Introduction
Cluster of differentiation 166 (CD166) is a cell
surface member of the immunoglobulin superfamily (1), which is overexpressed and regarded as
a valuable prognostic marker of disease progression and poor
survival in several types of epithelial tumors (2–4). Gene
silencing of CD166 decreases the concentration of Bcl-2 and
increases the level of apoptosis (PARP, active caspase-7) (5). We previously reported that the
activation of anti-apoptotic canonical NF-κB signaling greatly
induces CD166 expression in liver cancer cells after serum
deprivation (6), suggesting its
important roles in regulating apoptosis. Most recently, we revealed
that CD166 can exert its anti-apoptotic role by enhancing YAP
function, demonstrating that CD166 is an upstream regulator of YAP
(7). However, overexpression of YAP
cannot completely rescue the increased anti-carcinogenic effects
evoked by knockdown of CD166 (7).
Thus, the downstream regulation of the CD166 pro-carcinogenic
function needs to be further explored.
The forkhead box transcription factor superfamily
consists of 19 subclasses of FOX genes, FOXA-FOXS (8). The FOX transcription factors that
belong to the other (O) subfamily (FOXO) include four members
(FOXO1, 3, 4, 6) in mammals (8).
Overexpression of FOXO proteins inhibits tumor growth in
vitro and tumor size in vivo (9). The nuclear accumulation of FOXO
proteins was found to suspend cell cycle progression and promote
apoptosis in breast cancers (9,10).
Recent research efforts also provide new insights that FOXO
proteins appear to present antitumor properties in liver cancer,
including induction of the expression of pro-apoptotic genes, or
interfering with signaling cascades commonly altered in this
disease such as Wnt/β-catenin, PI3K/AKT/mTOR or MAPK pathways
(11). However, the upstream
regulation of FOXO functions, particularly those involving cell
membrane proteins are still largely unknown in liver cancer
cells.
In the present study, we found that CD166 exerts its
pro-carcinogenic role via the inhibition of FOXO proteins, i.e.
CD166 facilitates phosphorylation, cytosolic accumulation and
instability of FOXO proteins. The anti-carcinogenic function of
FOXO proteins can be reversed by CD166. Moreover, our data also
demonstrate that AKT is an inter-mediator between the upstream
regulator, CD166, and downstream effector, FOXO, in liver cancer
cells. Disruption of the relationship between CD166 and the
AKT/FOXO axis may serve as a novel therapeutic target for liver
cancer patients.
Materials and methods
Cell culture
Bel-7402, SMMC-7721, Chang Liver and HL-7702 cells
were cultured in DMEM supplemented with 5% fetal bovine serum
(FBS). Cells were treated by Wortmannin (50 μM; Cayman, Ann Arbor,
MI, USA), LY294002 [20 μM; Cell Signaling Technology (CST), Boston,
MA, USA], cycloheximide (CHX, 50 μg/ml; Sigma-Aldrich, St. Louis,
MO, USA) or MG132 (25 μM; Cayman) 1–24 h before harvest.
shRNA and protein expressing
plasmids
Lentiviral CD166-shRNA1 was purchased from Open
Biosystems (Huntsville, AL, USA; catalog no. TRCN0000150706).
CD166-shRNA2 and AKT-shRNA were cloned into pLKO.1 lentiviral
plasmid using the following primers: CD166-sh2-F,
CCGGTCAAGCAACCATCTAAACCTGCTCGAGCAGGTTTAGATGGTTGCTTGATTTTTG and
CD166-sh2-R,
AATTCAAAAATCAAGCAACCATCTAAACCTGCTCGAGCAGGTTTAGATGGTTGCTTGA;
AKT-shRNA-F,
CCGGAACTCCTCAAGAATGATGGCACTCGAGTGCCATCATTCTTGAGGAGTTTTTTTG and
AKT-shRNA-R,
AATTCAAAAAAACTCCTCAAGAATGATGGCACTCGAGTGCCATCATTCTTGAGGAGTT. AKT-Myc
was cloned into pGIPZ based lentiviral plasmid using the primers as
follows: AKT-Myc-F, GGTCGCTAGCAGCGACGTGGCTATTGTGAA and AKT-Myc-R,
GTATCCTGCAGGTTACAGATCTTCTTCAGAAATAAGTTTTTGTTCGGCCGTGCCGCTGGCCGAT.
The CD166-HA was cloned into pcDNA3.1(+) plasmid using the
following primers: CD166-HA-F, GTACGGATCCCACCAAGAAGGAGGAGGAAT and
CD166-HA-R, GTACCTCGAGTTAAGCGTAGTCTGGGACGTCGTATGGGTAGGCTTCAGTTTT.
The pTEN-HA-expressing plasmid was a gift from Dr Xuqian Fang
(Shanghai Jiaotong University, Shanghai, China), and the FOXO1 and
FOXO3a protein-expressing plasmids were purchased from OriGene
(Beijing, China).
Western blotting (WB)
Proteins were resolved on SDS-PAGE gels followed by
standard WB. Primary antibodies used were: anti-CD166 (Epitomics,
Burlingame, CA, USA; catalog no 3133), anti-HA [Cell Signaling
Technology (CST); catalog no. 3724], anti-Myc (CST; catalog no.
2278), anti-ubiquitin (CST; catalog no. 3936), anti-AKT (Epitomics,
catalog no. 2957), anti-p-AKT substrate (CST; catalog no. 9614),
anti-GAPDH (CST; catalog no. 5174), anti-p-FOXO3a (S253) (CST;
catalog no. 9466), anti-FOXO3a (CST; catalog no. 2497),
anti-p-FOXO1 (T24)/p-FOXO3a (T32) (CST; catalog no. 9464),
anti-p-FOXO1 (S256) (CST; catalog no. 9461), anti-FOXO1 (CST;
catalog no. 2880), anti-β-tublin (Epitomics; catalog no. 1879),
anti-p-AKT (CST; catalog no. 2965) and anti-histone H3 (Hua’an
Hangzhou, China; catalog no. R1105). For extracting nuclear and
cytosolic fractions of cells, a kit from Active Motif (Carlsbad,
CA, USA) was used.
Immunofluorescence (IF)
For IF, cells were fixed by 4% paraformaldehyde
(PFA) for 15 min, washed with PBS and blocking buffer (3% FBS + 1%
HISS + 0.1% Triton X-100) and then incubated overnight at 4°C in
primary antibodies against HA (CST; catalog no 2367) and FOXO3a
(Epitomics; catalog no. 3280). Alexa Fluor 488 or 555
fluorescent-conjugated secondary antibodies (Life Technologies,
Carlsbad, CA, USA) were used for detection.
Immunohistochemistry (IHC)
Following deparaffinization and rehydration of the
tissue sections, antigen retrieval was performed at 100°C for 2 h
with Tris-EDTA buffer, pH 6.0 (Beyotime). Endogenous peroxidase was
blocked with 3% peroxide for 20 min, followed by additional rinses
in PBS for 3×5 min. Sections were then blocked in a buffer
containing 5% BSA and 0.1% Triton X-100 and incubated overnight in
primary antibodies against CD166 (Epitomics, Burlingame, CA, USA;
catalog no. 3133) or FOXO3a (Epitomics; catalog no 3280). Signal
detection was accomplished using the Vectastain ABC kit (Vector
Laboratories, Burlingame, CA, USA).
Cell proliferation, caspase-3/7 activity
and soft agar assays
Cell proliferation was measured by an MTT-based
proliferation assay, as previously described (12). Caspase-3/7 activity was determined
using the Caspase-Glo 3/7 assay system (Promega, Madison, WI, USA).
Anchorage-independent soft agar growth assay and quantitative
RT-PCR was performed as previously described (12).
Xenograft mouse model
Bel-7402 cells (5×106) expressing
proteins as indicated were subcutaneously injected into athymic
nude mice (Bikai, Shanghai, China). Tumor size was measured every
six days using a caliper, and the tumor volume was calculated as
0.5 × L × W2, with L indicating length and W indicating
width. The mice were euthanized at 24 days after injection.
Results
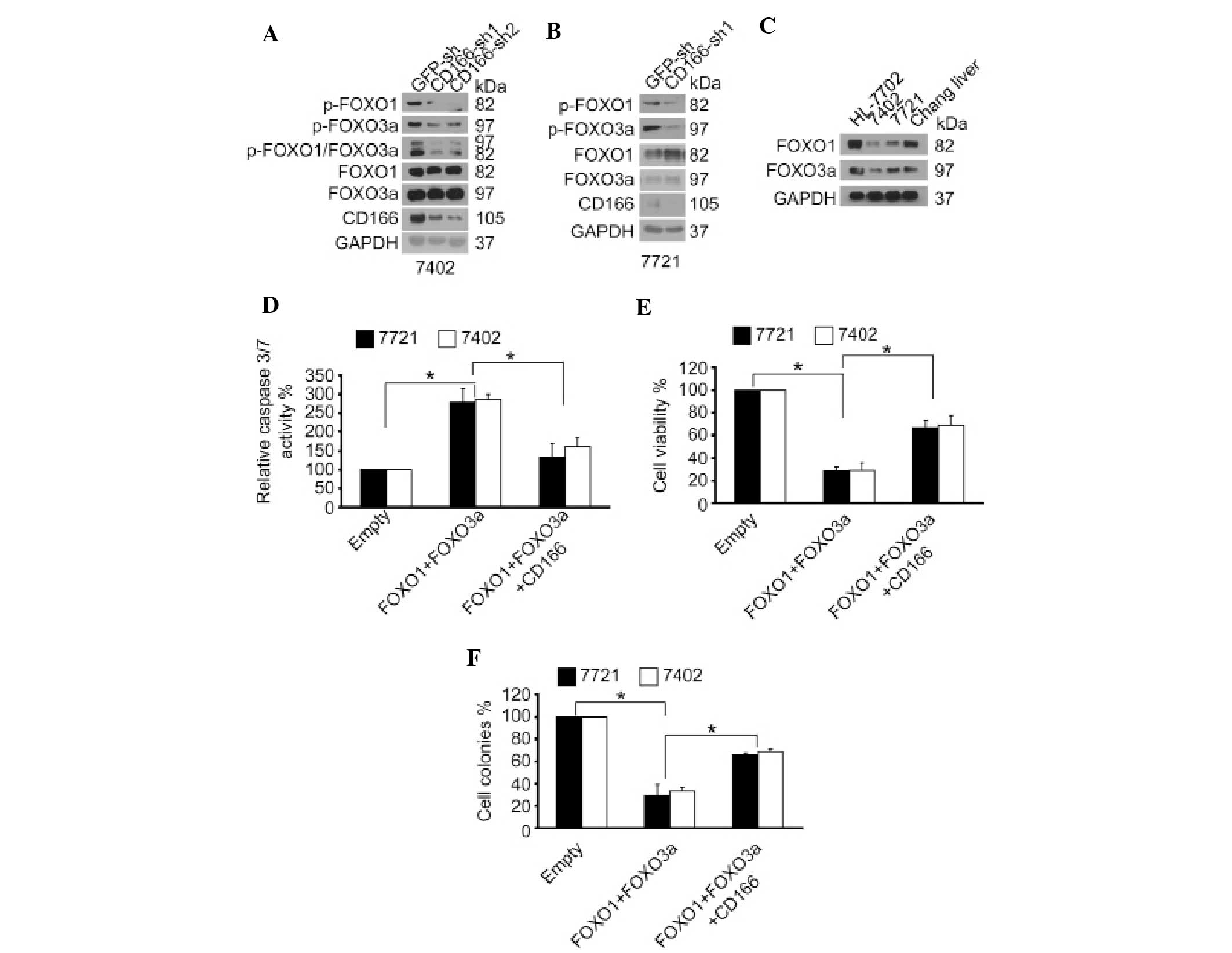
CD166 regulates phosphorylation of FOXO
proteins
We previously reported that silencing of CD166 can
induce apoptosis (7). Notably,
activation of FOXO proteins can also induce apoptosis (13). In addition, FOXO protein activity
can be inactivated through phosphorylation (8). Thereby, we tested whether CD166 has a
role in the regulation of phosphorylation of FOXO proteins.
Compared to the control, dephosphorylation of both FOXO1 and FOXO3a
was detected in Bel-7402 cells following CD166 knockdown (Fig. 1A). Similar results were also
obtained in SMMC-7721 cells (Fig.
1B). Moreover, it was demonstrated that both FOXO1 and FOXO3a
had relative higher levels in normal hepatic cell lines (HL-7702
and Chang Liver) compared to levels in the liver cancer cell lines
(Bel-7402 and SMMC-7721) (Fig. 1C),
suggesting that lower FOXO proteins levels in liver cancer cells
may lead to tumorigenesis. Collectively, the above data revealed
that CD166 exerts its pro-carcinogenetic role through inhibition of
FOXO proteins.
CD166 reverses the anti-carcinogenetic
effects induced by FOXO proteins
We examined whether CD166 plays a negative role on
FOXO proteins in liver cancer cells. We found that simultaneous
overexpression of FOXO1 and FOXO3a decreased cell proliferation
compared to the control, as measured by an MTT-based assay and
Ki-67 immunostaining (Fig. 1E and
data not shown). Furthermore, we found that overexpression of FOXO
proteins impaired the ability of these cells to form colonies in
soft agar (Fig. 1F), whereas
markedly increased apoptosis was noted, as shown by increased
caspase-3/7 activity and caspase-3 cleavage by immunostaining
(Fig. 1D and data not shown). In
addition, we observed that the reduced cell survival and
transformative phenotype induced by overexpression of FOXO proteins
could be partially rescued by simultaneous ectopic expression of
CD166 (Fig. 1D–F). These data
indicate that the inhibition of FOXO by CD166 is important for
human liver cancer cell growth and survival.
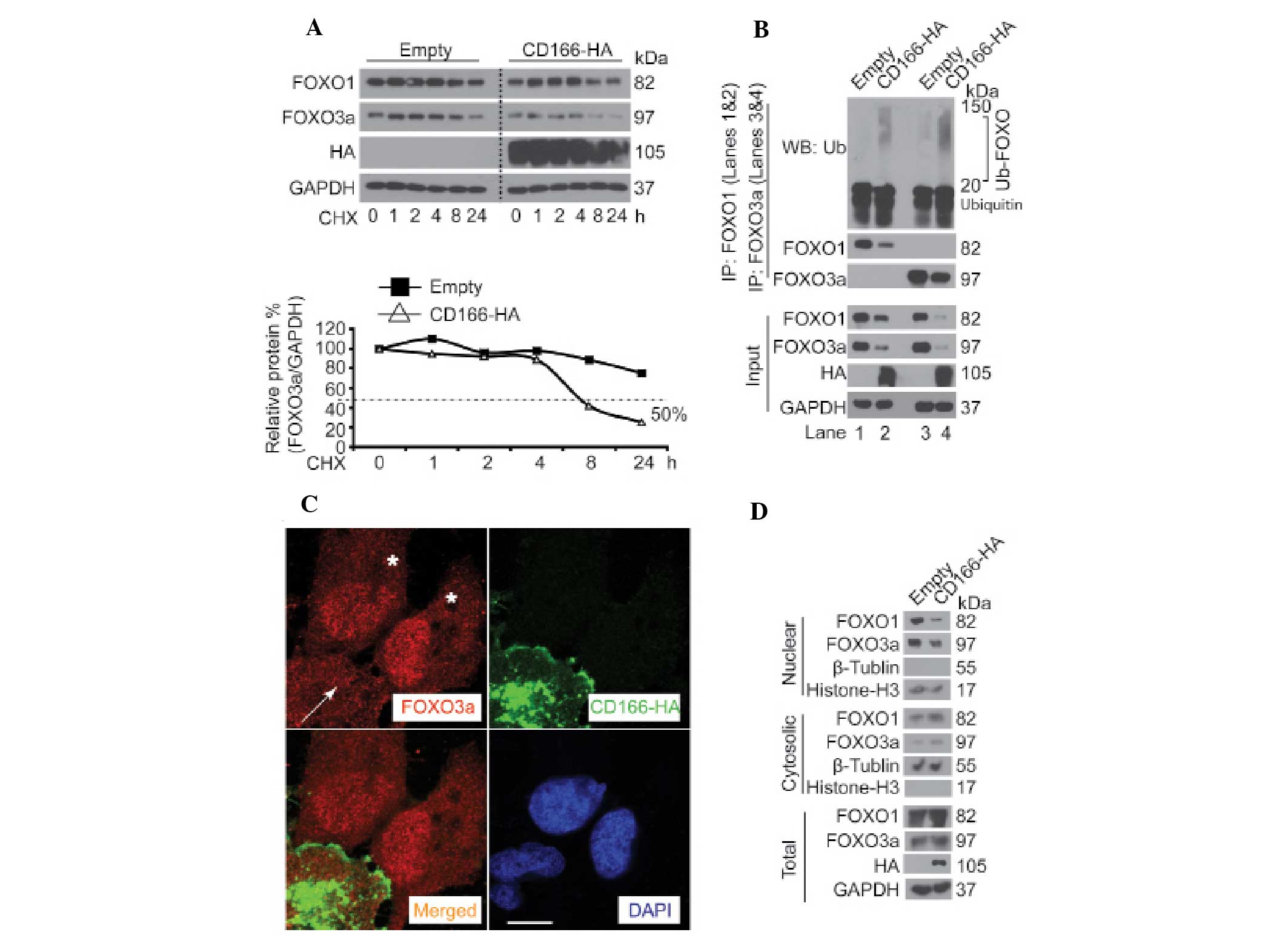
CD166 controls stability and subcellular
localization of FOXO proteins
Protein degradation is initialized by target protein
modification, such as phosphorylation (14,15).
Since CD166 regulated the phosphorylation of FOXO proteins
(Fig. 1A and B), we hypothesized
that CD166 also modulates degradation of FOXO proteins. It was
found that when Bel-7402 cells were treated with the protein
synthesis inhibitor, cycloheximide (CHX), the half-life time of
FOXO1 and FOXO3a was >8 h. However, FOXO1 and FOXO3a degraded
much more rapidly with a half-life time of ~8 h after
overexpression of CD166 (Fig. 2A,
upper panel). Compared to FOXO1, the effects of CD166 on FOXO3a
were much more obvious (Fig. 2A,
lower panel). In addition, in Bel-7402 cells with long exposure (72
h) to CD166, reduced expression accompanied by a greater
accumulation of ubiquitinated FOXO1 and FOXO3a was detected
compared to the control (for FOXO1, lane 2 vs. 1 and for FOXO3a,
lane 4 vs. 3) (Fig. 2B), suggesting
that CD166 is a regulator of ubiquitintation and degradation of
FOXO proteins. As known, inactivation of FOXO proteins leads to
their accumulation in the cytoplasm (8). To ascertain whether CD166 affects the
subcellular localization of FOXO, Bel-7402 cells were transfected
with CD166-HA-expressing plasmids. It was detected that nuclear
FOXO3a expression was significantly reduced in the Bel-7402 cells
with CD166-HA overexpression compared to the control (Fig. 2C). This observation was confirmed by
fractionation studies, which revealed that overexpression of CD166
facilitated FOXO protein localization from the nuclear fraction to
the cytosolic fraction (Fig. 2D).
Taken together, the data demonstrate that CD166 modulates FOXO
protein stability through alteration of their subcellular
localization.
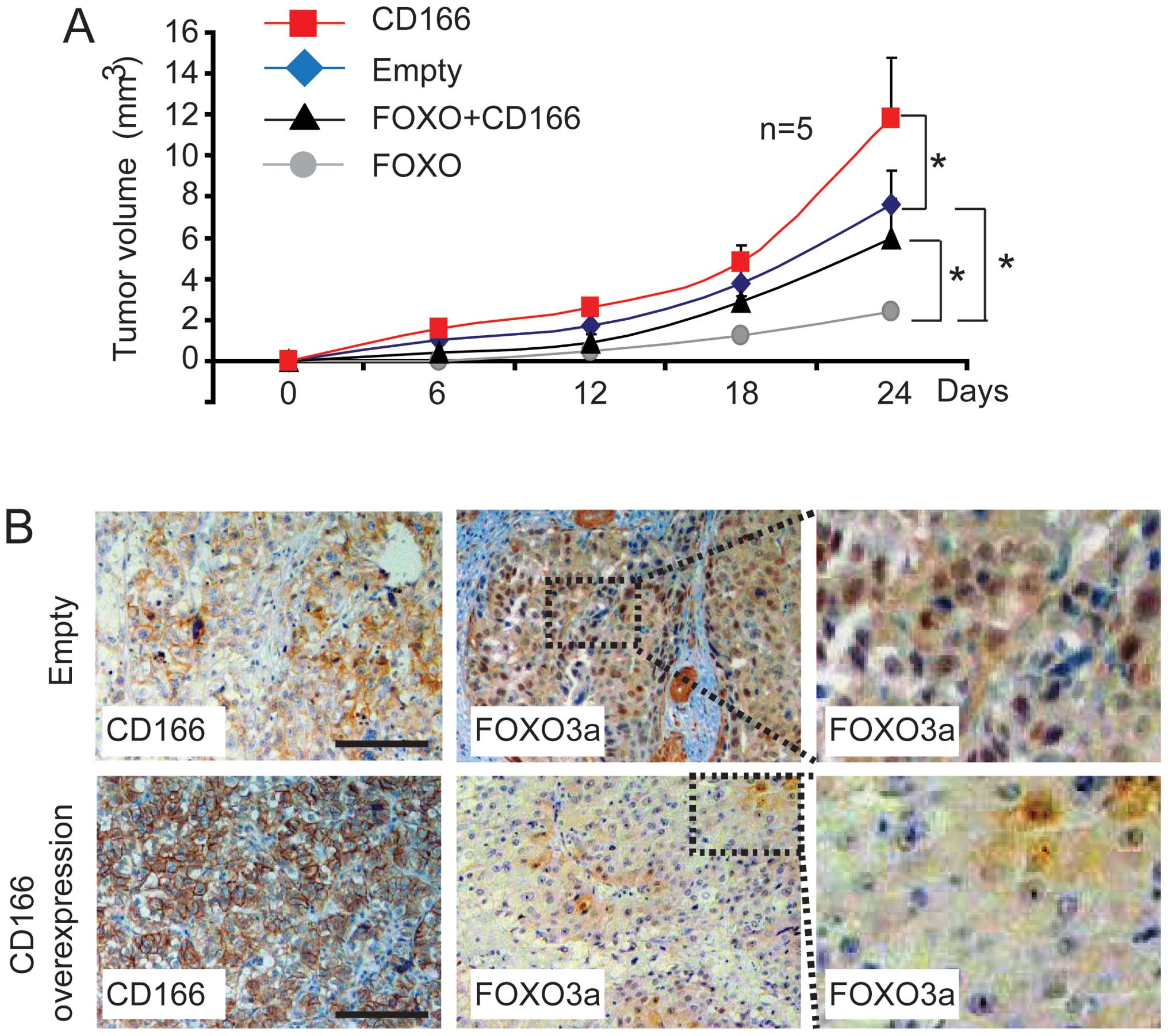
CD166 promotes liver cancer cell growth
through inhibition of FOXO in vivo
On the basis of the evidence that CD166 reduces
protein stability and expression (Fig.
2), we investigated the growth of Bel-7402 clones after
injection into athymic mice. Compared to the control (transfected
with the empty plasmid), Bel-7402 cells with CD166 overexpression
exhibited a relatively higher tumor growth rate (Fig. 3A). In comparison, Bel-7402 cells
with FOXO (FOXO1 and FOXO3a) overexpression effectively prevented
tumor growth, yet this effect was rescued by simultaneous
overexpression of CD166 (Fig. 3A),
thereby confirming the close relationship between FOXO and CD166
in vivo. To ascertain whether CD166 contributes to the
inhibition of FOXO in vivo, we stained sections from the
xenografts using anti-FOXO3a antibodies. Similar to the data shown
in Fig. 2B and C, the protein
expression of FOXO3a, particularly the nuclear fraction of FOXO3a
was significantly downregulated in xenograft tissue with CD166
overexpression compared to the control (transfected with the empty
plasmids) (Fig. 3B), suggesting
that long-term exposure to the overexpression of CD166 leads to
translocation from the nucleus to the cytoplasm and instability of
FOXO proteins.
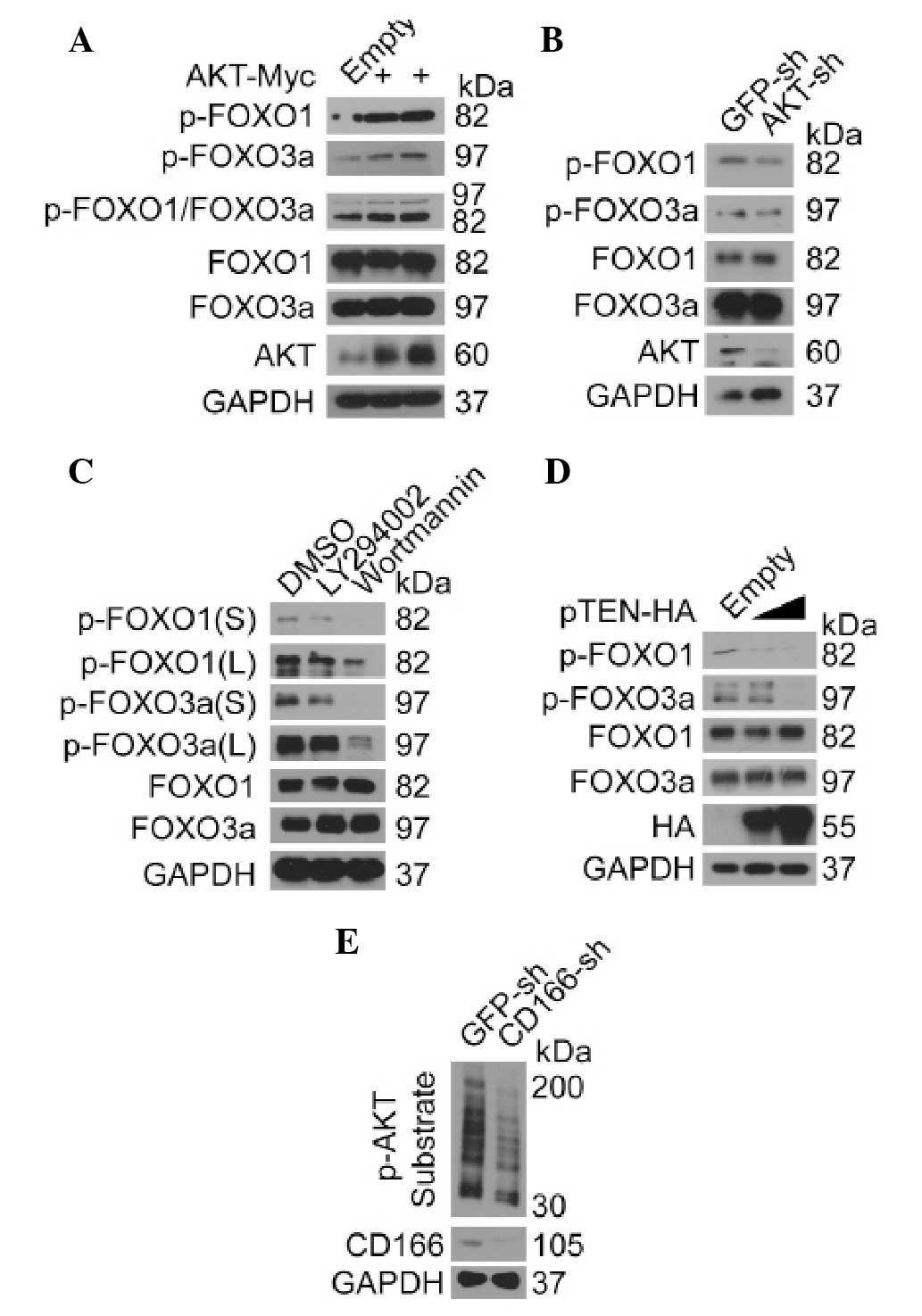
AKT regulates FOXO proteins in liver
cancer cells
Emerging evidence suggests that AKT controls the
activity of FOXO proteins (8,16,17).
However, to the best of our knowledge, there is no direct evidence
to support the conclusion that AKT regulates FOXO proteins in liver
cancer cells. Thus, we investigated the effect of AKT on FOXO in
Bel-7402 cells. Compare to the control, we found that
phosphorylation of FOXO1 and FOXO3a was greatly induced by
overexpression of AKT (Fig. 4A),
whereas it was reduced after knockdown of AKT (Fig. 4B). By using chemical PI3K/AKT
inhibitors, LY294002 and Wortmannin, respectively, we found that
phosphorylation of both FOXO1 and FOXO3a was markedly reduced
(Fig. 4C). Furthermore, when
endogenous PI3K/AKT inhibitor, pTEN, was overexpressed,
phosphorylation of FOXO1 and FOXO3a was also inhibited in a
dose-dependent manner (Fig. 4D),
suggesting that FOXO proteins are also regulated by AKT in liver
cancer cells. As shown in Fig. 4E,
we confirmed that AKT activity could be downregulated after
knockdown of CD166, as the phosphorylation of AKT substrates was
markedly reduced in the Bel-7402 cells with CD166 knockdown
compared to the control suggesting that the regulation of FOXO by
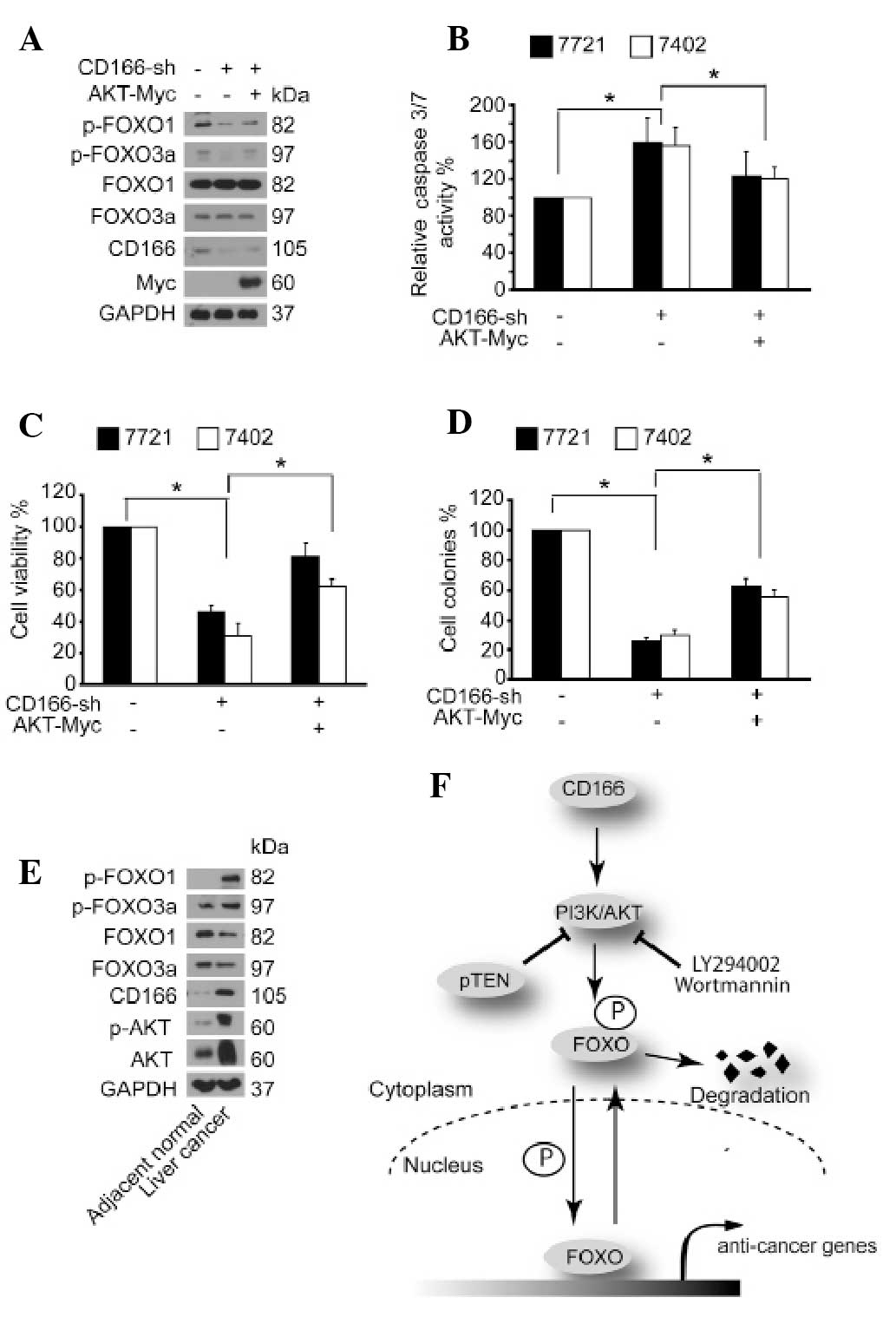
CD166 may be AKT dependent.
CD166 regulates FOXO proteins via
AKT
We then investigated whether the phosphorylation of
FOXO proteins controlled by CD166 is AKT dependent. It was found
that knockdown of CD166-induced dephosphorylation of FOXO proteins
could be rescued by simultaneous overexpression of AKT (Fig. 5A). Moreover, the induced caspase-3/7
activity, reduced cell proliferation and soft agar colony formation
by depletion of CD166 was partially reversed after overexpression
of AKT (Fig. 5B–D). Thus, we
proposed that AKT may act as an inter-mediator between the upstream
regulator, CD166, and the downstream effector, FOXO. Next, we
tested the protein expression patterns of FOXO, AKT and CD166 in
liver cancers and their adjacent normal liver tissues. Compared to
the adjacent normal liver tissues, higher expression levels of
CD166, p-AKT, total AKT and p-FOXO1/3a were correlated with lower
expression levels of total FOXO1/3a in liver cancer tissues
(Fig. 5E), suggesting that
upregulation of CD166 leads to induction of AKT, which in turn
facilitates phosphorylation and degradation of FOXO in liver
cancer.
Discussion
In the present study, we describe a close
relationship between CD166 and FOXO proteins (Fig. 5F). These two types of proteins play
opposing roles in the regulation of tumorigenesis in liver cancer
(6,7,11,18,19).
Nuclear localization of FOXO proteins induces expression of
anti-carcinogenic genes (11),
whereas CD166 facilitates translocation of FOXO proteins from the
nucleus to the cytoplasm (Fig. 2C and
D). CD166 maintains anti-apoptotic Bcl-2 protein expression,
suggesting that the anti-apoptotic function of CD166 is partially
dependent on Bcl-2 (7). Notably,
overexpression of Bcl-2 also diminishes death induced by expression
of FOXO proteins (20). We
previously reported that the anti-apoptotic function of CD166 is
via enhancement of both expression and activity of onco-protein,
YAP (7). The degradation of YAP can
be protected by TRIB2 (21–22), a protein that is also capable of
inhibiting and reducing nuclear FOXO proteins (23). Thus, we consider that CD166 plays a
similar role to that of TRIB2. YAP is a pro-carcinogenic protein
(7,12), while FOXO proteins are
anti-carcinogenic in liver cancer. However, whether and how YAP
antagonizes FOXO still remains unknown and needs further
exploration.
It has been reported that AKT phosphorylates FOXO3a
at T32 and S253 and FOXO1 at T24 and S256, respectively, which are
conserved from Caenorhabditis elegans to mammals (24). Notably, these phosphorylation sites
can also be regulated by CD166 (Fig. 1A
and B). Furthermore, the anti-carcinogenic effects and the
regulation of phosphorylation of FOXO proteins by knockdown of
CD166 can be reversed by AKT (Fig.
5A–D), providing another evidence that CD166 can regulate
AKT.
In summary, our data indicate that the CD166/AKT
axis modulates tumorigenesis via promotion of phosphorylation and
facilitation of degradation, ubiquitination and cytosolic
accumulation of FOXO proteins in liver cancer cells. Further
exploration of the interplay among these important signaling
pathways may lead to more effective therapeutic strategies for
liver cancer.
Acknowledgements
This study was supported by China National 973
Projects (grant nos. 20111812 and 20110402), the Natural Science
Foundation of China (grant nos. 81272292 and 81301689) and Climbing
Training Program (to J.W.) from Shanghai Tenth People’s
Hospital.
Abbreviations:
|
CD166
|
cluster of differentiation 166
|
|
CHX
|
cycloheximide
|
|
FOXO
|
forkhead box O
|
|
shRNA
|
small hairpin RNA
|
References
|
1
|
van Kempen LC, Nelissen JM, Degen WG,
Torensma R, Weidle UH, Bloemers HP, Figdor CG and Swart GW:
Molecular basis for the homophilic activated leukocyte cell
adhesion molecule (ALCAM)-ALCAM interaction. J Biol Chem.
276:25783–25790. 2001.PubMed/NCBI
|
|
2
|
Weichert W, Knösel T, Bellach J, Dietel M
and Kristiansen G: ALCAM/CD166 is overexpressed in colorectal
carcinoma and correlates with shortened patient survival. J Clin
Pathol. 57:1160–1164. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burkhardt M, Mayordomo E, Winzer KJ,
Fritzsche F, Gansukh T, Pahl S, Weichert W, Denkert C, Guski H and
Dietel MG: Cytoplasmic overexpression of ALCAM is prognostic of
disease progression in breast cancer. J Clin Pathol. 59:403–409.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kahlert C, Weber H, Mogler C, Bergmann F,
Schirmacher P, Kenngott HG, Matterne U, Mollberg N, Rahbari NN,
Hinz U, Koch M, Aigner M and Weitz J: Increased expression of
ALCAM/CD166 in pancreatic cancer is an independent prognostic
marker for poor survival and early tumour relapse. Br J Cancer.
101:457–464. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jezierska A, Matysiak W and Motyl T:
ALCAM/CD166 protects breast cancer cells against apoptosis and
autophagy. Med Sci Monit. 12:BR263–BR273. 2006.PubMed/NCBI
|
|
6
|
Wang J, Gu Z, Ni P, Qiao Y, Chen C, Liu X,
Lin J, Chen N and Fan Q: NF-κB P50/P65 hetero-dimer mediates
differential regulation of CD166/ALCAM expression via interaction
with micoRNA-9 after serum deprivation, providing evidence for a
novel negative auto-regulatory loop. Nucleic Acids Res.
39:6440–6455. 2011.
|
|
7
|
Ma L, Wang J, Lin J, Pan Q, Yu Y and Sun
F: Cluster of differentiation 166 (CD166) regulated by
phosphatidylinositide 3-kinases (PI3K)/AKT signaling to exert its
anti-apoptotic role via yes associated protein (YAP) in liver
cancer. J Biol Chem. Jan 30–2014.(Epub ahead of print). View Article : Google Scholar
|
|
8
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Medema RH, Kops GJ, Bos JL and Burgering
BM: AFX-like Forkhead transcription factors mediate cell-cycle
regulation by Ras and PKB through p27kip1. Nature.
404:782–787. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Urbich C, Knau A, Fichtlscherer S, Walter
DH, Brühl T, Potente M, Hofmann WK, de Vos S, Zeiher AM and
Dimmeler S: FOXO-dependent expression of the proapoptotic protein
Bim: pivotal role for apoptosis signaling in endothelial progenitor
cells. FASEB J. 19:974–976. 2005.PubMed/NCBI
|
|
11
|
Carbajo-Pescador S, Mauriz JL,
García-Palomo A and González-Gallego J: FoxO proteins: regulation
and molecular targets in liver cancer. Curr Med Chem. 21:1231–1246.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang J, Ma L, Weng W, Qiao Y, Zhang Y, He
J, Wang H, Xiao W, Li L, Chu Q, Pan Q, Yu Y and Sun F: Mutual
interaction between YAP and CREB promotes tumorigenesis in liver
cancer. Hepatology. 58:1011–1020. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
You H, Yamamoto K and Mak TW: Regulation
of transactivation-independent proapoptotic activity of p53 by
FOXO3a. Proc Natl Acad Sci USA. 103:9051–9056. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamashita M, Ying SX, Zhang GM, Li C,
Cheng SY, Deng CX and Zhang YE: Ubiquitin ligase Smurf1 controls
osteoblast activity and bone homeostasis by targeting MEKK2 for
degradation. Cell. 121:101–113. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sapkota G, Alarcon C, Spagnoli FM,
Brivanlou AH and Massague J: Balancing BMP signaling through
integrated inputs into the Smad1 linker. Mol Cell. 25:441–454.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fu Z and Tindall DJ: FOXOs, cancer and
regulation of apoptosis. Oncogene. 27:2312–2319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hales EC, Taub JW and Matherly LH: New
insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling
axis: targeted therapy of γ-secretase inhibitor resistant T-cell
acute lymphoblastic leukemia. Cell Signal. 26:149–161.
2014.PubMed/NCBI
|
|
18
|
Lin A, Yao J, Zhuang L, Wang D, Han J and
Lam EW; TCGA Research Network and Gan B. The FoxO-BNIP3 axis exerts
a unique regulation of mTORC1 and cell survival under energy
stress. Oncogene. Jul 15–2013.(Epub ahead of print). View Article : Google Scholar
|
|
19
|
Tao GZ, Lehwald N, Jang KY, Baek J, Xu B,
Omary MB and Sylvester KG: Wnt/β-catenin signaling protects mouse
liver against oxidative stress-induced apoptosis through the
inhibition of forkhead transcription factor FoxO3. J Biol Chem.
28:17214–17224. 2013.
|
|
20
|
Yusuf I, Zhu X, Kharas MGs, Chen J and
Fruman DA: Optimal B-cell proliferation requires phosphoinositide
3-kinase-dependent inactivation of FOXO transcription factors.
Blood. 104:784–787. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Zhang Y, Weng W, Qiao Y, Ma L,
Xiao W, Yu Y, Pan Q and Sun F: Impaired phosphorylation and
ubiquitination by p70 S6 kinase (p70S6K) and smad ubiquitination
regulatory factor 1 (Smurf1) promote tribbles homolog 2 (TRIB2)
stability and carcinogenic property in liver cancer. J Biol Chem.
288:33667–33681. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Park JS, Wei Y, Rajurkar M, Cotton
JL, Fan Q, Lewis BC, Ji H and Mao J: TRIB2 acts downstream of
Wnt/TCF in liver cancer cells to regulate YAP and C/EBPα function.
Mol Cell. 51:211–225. 2013.PubMed/NCBI
|
|
23
|
Zanella F, Renner O, García B, Callejas S,
Dopazo A, Peregrina S, Carnero A and Link W: Human TRIB2 is a
repressor of FOXO that contributes to the malignant phenotype of
melanoma cells. Oncogene. 29:2973–2982. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Manning BD and Cantley LC: AKT/PKB
signaling: navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|