Introduction
Cytokine, growth factor and integrin signaling are
regulated by SH2 domian-containing tyrosine phosphatase 2 (SHP2).
SHP2 has two SH2 domains within the N-terminus part, and a
phosphatase domain within the C-terminal part (1,2). SHP2
is expressed ubiquitously in mammalian tissue (3,4), and
regulates several cellular processes (5,6). In
general, SHP2 stimulates the release of cytokines and growth
factors, i.e. insulin, EGF, PDGF, FGF, IL-1 and IL-6 (7,4).
Activation of SHP2 may interfere with MAPK cellular signal
transduction via Erk, JNK/SAPK, p38/RK and BMK1/Erk5 (8). Furthermore, SHP2 is a bona fide
oncogene, as overexpression of SHP2 has been frequently observed in
adult human leukemia (9,10). Genetic analyses revealed that
germline mutations of PTPN11, which encodes SHP2 in humans, present
in nearly 50% of patients with developmental abnormalities, such as
leukemia and solid tumors (11–16).
Overexpression of SHP2 has been observed in most
breast cancer cell lines and in breast cancer tissues (8,17), and
is accompanied by lymph node metastasis. Inhibition of SHP2 in
breast cancer cell lines was found to abolish the growth and
decrease the survival of tumor cells, leading to the
differentiation of malignant cells into a normal breast epithelial
phenotype (12,18,19).
These observations suggest that SHP2 promotes tumor development
through increased tumor formation and metastasis.
However, the mechanism involved in the promotion of
the malignant potential of breast cancer by SHP2 is largely
unknown. To determine the influence of the SHP-2 signaling pathway
on the developmental process of breast cancer, we constructed an
SHP2 eukaryotic expression vector and transfected it into
MDA-MB-231 cells. Our results clearly showed that transfection of
SHP2 into MDA-MB-231 cells resulted in an altered phenotype. SHP2
overexpression was associated with significantly increased cell
proliferation and clone formation, and decreased chemotherapeutic
sensitivity in the SHP2-MB-231 group when compared with the control
groups. Anchorage-independent growth, migration and invasion of the
transfected cells in vitro were also increased. These
findings support the hypothesis that SHP2 is a cancer agonist;
overexpression of SHP2 contributes to the malignant progression of
breast cancers.
Materials and methods
Plasmids, cell culture and
transfection
The SHP2 mammalian expression vector was constructed
as described previously (18,20).
Briefly, SHP2 cDNA was obtained using RT-PCR from mouse embryonic
fibroblasts, and was cloned into the pcDNA3.1 vector. MDA-MB-231
human breast cancer cells were cultured in DMEM with 10%
heat-inactivated fetal bovine serum (FBS). MDA-MB-231 cells were
transfected with the pcDNA3.1 empty vector or the SHP2-pcDNA3.1
vector using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA,
USA). After 24 h, fresh DMEM was added which contained G418 (800
μg/μl). The culture medium was replaced thrice weekly until stably
transfected colonies were observed.
Immunoblotting
Immunoblotting was performed as previously described
(18). In brief, 15 μg of the
protein samples was separated on an SDS-polyacrylamide gel by
electrophoresis. The separated proteins were transferred to
nitrocellulose membranes. The membranes were blocked in PBST (5%
BSA, 0.05% Tween-20, pH 7.4) at room temperature for 1 h.
Subsequently, the membranes were probed with primary antibodies
directed against target proteins overnight at 4°C. After three
washes in PBST, the membranes were incubated with the secondary
antibody conjugated to horseradish peroxidase (Amersham, Arlington
Heights, IL, USA). Signals were detected using a chemiluminescence
method (Amersham). The same membranes were stripped with stripping
buffer for 15 min at room temperature and immunoblotted with
anti-acting antibody. Digitalization of films was performed using
ImageJ 1.43G (NIH, Bethesda, MD, USA).
Cell adhesion assay
MB-231, vector-MB-231 and SHP2-MB-231 cells were
collected upon trypsinization, and washed in serum-free DMEM (0.2%
trypsin inhibitor). Cells were resuspended at 105
cells/ml in DMEM (10% FBS), and cultured in fibronectin (FN) (10
mg/ml)-pre-coated 96-well plates (100 μl suspension/well).
After 30 min, 1 h and 2 h, non-adherent cells were
removed by washing the culture plate with PBS. Attached cells were
stained with 0.1% crystal violet in 20% methanol. The crystal
violet staining was eluted with 0.1 M sodium citrate (pH 4.2), and
the plate was measured at 490 nm using a microplate reader.
Cell migration assay
Cell migration was analyzed using Transwell System
(Costar Corp., Acton, MA, USA). MDA-MB-231 cells (SHP2- or
vector-transfected) were trypsinized, and 1×105 cells
were added to the top chambers of the Transwell (8-μm pore size; BD
Bioscience, Bedford, MA, USA), and assay medium was added to the
bottom chambers and incubated at 37°C in 5% CO2 for 12
h. The membrane was fixed with methanol, and the cells remaining on
the upper chamber were removed. The migrated cells were stained
with Giemsa and were counted. For each well, five microscopic
fields were counted randomly. Data obtained from triplicate wells
were analyzed.
Wound-healing assay
Wound-healing assay was performed as previously
described (21). Briefly, MB-231,
vector-MB-231 and SHP2-MB-231 cells were seeded in 6-well dishes
(1×105 cells/well) and starved overnight. A single
scratch wound was generated using a pipette tip. The cells were
cultured in DMEM with 5% FBS. After 0, 24 and 48 h, images were
captured using an inverted microscope, and the width of the wounded
area was measured. The relative migration distance was calculated
with the following formula: Migration distance = [(width at 0 h -
width at observation point)/width at 0 h] × 100%
Cell growth assay
Anchorage-independent growth was assessed by a soft
agar clonogenic assay. Cells were detached and plated in 0.6%
agarose with a 1.2% agarose underlay (1×103 cells per
well in 6-well plates). The number of foci was counted after 18
days. For the focus formation assay, the cells were reseeded and
cultured for 4 weeks in DMEM with 10% FBS. The cells were fixed
with formalin and stained with 0.1% crystal violet.
Cell proliferation assay
MB-231, vector-MB-231 and SHP2-MB-231 cells were
plated at 1×104 cells/well in 96-well plates. After 24
h, cisplatin (4 μg/ml) or DMSO was added to the culture medium. The
cells were harvested at 24, 48 and 72 h and were counted using a
hemocytometer. The experiments were repeated in triplicate. The
proliferation rate was calculated using the following formula:
Proliferation rate = [(cell number at harvest time - cell number at
0 h)/cell number at 0 h] × 100%.
In vivo tumorigenicity assay
Tumor cells (2×106/100 μl PBS) were
injected subcutaneously into the mid-dorsum of BALB/c nude mice
(4–6 weeks old) in a total volume of 100 μl. Mice were monitored
weekly for tumor development. Tumor sizes were measured using
vernier calipers, and the tumor volume was calculated according to
the formula: length × width2 ×10, which approximates the
volume of an elliptical solid. Mice were sacrificed 50 days after
injection, and the tumor tissues were harvested. Upon removal of
the tumors, the tumor volume was calculated using the equation:
Tumor volume = (length × width2)/2. Tumor samples were
fixed in 10% formalin for further analysis.
Histological staining
Immunohistochemical procedure was performed as
previously described (21). Tumor
samples fixed in 10% neutral buffered formalin for at least 24 h.
Paraffin embedding was performed, and sections (3-μm) were cut and
stained with hematoxylin and eosin (H&E).
CD31 immunohistochemistry
Sections (4 μm) were blocked with 1% BSA and 0.01%
TritonX-100 for 1 h at room temperature, and incubated with the
primary antibody (anti-CD31, PECAM, 1:50) overnight at 4°C.
Negative controls were performed by either omitting the primary
antibody or incubating sections with normal rat IgG. Detection was
performed by incubation with biotinylated secondary antibodies
(Vector Laboratories Inc., Burlingame, CA, USA) for 1 h at room
temperature. The sections were incubated with the
avidin-biotin-peroxidase complex (Vector Laboratories Inc.; 1:100
in PBS) for 1 h and developed in 0.05% 3,3′-diaminobenzidine
(Sigma) containing 0.003% H2O2 in PBS. To
determine the average microvessel density (AMVD), the number of
CD31-positive microvessels was counted in 10 randomly chosen visual
fields under microscopy. The AMVD was calculated and expressed as
the number of microvessels per mm2 area.
Data and statistical analyses
All experiments were repeated a minimum of 3 times.
Data are shown as mean values ± standard deviation. Groups were
compared by one-way ANOVA followed by Pearson coefficient analysis
for bivariate correlation. A P-value of <0.05 was considered to
indicate a statistically significant result.
Results
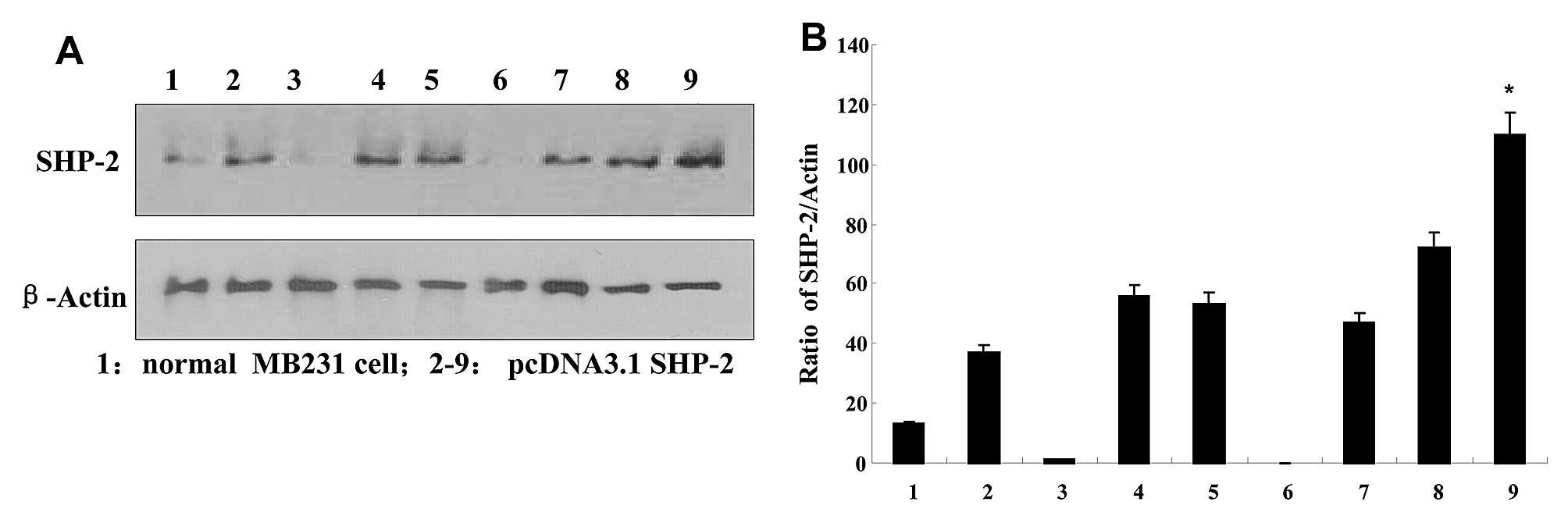
SHP2 protein expression in the
transfected breast cancer cells
The expression of SHP2 was examined in each of the
transduced populations. After transfection with pcDNA3.1-SHP2,
G418-resistant MDA-MB-231 clones were expanded as monoclonal
populations. Four weeks later, 15 clones were selected to examine
SHP2 expression by performing western blotting. As shown in
Fig. 1, high expression of SHP2 was
detected in the respective clones, when compared with the
non-transfected MB231 cells. Colonies of line 9 were chosen for
subsequent experiment, and named SHP2-MB-231. The expression of
SHP2 was normalized to the constitutively expressed β-actin protein
using densitometry.
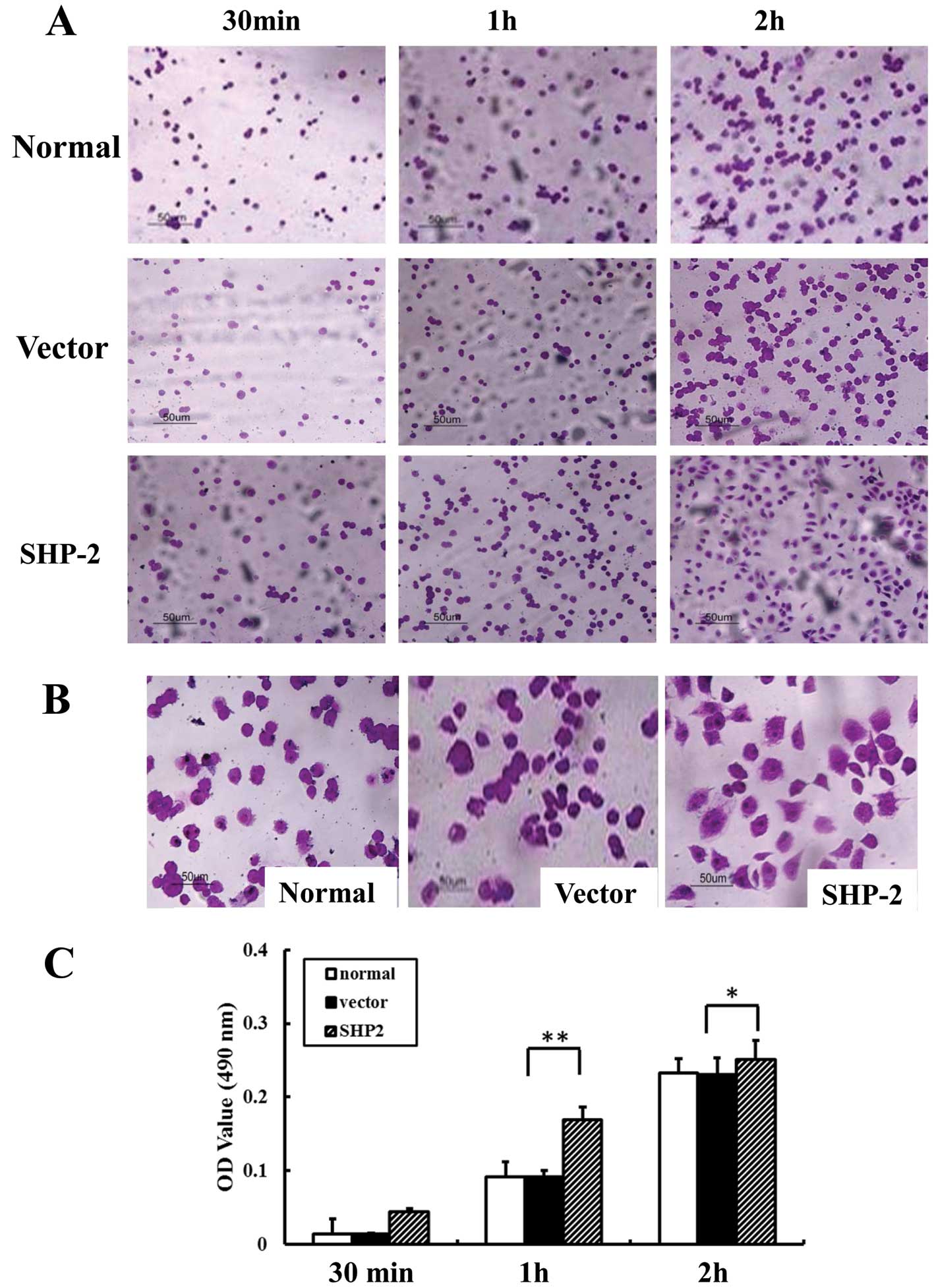
Overexpression of SHP2 is associated with
the increased adhesion of MB231 cells to FN
We examined the adhesive ability of the MB-231,
vector-MB-231 and SHP2-MB-231 cells to FN. As shown in Fig. 2, the number of adhesive cells in the
SHP2-MB-231 group was significantly increased when compared with
the MB-231 and vector-MB-231 groups (Fig. 2A). Of note, after 2 h of incubation,
SHP2-transfected cells exhibited an extensively elongated
appearance, while most cells in both control groups appeared round
(Fig. 2B). Moreover, the absorbance
value in the SHP2-MB-231 group was increased 3- and 2-fold at 30
min and 1 h, respectively, when compared with the control group
(Fig. 2C).
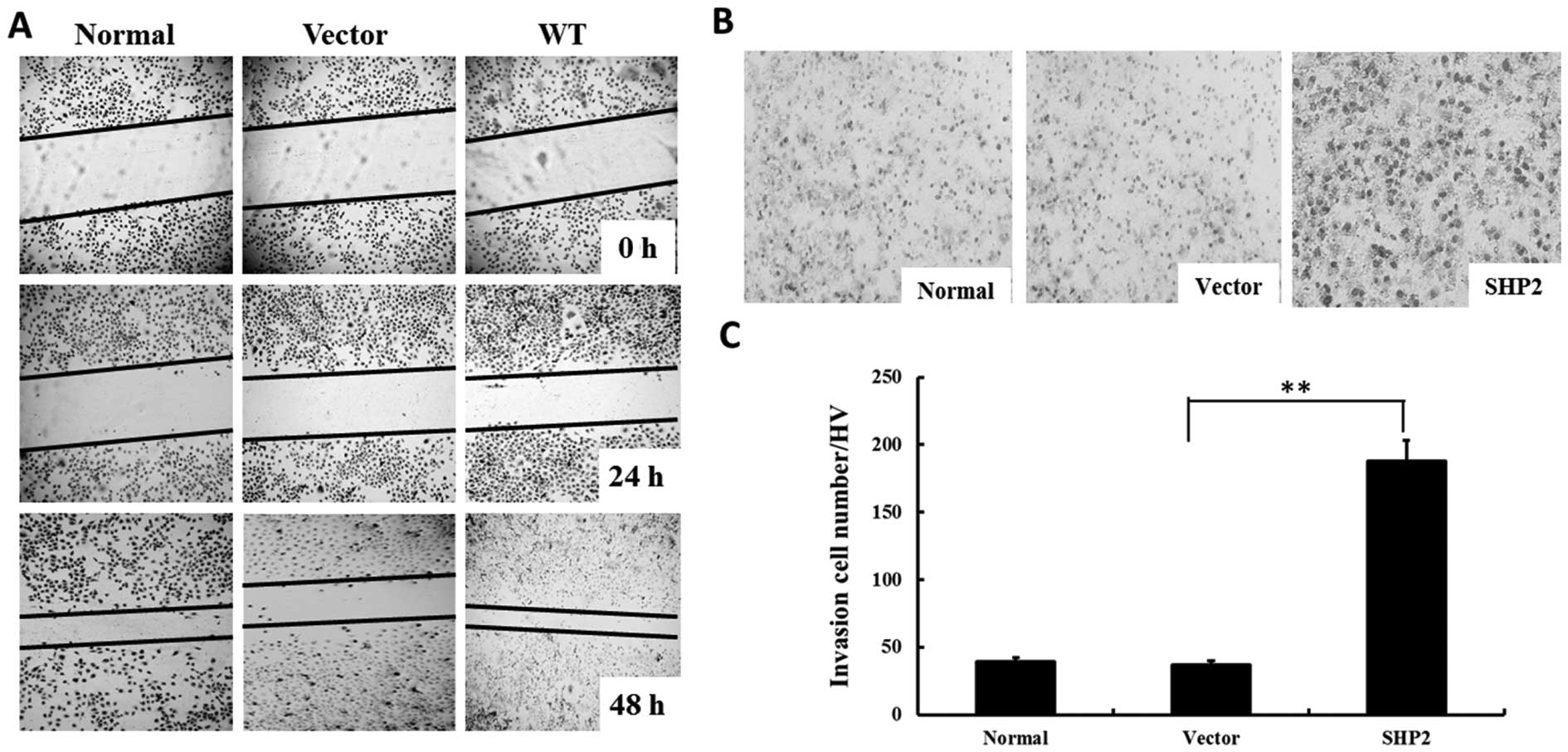
Overexpression of SHP2 enhances migration
and invasion of MDA-MB-231 cells
A wound-healing assay was performed to investigate
whether SHP2 overexpression affects tumor migration in
vitro. As shown in Fig. 3A,
overexpression of SHP2 significantly increased the migration of
MB-231 cells, as the width of the scratch was markedly decreased
after 48 h. In contrast, the width of the scratch was only slightly
decreased in the MB-231 control group, as well as the vector-MB-231
group. Moreover, overexpression of SHP2 in the SHP2-MB-231 group
showed a similar pattern in the transwell experiment (Fig. 3B). After 12 h of incubation, the
invasive cells in the vector-MB-231 group was 4-fold higher when
compared with the control groups (Fig.
3C).
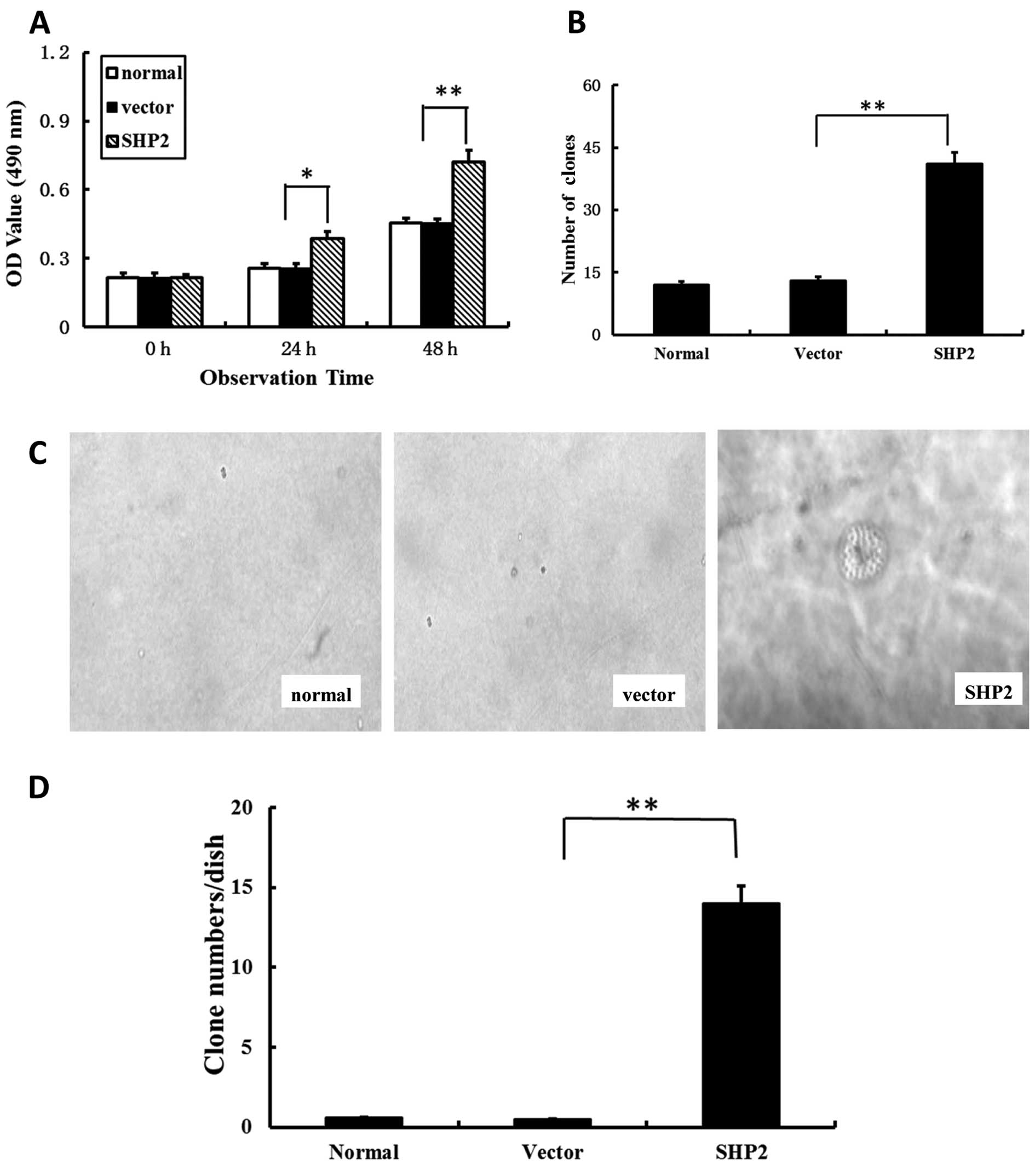
Overexpression of SHP2 accelerates the
proliferation of MDA-MB-231 cells
To determine whether the increase in cell migration,
wound closure and invasion in breast cancer cells was due to
increased cell growth, the proliferation was assessed by MTS assay.
As shown in Fig. 4A, a
significantly higher absorbance value at 490 nm was observed in the
SHP2-MB-231 group when compared with both control groups. These
data suggest that the differences in actual cell migration were due
to differential cell growth.
To further characterize the effect of SHP2 on
MDA-MB-231 cells, tumorigenesis was investigated. The focus
formation experiment was performed, which reflected an increase in
density-dependent or anchorage-independent growth in soft agar. The
transfected MB-231 cells with SHP2 showed a significantly enhanced
focus formation ratio. The number of clones in the SHP2-MB-231
group was markedly higher in comparison with the vector-transfected
and non-transfected control groups (Fig. 4C and D). Anchorage-independent
growth, as assessed by colony growth in soft agar, resulted in an
up to 30-fold increase in the SHP2-MB-231 group than in control
groups (Fig. 4D).
Overexpression of SHP2 increases
resistance to chemotherapeutic agent cisplatin
To investigate the influence of SHP2 overexpression
on cell proliferation, MST assay was performed after incubation of
MDA-MB-231 cells with cisplatin. Cisplatin is a DNA-reactive agent
which is commonly used in chemotherapy protocols for the treatment
of breast cancer. As shown in Fig.
5A, in the control group, 4 μg/ml cisplatin inhibited the
proliferation and increased the apoptosis of MB-231 cells in the
control groups. In contrast, a significantly high proliferative
rate and decreased apoptosis was observed in the
SHP2-overexpressing MB-231 cells (Fig.
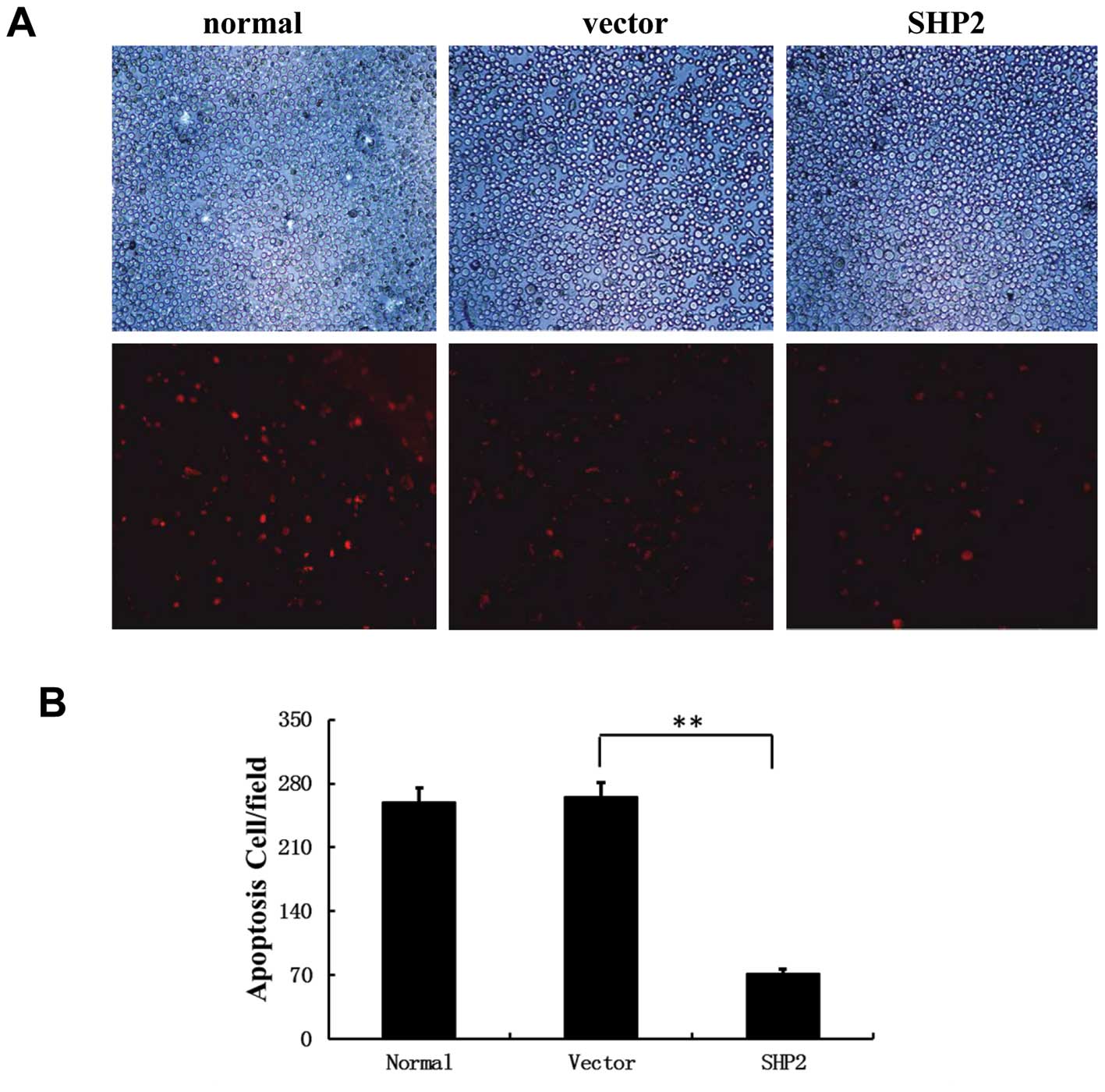
5A). In addition, fluorescence microscopy showed clear evidence
of fragmented nuclei in the MDA-MB-231/empty vector control cells
(Fig. 5A and B), which was markedly
decreased in the SHP2-overexpression group. These data confirmed a
positive role of SHP2 overexpression in MB-231 cell survival and
proliferation.
Overexpressing of SHP2 promotes mammary
tumor growth in mice
We demonstrated that the overexpression of SHP2 in
MB-231 cells was associated with tumor invasion, proliferation and
clone formation in an in vitro experiment. We next aimed to
test whether SHP2 promotes the growth of MB-231 cells in an in
vivo experiment. Therefore, a tumor xenograft model was
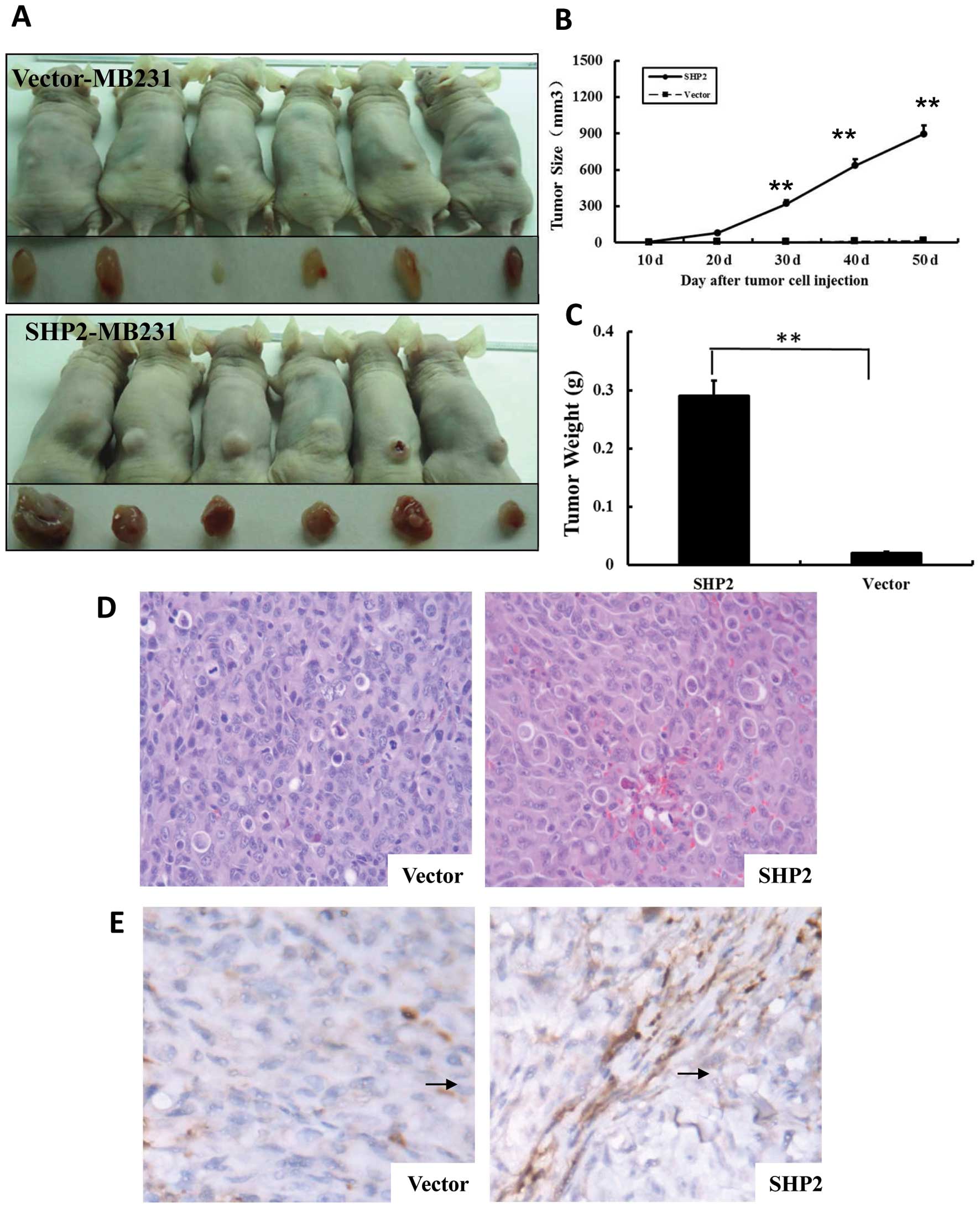
established by subcutaneously implantation of MB-231 cells. Mammary
tumors were detected 10 days after implantation (Fig. 6) in the vector-MB-231 and
SHP2-MB-231 groups. The tumor size in the SHP2-MB-231 group
continuously increased until day 50 after implantation (harvest
point). In contrast, the tumor size in the vector-MB-231 control
group remained at similar levels as day 10 until the end of the
observation period. As shown in Fig.
6B, the size of solid tumors was significantly higher in the
SHP2-MB-231 group than in the vector-MB231 control group. Moreover,
the tumor weight in the SHP2-MB-231 group was 20-fold higher when
compared with the vector-MB-231 control group (P<0.01) (Fig. 6C).
SHP2 overexpression enhances tumor
angiogenesis in mice
Since tumorigenesis is dependent on the sustained
formation of new vessels, we determined the effect of SHP2
overexpression on tumor angiogenesis in the tumor xenograft model
by performing CD31 IHC staining. As shown in Fig. 6E, there was an approximately
multiple increase in AMVD in the tumor tissues from the SHP2-MB-231
group when compared with the tissue sections from the vector-MB-231
control groups. Similar observation was confirmed from
morphological evaluations. These results suggest that SHP2
overexpression enhances tumor growth by promoting angiogenesis.
Overexpression of SHP2 contributes to
elevated phospho-Erk/AKT activation in breast cancer cells
SHP2 is a positive mediator of ligand-stimulated Ras
activation. Previous studies have demonstrated that the positive
effect of SHP2 on Ras activation is at least partially via the
promotion of the phosphorylation of downstream ligand (8,22,23).
SHP2 is required in signal transduction to downstream MEK/ERK and
PI-3K/AKT pathways (8,23). Therefore, we examined the activation
of MEK/ERK and PI-3K/AKT dependent kinase following SHP2
overexpression. Western blotting was performed to examine
phosphorylated Erk and AKT, and their counterparts, respectively.
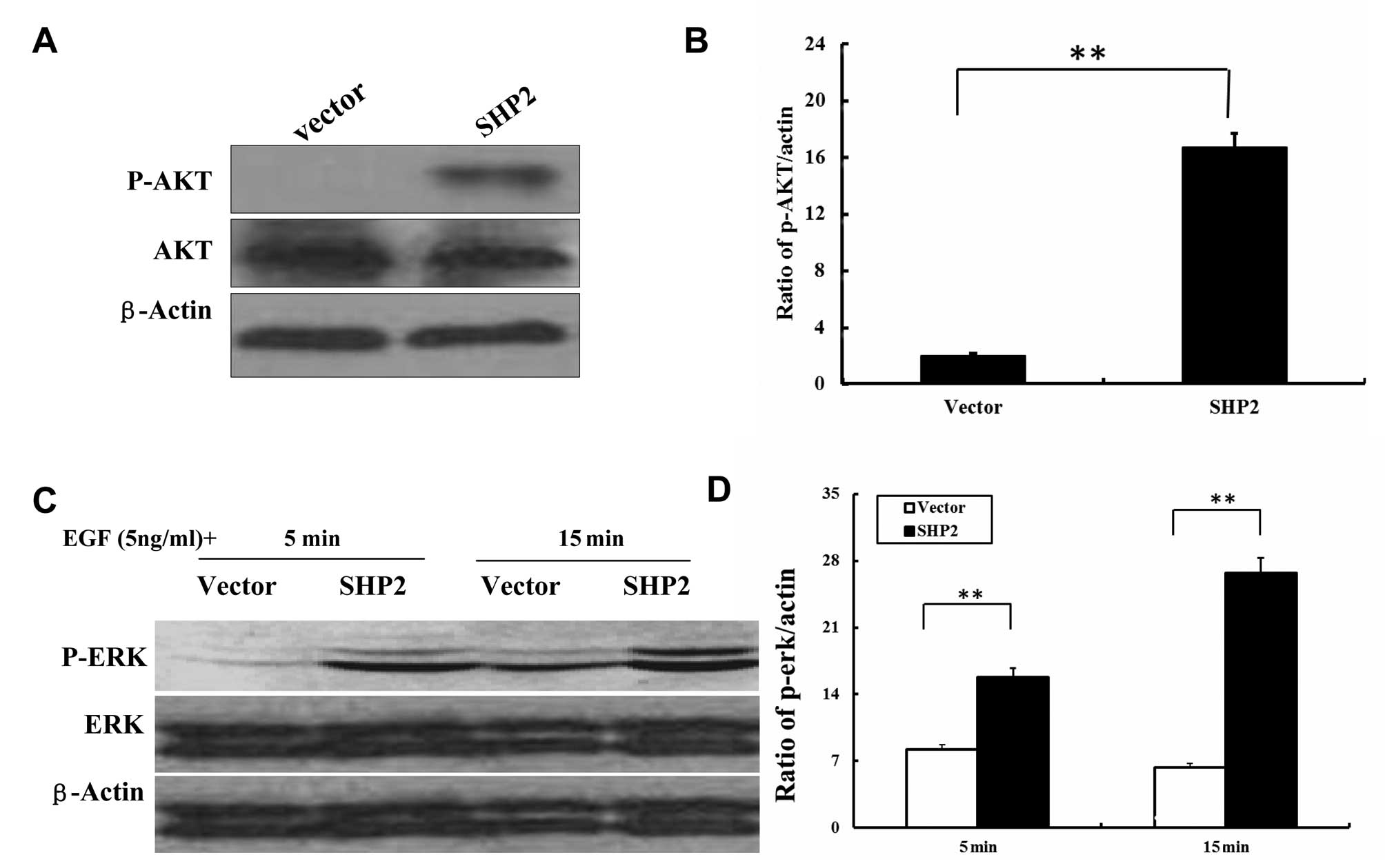
In the SHP2-MB-231 cells, SHP2 overexpression resulted in
significantly increased AKT phosphorylation (Fig. 7A and B). In addition, stimulation of
MB-231 cells with EGF caused phosphorylation of ERK 5 and 15 min
after incubation, respectively (Fig. 7C
and D). Taken together, these data suggest that SHP2 regulates
its downstream MEK/ERK and PI-3K/Akt signaling pathways in a
distinct manner in these cell lines and therefore may perturb cell
cycle progression at different check-points.
Discussion
Upregulation of SHP2 has been widely observed in
human breast cancer. The present study was designed to investigate
the effect of SHP2 on the malignant phenotype of human breast
cancer. Our results indicated that overexpression of SHP2 was
associated with increased cell proliferation, clone formation and
decreased chemotherapeutic sensitivity. Transfection of SHP2 into
breast cancer cells significantly promoted the tumor growth in a
mouse xenograft model. Moreover, the mechanism of the promotion of
tumorigenesis by SHP2 appears to involve its ability to increase
the activity of ERK/AKT-mediated signaling pathways.
Upregulation of SHP2 expression is observed in
advanced tumors (9,20,24,25),
and is accompanied by increased intracellular adhesion, and is
essential for tumor metastasis (18). Therefore, overexpression of SHP2 in
cancer has been considered as a mechanism mediating tumor
metastasis (11–14). Our results indicate that
upregulation of SHP2 expression in breast cancer cells promotes
tumor invasiveness. Inhibition of SHP2 in breast cancer cells was
found to cause transition of mesenchymal cells to epithelial cells,
leading to decreased anchorage-independent cell growth (19). These observations suggest that SHP2
may play a central role in tumor malignancy. We found that the
capabilities of adhesion and extension on fibronectin were
significantly promoted in breast cancer cells following SHP2
transfection. This observation supports the hypothesis that
overexpression of SHP2 accelerates the malignant potential of these
cells. Formation of large, invasive breast tumors in mice after
injection of the SHP2-MB-231 human breast tumor cells clearly
demonstrates a critical role of SHP2 in the rapid development of
breast tumors after tumor cell implantation.
Tumor angiogenesis is a crucial event in the growth
of solid tumors. In the present study, angiogenesis in the solid
tumors was increased after tumor cell implantation in the SHP2
overexpression group. Studies have demonstrated that a phenotype of
tumor angiogenesis is switched on from the very early stages of
tumor progression, in order to supply nutrients and oxygen. One
marker of new blood vessel formation is CD31, a member of the
immunoglobulin superfamily. Microvessel density can be quantitated
as a representation of active tumor-associated angiogenesis by
performing CD31 staining. Typically, microvessels are localized at
the peripheral region of the solid tumor (26), which is consistent with our
findings. In the present study, mass necrosis was observed in the
central part of the solid tumors derived from the injection of
MB-231 cells. A previous study showed a correlation between hypoxia
and necrosis of solid tumors (27),
revealing that the oxygen supply as well as angiogenesis might be
important factors in tumor growth. Of note, no necrosis was
observed in the solid tumors induced by implantation of the
SHP2-overexpressing MB-231 cells. Moreover, significantly increased
CD31 expression was observed as well in the same group. Our
observations suggest that the angiogenic phenotype of the
SHP2-overexpressing human tumor xenografts provides a survival
advantage to the interior tumor epithelial cells.
Apoptosis is an essential factor to maintain
homeostasis, and loss of apoptosis is an important characteristic
of oncogenesis (17). Research
indicates that SHP2 overexpression is associated with decreased
apoptosis of tumor cells in a 3D culture model (21,22).
Our results indicated that MB-231 cells with SHP2 overexpression
showed a significantly lower apoptotic rate following treatment
with cisplatin. The transfection of SHP2 resulted in decreased cell
death in the MB-231 cells and induced colony-formation ability in
either 2D or 3D culture, suggesting that overexpression of SHP2 may
be a causative factor in tumorigenesis.
Noteworthy, downregulation of most members of the Sr
family is observed in tumors (8).
This discrepancy may be due to tissue-specific molecular mechanisms
(28). The function of SHP2 in the
development of breast cancer needs further investigation.
SHP2 is required in signal transduction to
downstream MEK/ERK and PI-3K/AKT pathways (4,8,29). It
is reported, that SHP2 interferes with MAPK cellular signal
transduction via Erk, JNK/SAPK, p38/RK and BMK1/Erk5 in leukemic
cell and in breast cancer MCF-7 cells (17). In the present study, we examined the
activation of MEK/ERK and PI-3K/AKT dependent kinase following SHP2
activation. We found that overexpression of SHP2 was associated
with the activation of these signaling pathways in MB-231 cells.
Therefore, overexpression of SHP2 may be necessary for mediating
mammary tumorigenesis through promotion of Erk signaling or
selective activation of PI3K signaling. These results reveal a
critical role of SHP2 in breast cancer.
Taken together, our results indicate that
overexpression of SHP2 in breast cancer cells leads to malignant
transformation, and SHP2 appears to be a novel candidate for
establishing new therapeutic strategies against breast cancer. Our
results suggest that SHP2 may represent a key molecule in
tumorigenesis, and blockade of SHP2 may be an effective
immunotherapeutic strategy.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (codes: 30873046,
30973424, 81072663 and 81272258) and by the Fundamental Research
Funds for the Central Universities (2242014K40004).
Abbreviations:
|
SHP2
|
Src homology phosphotyrosine
phosphatase 2
|
References
|
1
|
Bentires-Alj M, Paez JG, David FS, et al:
Activating mutations of the Noonan syndrome-associated SHP2/PTPN11
gene in human solid tumors and adult acute myelogenous leukemia.
Cancer Res. 64:8816–8820. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ben-Jonathan N, Liby K, McFarland M and
Zinger M: Prolactin as an autocrine/paracrine growth factor in
human cancer. Trends Endocrinol Metab. 13:245–250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chan G, Kalaitzidis D and Neel BG: The
tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis
Rev. 27:179–192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martinelli S, Carta C, Flex E, et al:
Activating PTPN11 mutations play a minor role in pediatric and
adult solid tumors. Cancer Genet Cytogenet. 166:124–129. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miyamoto D, Miyamoto M, Takahashi A,
Yomogita Y, Higashi H, Kondo S and Hatakeyama M: Isolation of a
distinct class of gain-of-function SHP-2 mutants with oncogenic
RAS-like transforming activity from solid tumors. Oncogene.
27:3508–3515. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grossmann KS, Rosario M, Birchmeier C and
Birchmeier W: The tyrosine phosphatase Shp2 in development and
cancer. Adv Cancer Res. 106:53–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chan RJ and Feng GS: PTPN11 is the first
identified proto-oncogene that encodes a tyrosine phosphatase.
Blood. 109:862–867. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Serra V, Scaltriti M, Prudkin L, et al:
PI3K inhibition results in enhanced HER signaling and acquired ERK
dependency in HER2-overexpressing breast cancer. Oncogene.
30:2547–2557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tartaglia M, Mehler EL, Goldberg R, et al:
Mutations in PTPN11, encoding the protein tyrosine phosphatase
SHP-2, cause Noonan syndrome. Nat Genet. 29:465–468. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Edouard T, Combier JP, Nedelec A, et al:
Functional effects of PTPN11 (SHP2) mutations causing LEOPARD
syndrome on epidermal growth factor-induced phosphoinositide
3-kinase/AKT/glycogen synthase kinase 3beta signaling. Mol Cell
Biol. 30:2498–2507. 2010. View Article : Google Scholar
|
|
11
|
Masunaga R, Kohno H, Dhar DK, et al:
Cyclooxygenase-2 expression correlates with tumor
neovascularization and prognosis in human colorectal carcinoma
patients. Clin Cancer Res. 6:4064–4068. 2000.PubMed/NCBI
|
|
12
|
Zhou X, Coad J, Ducatman B and Agazie YM:
SHP2 is up-regulated in breast cancer cells and in infiltrating
ductal carcinoma of the breast, implying its involvement in breast
oncogenesis. Histopathology. 53:389–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong Q, Siminovitch KA, Fialkow L,
Fukushima T and Downey GP: Negative regulation of myeloid cell
proliferation and function by the SH2 domain-containing tyrosine
phosphatase-1. J Immunol. 162:3220–3230. 1999.PubMed/NCBI
|
|
14
|
Agarwal R, D’Souza T and Morin PJ:
Claudin-3 and claudin-4 expression in ovarian epithelial cells
enhances invasion and is associated with increased matrix
metalloproteinase-2 activity. Cancer Res. 65:7378–7385. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Frearson JA and Alexander DR: The
phosphotyrosine phosphatase SHP-2 participates in a multimeric
signaling complex and regulates T cell receptor (TCR) coupling to
the Ras/mitogen-activated protein kinase (MAPK) pathway in Jurkat T
cells. J Exp Med. 187:1417–1426. 1998. View Article : Google Scholar
|
|
16
|
Hadari YR, Kouhara H, Lax I and
Schlessinger J: Binding of Shp2 tyrosine phosphatase to FRS2 is
essential for fibroblast growth factor-induced PC12 cell
differentiation. Mol Cell Biol. 18:3966–3973. 1998.PubMed/NCBI
|
|
17
|
Carver KC, Piazza TM and Schuler LA:
Prolactin enhances insulin-like growth factor I receptor
phosphorylation by decreasing its association with the tyrosine
phosphatase SHP-2 in MCF-7 breast cancer cells. J Biol Chem.
285:8003–8012. 2010. View Article : Google Scholar
|
|
18
|
Zhou XD and Agazie YM: Inhibition of SHP2
leads to mesenchymal to epithelial transition in breast cancer
cells. Cell Death Differ. 15:988–996. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou X and Agazie YM: Molecular mechanism
for SHP2 in promoting HER2-induced signaling and transformation. J
Biol Chem. 284:12226–12234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tartaglia M, Niemeyer CM, Fragale A, et
al: Somatic mutations in PTPN11 in juvenile myelomonocytic
leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat
Genet. 34:148–150. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eklund EA, Jalava A and Kakar R: PU.1,
interferon regulatory factor 1, and interferon consensus
sequence-binding protein cooperate to increase gp91(phox)
expression. J Biol Chem. 273:13957–13965. 1998. View Article : Google Scholar
|
|
22
|
Bouyain S and Watkins DJ: Identification
of tyrosine phosphatase ligands for contactin cell adhesion
molecules. Commun Integr Biol. 3:284–286. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang S, Yu WM, Zhang W, McCrae KR, Neel BG
and Qu CK: Noonan syndrome/leukemia-associated gain-of-function
mutations in SHP-2 phosphatase (PTPN11) enhance cell migration and
angiogenesis. J Biol Chem. 284:913–920. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tartaglia M, Kalidas K, Shaw A, et al:
PTPN11 mutations in Noonan syndrome: molecular spectrum,
genotype-phenotype correlation, and phenotypic heterogeneity. Am J
Hum Genet. 70:1555–1563. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tartaglia M, Martinelli S, Iavarone I, et
al: Somatic PTPN11 mutations in childhood acute myeloid leukaemia.
Br J Haematol. 129:333–339. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen H, Pimienta G, Gu Y, et al: Proteomic
characterization of Her2/neu-overexpressing breast cancer cells.
Proteomics. 10:3800–3810. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nijsten T, Colpaert CG, Vermeulen PB,
Harris AL, Van ME and Lambert J: Cyclooxygenase-2 expression and
angiogenesis in squamous cell carcinoma of the skin and its
precursors: a paired immunohistochemical study of 35 cases. Br J
Dermatol. 151:837–845. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van IC, Rahner C and Anderson JM:
Regulated expression of claudin-4 decreases paracellular
conductance through a selective decrease in sodium permeability. J
Clin Invest. 107:1319–1327. 2001. View
Article : Google Scholar
|
|
29
|
Montagner A, Yart A, Dance M, Perret B,
Salles JP and Raynal P: A novel role for Gab1 and SHP2 in epidermal
growth factor-induced Ras activation. J Biol Chem. 280:5350–5360.
2005. View Article : Google Scholar : PubMed/NCBI
|