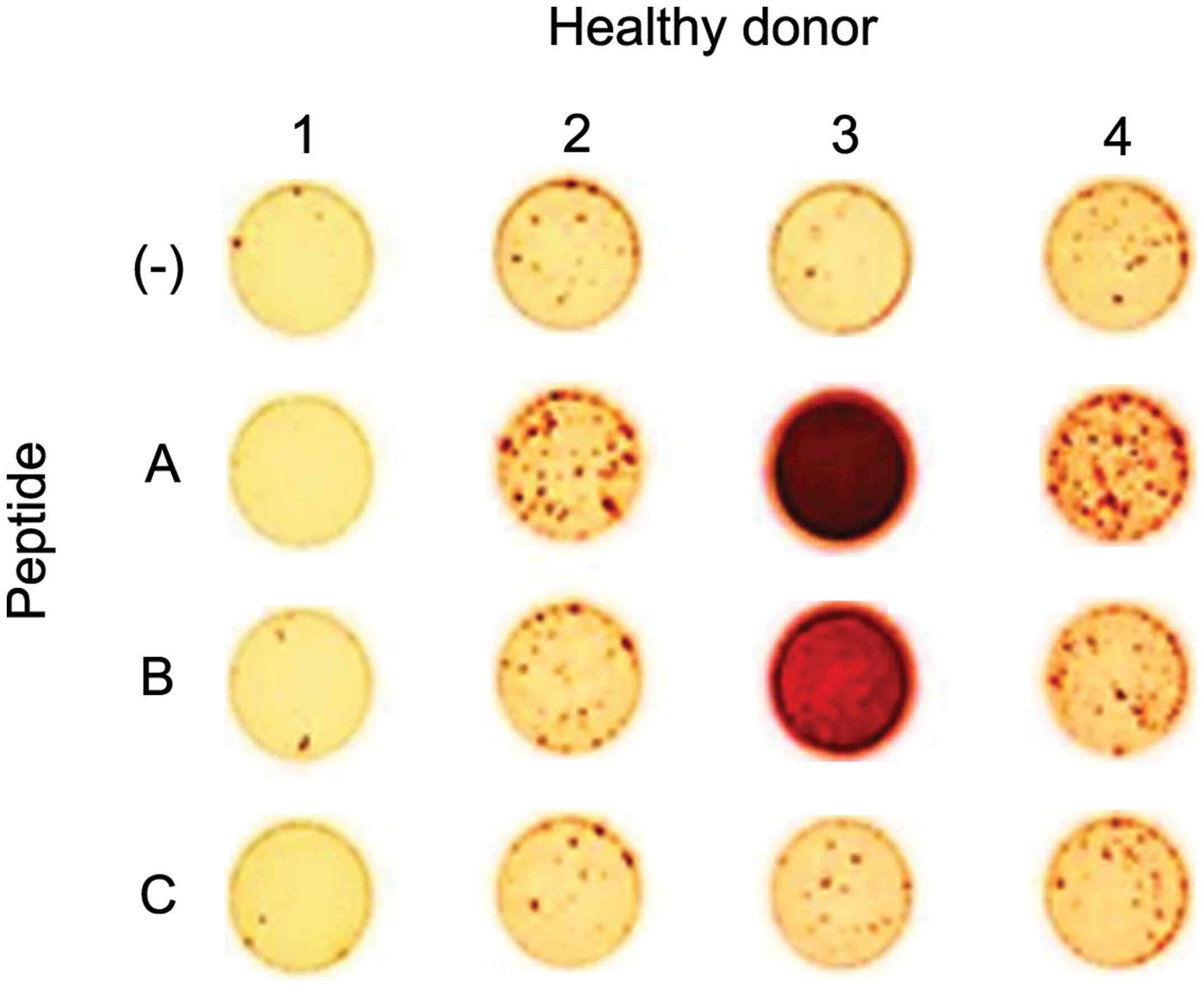
ELISPOT assay. Peptides A, B and C could induce peptide-specific
CTLs that were able to specifically recognize T2 cells pulsed with
each peptide, but not T2 cells without peptides (Fig. 2). Peptides B and C were able to
induce CTLs from only one donor (healthy donor 3 for peptide B and
healthy donor 4 for peptide C), but peptide A was able to induce
CTLs in three of four donors (healthy donors 2, 3 and 4). Based on
this result, we used peptide A for further examinations.
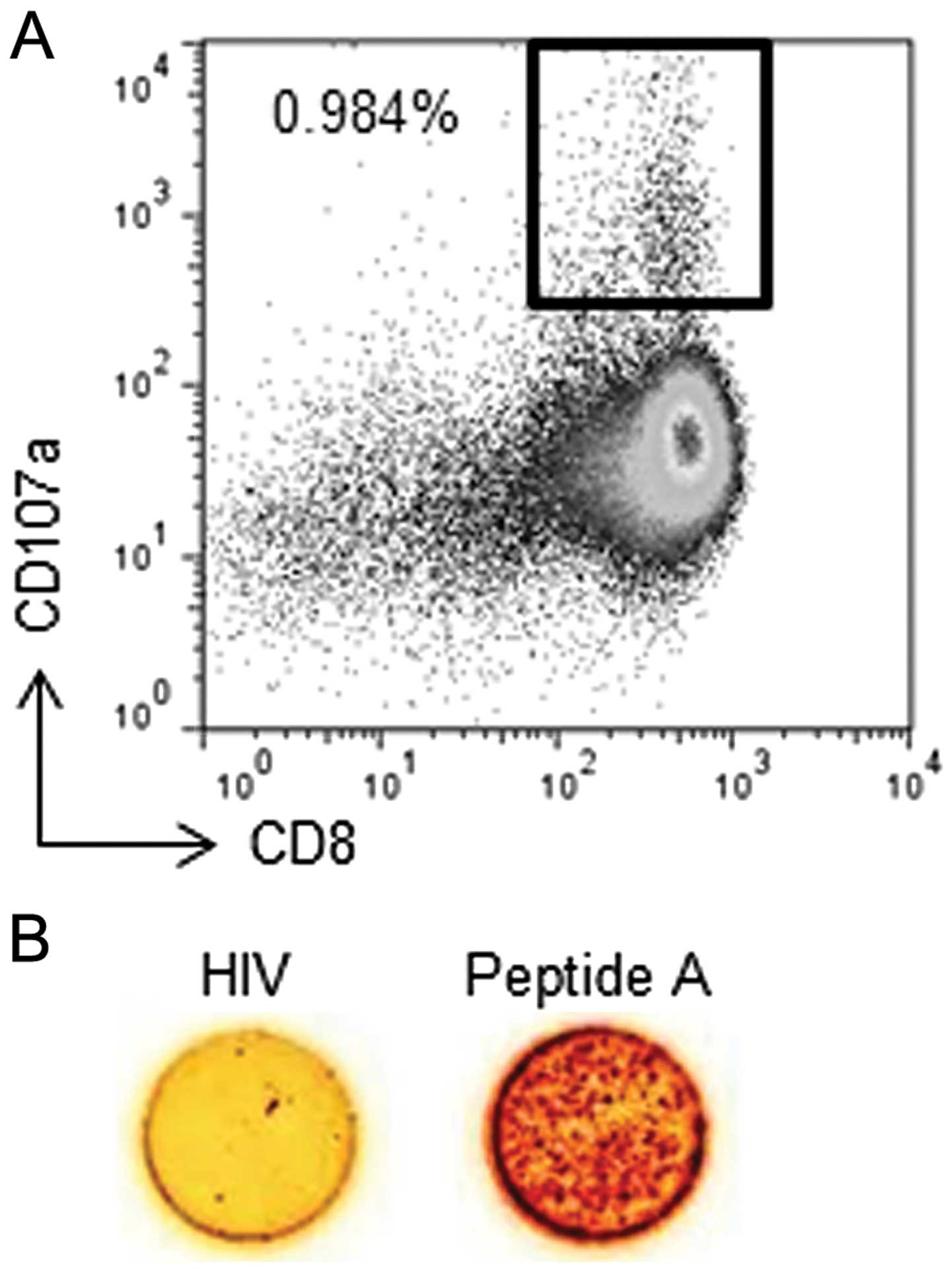
Next, we obtained one CTL clone from peptide
A-specific CTLs that was able to specifically recognize T2 cells
pulsed with peptide A, but not T2 cells pulsed with an irrelevant
HIV-gag peptide, using single cell sorting with a CD107a antibody.
The population of CD8+CD107a+ cells
represented 0.984% of all stimulated cells (Fig. 3A). These cells were sorted as single
cells in each well of a 96-well plate. Twenty-one days after cell
sorting, peptide specificity was assessed using the IFN-γ ELISPOT
assay (Fig. 3B). The established
clone reacted to the T2 cells pulsed with peptide A, but not to T2
cells pulsed with the irrelevant HIV-gag peptide. These results
indicate that a peptide A-specific CTL clone was successfully
established from PBMCs from a healthy donor.
The EML4-ALK-specific CTL clone
recognizes HLA-A*02:01+ lung carcinoma cells
with the EML4-ALK variant 3a/b incubated with IFN-γ
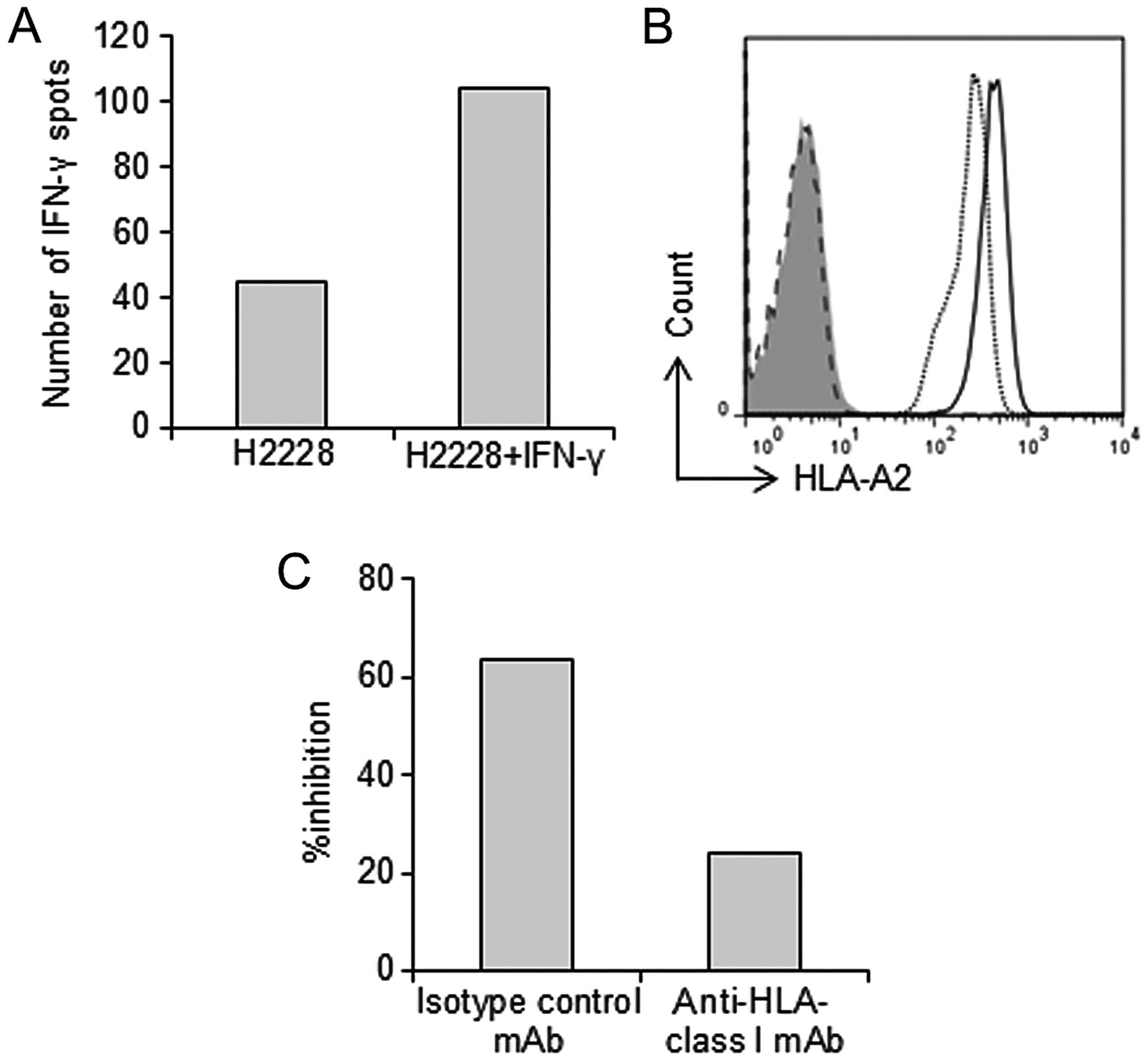
We next evaluated the ability of the
EML4-ALK-specific CTL clone to recognize the cancer cell line
H2228, which expresses HLA-A*02:01 and EML4-ALK, using
the IFN-γ ELISPOT assay. Even though the EML4-ALK-specific CTL
clone failed to recognize H2228 cells, it did recognize those
pretreated with 100 U/ml IFN-γ 48 h prior to examination (Fig. 4A). We examined the effect of IFN-γ
on H2228 cells. Incubating target cells with IFN-γ for 48 h
increased the expression of MHC class I molecules on the cell
surface (Fig. 4B). This result
indicates that the peptide A-specific CTL clone was able to
recognize H2228 cells because of increased expression of MHC-class
I on the H2228 cell surface.
Specific IFN-γ production by the peptide A-specific
CTL clone was detectable in H2228 cells treated with IFN-γ. The
specificity was abolished by an anti-HLA-class I mAb, but not by an
isotype control, suggesting that the observed production was HLA-A2
restricted (Fig. 4C).
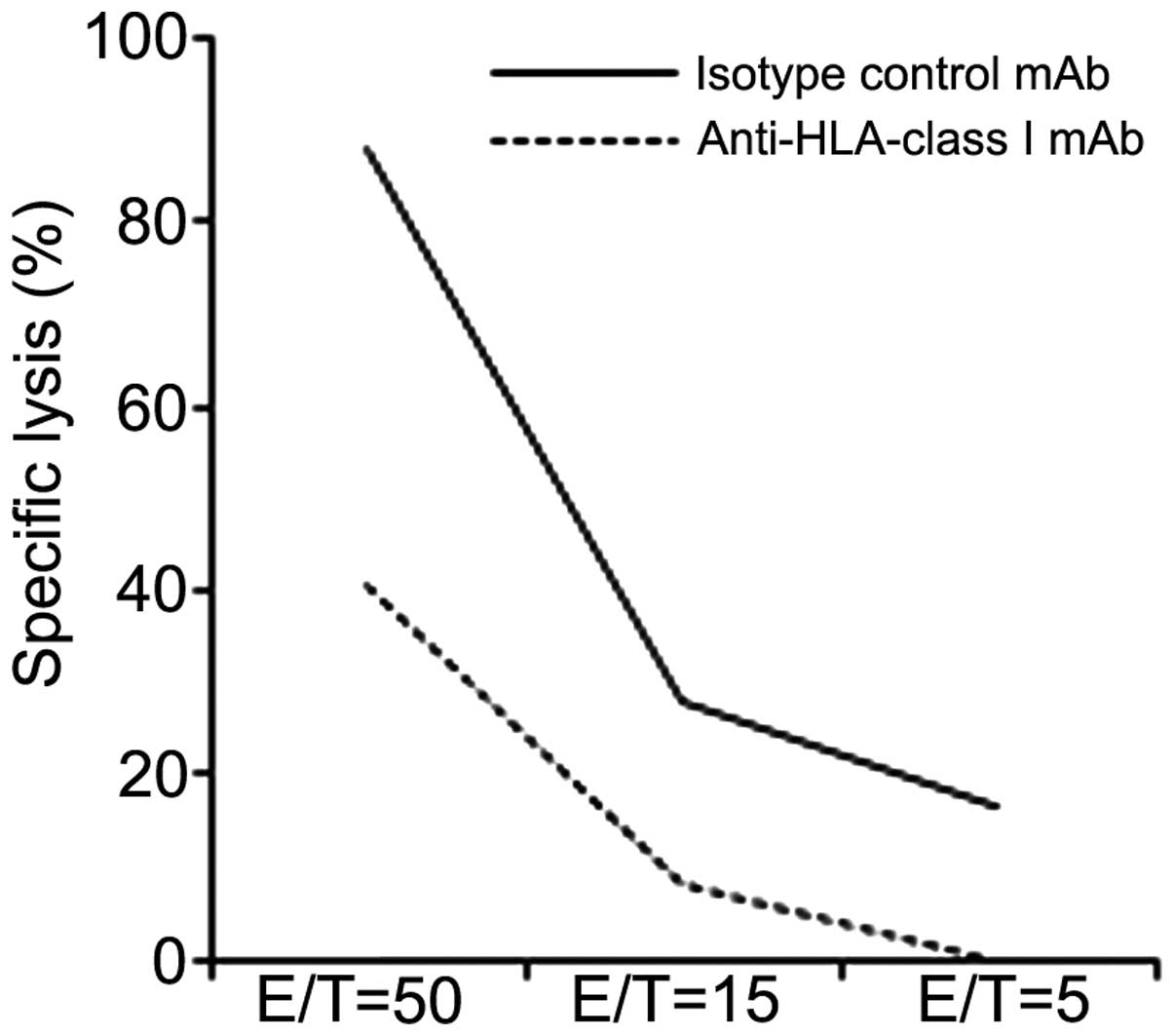
A cytotoxicity assay was also performed. The peptide
A-specific CTL clone was able to specifically lyse H2228 cells
pretreated with IFN-γ 48 h prior to examination. This specific
lysis was blocked by the anti-HLA-class I mAb, but not by the
isotype control. These results indicate that the peptide A-specific
CTL clone showed cytotoxicity and the ability to produce IFN-γ
against HLA-A*02:01+ EML4-ALK+
NSCLC cell lines (Fig. 5).
Discussion
In the present study, we identified a new
tumor-associated CTL epitope (peptide A) derived from EML4-ALK,
which binds to HLA-A*02:01 molecules, and we were able
to establish a peptide-specific CTL clone from human PBMCs that
specifically recognized cognate peptide-pulsed T2 cells and
HLA-A*02:01 tumor cells expressing EML4-ALK that had
been pretreated with IFN-γ.
EML4-ALK-positive lung cancers are highly sensitive
to ALK inhibition. However, as with trastuzumab or gefitinib
(30,31), patients typically gain resistance
within 1 to 2 years of starting therapy (23). We aimed to overcome these
difficulties with immunotherapy.
We identified a glypican-3 (GPC3)-derived peptide
and showed that GPC3-specific CTL frequency after vaccination
correlated with OS. OS was significantly longer in patients with
high GPC3-specific CTL frequencies than in those with low
frequencies (32). This indicates
that the ability to induce a peptide-specific CTL clone is
important for effective immunotherapy. We also revealed that GPC3
is an ideal target for anticancer immunotherapy since it is
specifically overexpressed in hepatocellular carcinoma (HCC)
(33–35).
In the present study, we chose a peptide array from
EML4-ALK, from which we were able to induce a peptide-specific CTL
clone. EML4-ALK is a strong oncogene overexpressed in cancer cells
of NSCLC, breast cancer, kidney cancer and colon cancer (17). We performed RT-PCR and assayed the
EML4 DNA levels of certain lung cancer cell lines. H2228 cells
express EML4 moderately but at higher levels than other lung cancer
cell lines. EML4 expression has been reported as highly expressed
in CD8+ T cells. RT-PCR showed that EML4 DNA levels were
high in PBMCs and CD8+ T cells. Because of a lack of
suitable antibodies, we could not perform western blotting.
However, our success at inducing a peptide A-specific CTL clone
from CD8+ T cells indicated that the CTL clone had no
cytotoxicity against CD8+ T cells.
This CTL clone could not recognize cancer cell lines
without the ability to increase the amount of HLA class I presented
on cell surfaces. Further examination is needed to achieve higher
tumor reactivity. Combination chemotherapy or radiation therapy
plus immunotherapy was recently reported to have a synergistic
effect (36). Moreover, some
mechanisms of synergy between radiation therapy, chemotherapy and
immunotherapy have been revealed (37). In one of the mechanisms, these
therapies upregulated tumor antigens and MHC moieties. These
results suggest that combination therapy could be used to make
tumor cell lines more susceptible to this peptide A-specific CTL
clone-mediated cytolysis (38–41).
In addition, this treatment may be able to overcome
resistance to ALK inhibition. Some resistance mechanisms for
targeting drugs have been examined. The most commonly identified
causes of resistance are point mutations such as L1196M (42–44),
G1269A (22) and S1206Y (21). These point mutations occur in the
tyrosine kinase domain, which plays an important role in
oncogenesis. Our peptide array was selected from EML4, which has no
correlation with these point mutations. It is possible that this
treatment is effective for tumor cells resistant to ALK
inhibitors.
In this study, we identified a new epitope peptide
derived from the EML4-ALK fusion gene. We successfully induced an
HLA-A*02:01-restricted peptide-specific CTL clone that
demonstrated cytotoxicity for EML4-ALK-positive tumor cells. This
is a new epitope-based vaccine therapy design for EML4-ALK-positive
cancer cells. In order to obtain a stronger effect, further
analysis is needed.
Acknowledgements
We thank Professor S. Yano for providing the H2228
cell line, which possesses the EML4-ALK fusion gene, Professor H.
Mano for providing the EML4-ALK fusion DNA and Professor N. Hirano
for providing artificial APCs. This study was supported in part by
Health and Labor Science Research Grants for Clinical Research and
Third Term Comprehensive Control Research for Cancer from the
Ministry of Health, Labor and Welfare, Japan and the National
Cancer Center Research and Development Fund (25-A-7).
References
|
1
|
Silvestri GA, Tanoue LT, Margolis ML, et
al: The noninvasive staging of non-small cell lung cancer: the
guidelines. Chest. 123:147S–156S. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reck M: What future opportunities may
immuno-oncology provide for improving the treatment of patients
with lung cancer? Ann Oncol. 23(Suppl 8): viii28–viii34. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang SC, Chang CY and Shih JY: The role
of epidermal growth factor receptor mutations and epidermal growth
factor receptor-tyrosine kinase inhibitors in the treatment of lung
cancer. Cancers. 3:2667–2678. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gridelli C, Peters S, Sgambato A, Casaluce
F, Adjei AA and Ciardiello F: ALK inhibitors in the treatment of
advanced NSCLC. Cancer Treat Rev. 40:300–306. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hall RD, Gray JE and Chiappori AA: Beyond
the standard of care: a review of novel immunotherapy trials for
the treatment of lung cancer. Cancer Control. 20:22–31.
2013.PubMed/NCBI
|
|
6
|
Jackman DM, Miller VA, Cioffredi LA, et
al: Impact of epidermal growth factor receptor and KRAS mutations
on clinical outcomes in previously untreated non-small cell lung
cancer patients: results of an online tumor registry of clinical
trials. Clin Cancer Res. 15:5267–5273. 2009. View Article : Google Scholar
|
|
7
|
West H, Oxnard GR and Doebele RC: Acquired
resistance to targeted therapies in advanced non-small cell lung
cancer: new strategies and new agents. Am Soc Clin Oncol Educ Book.
2013, View Article : Google Scholar : http://meetinglibrary.asco.org/content/198-132.
|
|
8
|
Wu YL, Park K, Soo RA, et al: INSPIRE: a
phase III study of the BLP25 liposome vaccine (L-BLP25) in Asian
patients with unresectable stage III non-small cell lung cancer.
BMC Cancer. 11:4302011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tyagi P and Mirakhur B: MAGRIT: the
largest-ever phase III lung cancer trial aims to establish a novel
tumor-specific approach to therapy. Clin Lung Cancer. 10:371–374.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Quoix E, Ramlau R, Westeel V, et al:
Therapeutic vaccination with TG4010 and first-line chemotherapy in
advanced non-small-cell lung cancer: a controlled phase 2B trial.
Lancet Oncol. 12:1125–1133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brahmer JR, Tykodi SS, Chow LQ, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lynch TJ, Bondarenko I, Luft A, et al:
Ipilimumab in combination with paclitaxel and carboplatin as
first-line treatment in stage IIIB/IV non-small-cell lung cancer:
results from a randomized, double-blind, multicenter phase II
study. J Clin Oncol. 30:2046–2054. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Topalian SL, Hodi FS, Brahmer JR, et al:
Safety, activity, and immune correlates of anti-PD-1 antibody in
cancer. N Engl J Med. 366:2443–2454. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soda M, Choi YL, Mano H, et al:
Identification of the transforming EML4-ALK fusion gene in
non-small-cell lung cancer. Nature. 448:561–566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bonanno L, Favaretto A, Rugge M, et al:
Role of genotyping in non-small cell lung cancer treatment: current
status. Drugs. 71:2231–2246. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fukui T, Yatabe Y, Mitsudomi T, et al:
Clinicoradiologic characteristics of patients with lung
adenocarcinoma harboring EML4-ALK fusion oncogene. Lung Cancer.
77:319–325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin E, Li L, Guan Y, et al: Exon array
profiling detects EML4-ALK fusion in breast, colorectal, and
non-small cell lung cancers. Mol Cancer Res. 7:1466–1476. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Robertson FM, Petricoin EF III,
Cristofanilli M, et al: Presence of anaplastic lymphoma kinase in
inflammatory breast cancer. Springerplus. 2:4972013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sasaki T, Rodig SJ, Jänne PA, et al: The
biology and treatment of EML4-ALK non-small cell lung cancer. Eur J
Cancer. 46:1773–1780. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kwak EL, Bang YJ, Iafrate AJ, et al:
Anaplastic lymphoma kinase inhibition in non-small-cell lung
cancer. New Engl J Med. 363:1693–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Katayama R, Shaw AT, Engelman JA, et al:
Mechanisms of acquired crizotinib resistance in ALK-rearranged lung
cancers. Sci Transl Med. 4:120ra172012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Doebele RC, Pilling AB, Camidge DR, et al:
Mechanisms of resistance to crizotinib in patients with ALK gene
rearranged non-small cell lung cancer. Clin Cancer Res.
18:1472–1482. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shaw AT and Engelman JA: ALK in lung
cancer: past, present, and future. J Clin Oncol. 31:1105–1111.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sasaki T, Koivunen J, Jänne PA, et al: A
novel ALK secondary mutation and EGFR signaling cause resistance to
ALK kinase inhibitors. Cancer Res. 71:6051–6060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Latif M, Saeed A and Kim SH: Journey of
the ALK-inhibitor CH5424802 to phase II clinical trial. Arch Pharm
Res. 36:1051–1054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Passoni L, Scardino A,
Gambacorti-Passerini C, et al: ALK as a novel lymphoma-associated
tumor antigen: identification of 2 HLA-A2.1-restricted
CD8+T-cell epitopes. Blood. 99:2100–2106. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hirohashi Y, Torigoe T, Maeda A, et al: An
HLA-A24-restricted cytotoxic T lymphocyte epitope of a
tumor-associated protein, survivin. Clin Cancer Res. 8:1731–1739.
2002.PubMed/NCBI
|
|
28
|
Hirano N, Butler MO, Xia Z, et al:
Engagement of CD83 ligand induces prolonged expansion of
CD8+T cells and preferential enrichment for antigen
specificity. Blood. 107:1528–1536. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yoshikawa T, Nakatsugawa M, Sakemura N, et
al: HLA-A2- restricted glypican-3 peptide-specific CTL clones
induced by peptide vaccine show high avidity and antigen-specific
killing activity against tumor cells. Cancer Sci. 102:918–925.
2011. View Article : Google Scholar
|
|
30
|
Robinson KW and Sandler AB: EGFR tyrosine
kinase inhibitors: difference in efficacy and resistance. Curr
Oncol Rep. 15:396–404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lesniak D, Sabri S, Abdulkarim B, et al:
Spontaneous epithelial- mesenchymal transition and resistance to
HER-2-targeted therapies in HER-2-positive luminal breast cancer.
PLoS One. 8:e719872013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sawada Y, Yoshikawa T, Nakatsura T, et al:
Phase I trial of a glypican-3-derived peptide vaccine for advanced
hepatocellular carcinoma: immunologic evidence and potential for
improving overall survival. Clin Cancer Res. 18:3686–3696. 2012.
View Article : Google Scholar
|
|
33
|
Nakatsura T, Yoshitake Y, Nishimura Y, et
al: Glypican-3, overexpressed specifically in human hepatocellular
carcinoma, is a novel tumor marker. Biochem Biophys Res Commun.
306:16–25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Okabe H, Satoh S, Nakamura Y, et al:
Genome-wide analysis of gene expression in human hepatocellular
carcinomas using cDNA microarray: identification of genes involved
in viral carcinogenesis and tumor progression. Cancer Res.
61:2129–2137. 2001.
|
|
35
|
Saito-Hisaminato A, Katagiri T, Nakamura
Y, et al: Genome-wide profiling of gene expression in 29 normal
human tissues with a cDNA microarray. DNA Res. 9:35–45. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Weir GM, Liwski RS and Mansour M: Immune
modulation by chemotherapy or immunotherapy to enhance cancer
vaccines. Cancers. 3:3114–3142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hodge JW, Ardiani A, Gameiro SR, et al:
The tipping point for combination therapy: cancer vaccines with
radiation, chemotherapy, or targeted small molecule inhibitors.
Semin Oncol. 39:323–339. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Garnett CT, Palena C, Hodge JW, et al:
Sublethal irradiation of human tumor cells modulates phenotype
resulting in enhanced killing by cytotoxic T lymphocytes. Cancer
Res. 64:7985–7994. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gelbard A, Garnett CT, Hodge JW, et al:
Combination chemotherapy and radiation of human squamous cell
carcinoma of the head and neck augments CTL-mediated lysis. Clin
Cancer Res. 12:1897–1905. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaneno R, Shurin GV, Shurin MR, et al:
Chemotherapeutic agents in low noncytotoxic concentrations increase
immunogenicity of human colon cancer cells. Cell Oncol. 34:97–106.
2011. View Article : Google Scholar
|
|
41
|
Ramakrishnan R, Assudani D, Gabrilovich
DI, et al: Chemotherapy enhances tumor cell susceptibility to
CTL-mediated killing during cancer immunotherapy in mice. J Clin
Invest. 120:1111–1124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Choi YL, Soda M, Yamashita Y, et al:
EML4-ALK mutations in lung cancer that confer resistance to ALK
inhibitors. N Engl J Med. 363:1734–1739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Katayama R, Khan TM, Benes C, et al:
Therapeutic strategies to overcome crizotinib resistance in
non-small cell lung cancers harboring the fusion oncogene EML4-ALK.
Proc Natl Acad Sci USA. 108:7535–7540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lovly CM and Pao W: Escaping ALK
inhibition: mechanisms of and strategies to overcome resistance.
Sci Transl Med. 4:120ps22012. View Article : Google Scholar : PubMed/NCBI
|