Introduction
Nucleostemin (NS) is a protein related to cell
proliferation which was originally identified in 2002, and located
in the nucleolus; it was first found in some early pluripotent
cells such as embryonic, neural and bone marrow-derived stem cells
in rat, but was not expressed in terminally differentiated cells
(1,2). NS is highly expressed in several types
of tumor tissues and cells, such as prostate and esophageal cancer
(3,4). After knocking down NS expression in
tumor cells, its proliferation became weak, and most studies
suggest that NS regulates cell proliferation through blocking the
cells at the G0/G1 or G2/M point and then leads the cells out of
the cell cycle (5–7). These studies indicated that NS is
essential to maintain proliferation and undifferentiated state of
stem and tumor cells.
Most traditional studies on NS action mechanism
suggest that NS could combine with p53 to inhibit its tumor
suppressor function, and then lead to tumorigenesis (1). In addition, NS could protect telomere,
its overexpression could regulate the telomere length negatively
and then delay cell senescence; however, the absence of NS could
increase the probability of telomere damage and mutation and then
result in cell senescence (8). This
may also be one type of mechanism of NS in tumorigenesis and tumor
development.
However, in our previous studies, we used human
acute myeloid leukemia cell line HL-60 which was p53-null as the
research material. After inhibiting NS expression, HL-60 cells
presented more apoptosis (9). Thus,
we speculated that NS may also play its role independent of p53.
Similar views have been reported by others, including Beekman et
al (10), Jafamejad et
al (11) and Nikpour et
al (12). However, to date,
only few studies have focused on the detailed mechanism of this
pathway.
Clinically, the therapeutic effect of tumors is
closely associated with p53. P53-null or mutate is one of the main
reasons for drug-resistant, poor prognostic and therapeutic
effects. Thus, exploring effective treatment targets and measures
for p53-null and p53-mutate tumors is a critical issue that
requires immediate attention. The present study analyzed the gene
expression profiling changes of HL-60 cells after knocking down NS,
aiming to explore the detailed mechanism of NS p53-independent
pathway. These findings may lay the foundations for drug
development for p53-null leukemia and even p53-null tumors.
Materials and methods
Cell culture
HL-60 cells were cultured in RPMI-1640 medium
(Gibco-BRL) supplemented with 10% fetal bovine serum (FBS;
Gibco-BRL), 100 U/ml of penicillin, 100 μg/ml streptomycin and were
incubated at 37°C with 5% humidified CO2. The cells were
changed medium every 2 days.
Construction of NS-siRNA lentiviral
vector
RNA interference (RNAi) sequence was designed
corresponding to NS gene (Genbank NM_014366). Two single-stranded
DNA oligonucleotides containing NS-RNAi sequence, loop circle,
AgeI/EcoRI enzyme cutting site and termination signal
sequence were synthesized (Table I)
and annealed to a double-stranded DNA template. Subsequently, the
template was inserted into the AgeI and EcoRI enzyme
sites of GV248 lentiviral vector (Genechem, Shanghai, China) with
catalysis by T4 DNA ligase to construct recombinant vectors
expressing NS-shRNA. The recombinant vectors were transformed into
competent E.coli and then confirmed by DNA sequencing. The
recombinant vector and packaging vectors were co-transfected into
293T cells, and the supernatants containing packaged vectors were
harvested 48 h later. Subsequent purification using
ultracentrifugation was performed and the titer of lentiviruses was
determined.
 | Table ISequences of two single-stranded DNA
oligonucleotides. |
Table I
Sequences of two single-stranded DNA
oligonucleotides.
| No. | 5′ | STEM | Loop | STEM | 3′ |
|---|
| NS-RNAi-1 | ccgg |
agCAAGTATTGAAGTAGTAAA | CTCGAG |
TTTACTACTTCAATACTTGCT | TTTTTg |
| NS-RNAi-2 | aattcaaaaa |
agCAAGTATTGAAGTAGTAAA | CTCGAG |
TTTACTACTTCAATACTTGCT | |
Validation of the RNA-interference
effect
Fresh medium for HL-60 cells was changed 24 h before
transfection to ensure the cells in logarithmic phase at
transfection. At the time of transfection, the cells were harvested
and centrifuged, and then suspended in fresh medium with 10% FBS
and seeded into a 6-well plate. A certain amount of NS-shRNA
lentivirus was added to HL-60 cells according to lentiviral titer
and MOI of HL-60 cells. At the same time, to exclude the influence
of blank lentiviral vector and off-target silencing effect mediated
by specific-shRNA, the blank control and the negative control group
were also set. The same amount of blank lentiviral vectors and
negative control lentivirus was added to HL-60 cells as the blank
control group and negative control group, respectively. The final
volume in each well was 2 ml. Sixteen hours after transfection, the
medium was changed and continued to culture. Seventy-two hours
after transfection, the plate was placed under inverted
fluorescence microscope to observe the transfection efficiency.
Cells (5–10×106) in each group were
harvested 96 h after transfection. Total-RNA was extracted from
cells using TRIzol reagent (Invitrogen). The reverse transcription
reaction was performed with PrimeScript® RT reagent Kit
with gDNA Eraser (Takara). Expression of NS mRNA was detected by
ABI 7500 real-time PCR instrument using SYBR® Premix Ex
TapTMII (Tli RNaseH Plus) Kit (Takara). The sequence of primers for
NS and GAPDH are shown in Table
II. Relative gene expression was quantified to calculate the
inhibition efficiency.
| Table IIPrimer pairs used for quantitative
RT-PCR. |
Table II
Primer pairs used for quantitative
RT-PCR.
| Gene | Primer pairs |
|---|
| PIK3CD | F: 5′-TTT CTC ATG GCT
GTC CTT CAG-3′
R: 5′-CAG GAG AAT CTA ACG GAT GC-3′ |
| AKT2 | F: 5′-CAT CAC ATC TGG
TTT CCT TGG-3′
R: 5′-AAC TGG AAA TGT AAT TTT GGG-3′ |
| STAT3 | F: 5′-ACC AGC AAT ATA
GCC GAT TCC-3′
R: 5′-CCA TTG GCT TCT CAA GAT ACC-3′ |
| STAT5A | F: 5′-ATT ATC TCA GCC
CTG GTG ACC-3′
R: 5′-CTG CTG CTC ACT GAT GAT GGT-3′ |
| GRB2 | F: 5′-GGA CAT CCT CAA
GGT TTT GAA C-3′
R: 5′-CGC TCT CAC TCT CTC GGA TAA G-3′ |
| HRAS | F: 5′-AGC TGA TCC AGA
ACC ATT TTG T-3′
R: 5′-GTT GAT GGC AAA CAC ACA CAG-3′ |
| MAPK9 | F: 5′-CTG CGT CAC CCA
TAC ATC AC-3′
R: 5′-CTT TCT TCC AAC TGG GCA TC-3′ |
| MAPK13 | F: 5′-AGG TCT CTG GGG
GTT GAG TTG GG-3′
R: 5′-AGG GGC AGC AAC GTC TCA TTG C-3′ |
| GAPDH | F: 5′-TGA CTT CAA CAG
CGA CAC CCA-3′
R: 5′-CAC CCT GTT GCT GTA GCC AAA-3′ |
| NS | F:
5′-TAGAGGTGTTGGATGCCAGAG-3′
R: 5′-CACGCTTGGTTATCTTCCCTTTA-3′ |
Microarray hybridization
Total RNA was purified using the RNase Mini Kit
(Qiagen p/n 74104), according to the protocol for Quick Amp
Labeling Kit, One-Color (Agilent p/n 5190-0442), reverse transcript
total-RNA to cDNA, and then further transcripted cDNA to cRNA with
Cy3 labeling and purifying it. Next, the purified Cy3-labeled cRNA
was used for hybridization on Agilent 4×44K Human Whole-Genome
60-mer oligonucleotide microarrays following the protocol for
Agilent Gene Expression Hybridization Kit (p/n 5188–5242).
Microarray data acquisition, processing
and analysis
Microarrays were scanned with Agilent DNA Microarray
Scanner (Agilent p/n G2565BA), using the setting parameters
recommended by Agilent Technologies (Green PMT were set at XDR Hi
100% and XDR Lo 10%; scan resolution was set to 5 μm). Then, the
Agilent Feature Extraction software v11.0.1.1 was used to analyze
acquired microarray images, and the resulting text files extracted
from it were imported into the GeneSpring GX v12.0 software package
(Agilent Technologies) for further analysis. After quantile
normalization of the raw data, genes that at least 2/2 samples have
flags in Detected (‘All Targets Value’) were chosen for further
data analysis. Differentially expressed genes with statistical
significance were identified through fold-change filtering for 2
compared samples. Pathway analysis of the differentially expressed
genes was performed using the KEGG Pathway Database (http://www.genome.jp/kegg).
Real-time quantitative PCR
Reverse transcription was performed using
PrimeScript® RT reagent Kit with gDNA Eraser (Takara).
Quantification of gene expression was performed by ABI 7500
real-time PCR instrument using SYBR® Premix Ex TapTMII
(Tli RnaseH Plus) Kit (Takara). The expression level of GAPDH was
used as an internal control. The expression of the following genes
was analyzed: PIK3CD, AKT2, STAT3, STAT5A, GRB2, HRAS, MAPK9,
MAPK13. The primers are listed in Table II.
Statistical analysis
The experiments were repeated at least three times
and the data are presented as means ± SD (standard deviation).
Statistical software SPSS 17.0 was used for the assessment. The
Student’s t-test was used to compare means of two groups. Fisher’s
exact test was used to analyze the significance of the pathway.
P<0.05 was considered to indicate statistically significant
differences.
Results
Downregulation of NS in HL-60 cells after
NS-shRNA lentivirus transfection
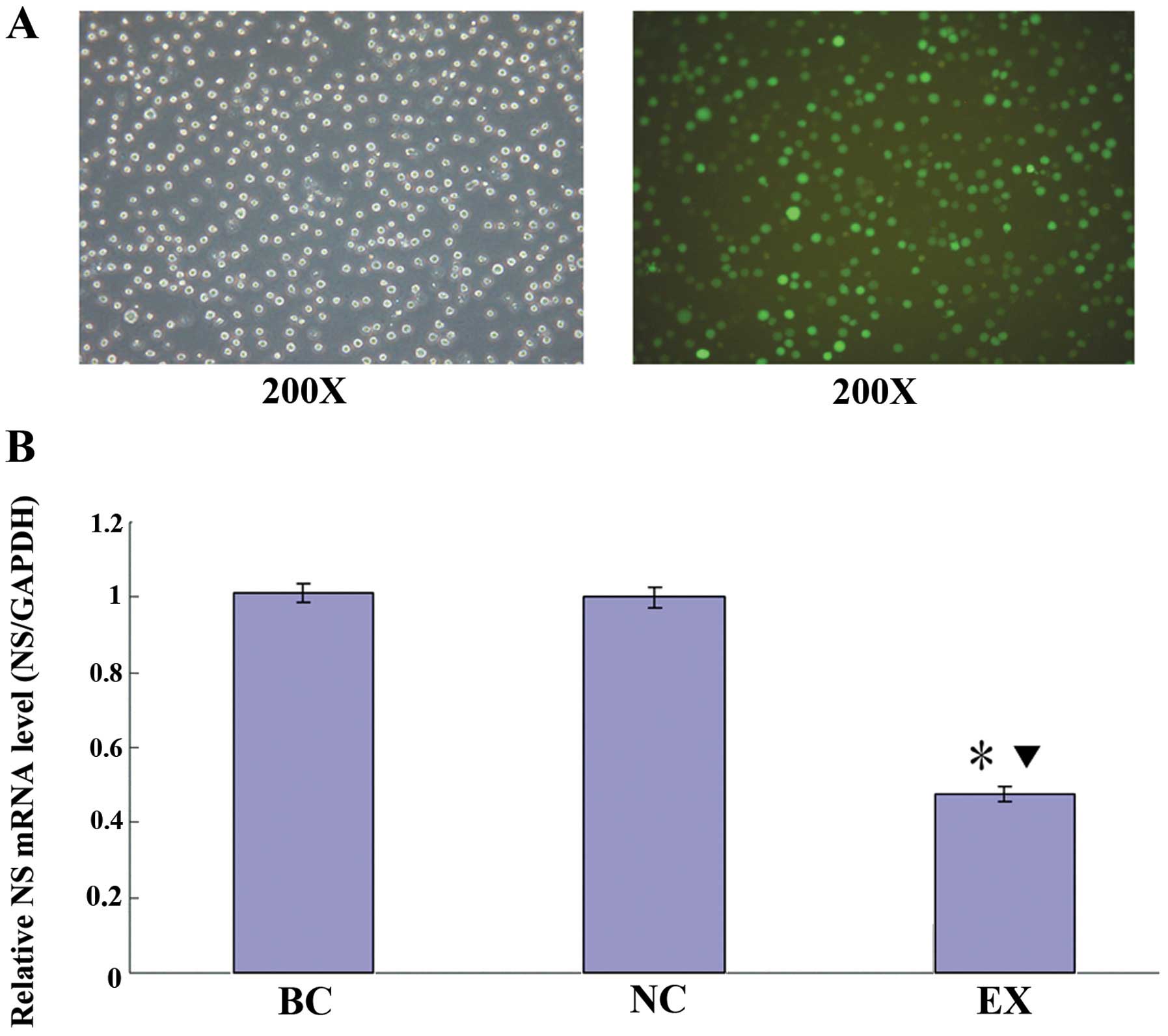
The lentiviral vectors transfected HL-60 cells
successfully and the transfection efficiency was >80% which
could be observed under an inverted fluorescence microscope
(Fig. 1A). The NS mRNA expression
was determined by real-time qPCR. As shown in Fig. 1B, there was no significant
difference between the blank control group (BC) and the negative
control group (NC; P>0.05); compared with the BC and the NC
group, the levels of NS mRNA in the experimental group (EX) were
significantly reduced by 52.9 and 52.3%, respectively (P<0.05).
To maximize reducing the off-target effect, we chose the NC and the
EX group for further DNA microarray analysis.
Data analysis: general features
Total-RNA isolated from the EX and NC group cells
was used to synthesize cDNA. Probe labeling, hybridization and
scanning were performed by Shanghai Kangcheng Co., Ltd (China). To
identify differentially expressed genes with statistical
significance, a fold change filtering between two samples was
performed and the default threshold was ≥2.0 fold-change. Results
showed that 2,628 genes, 818 upregulated and 1,810 downregulated,
were differentially expressed in HL-60 cells following
downregulation of NS.
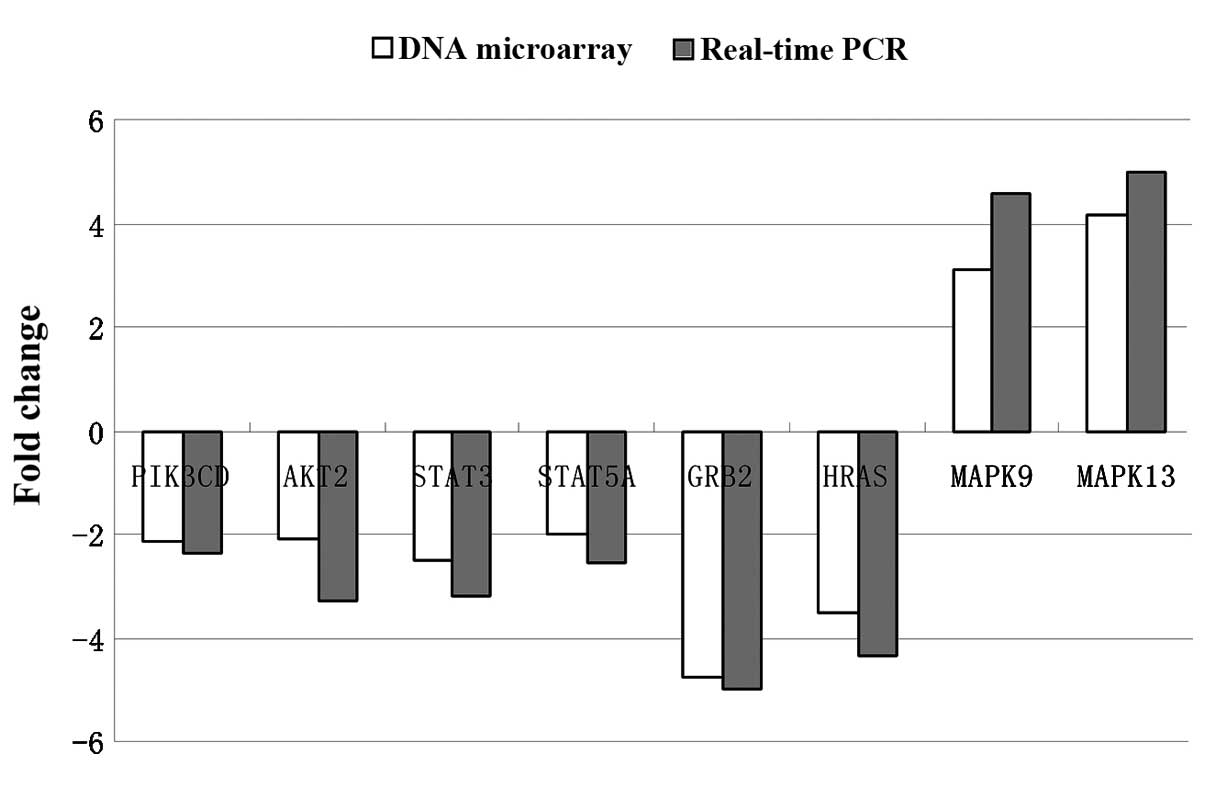
Real-time qPCR validation
To evaluate the reliability of the array results,
eight differentially expressed genes (PIK3CD, AKT2, STAT3, STAT5A,
GRB2, HRAS, MAPK9, MAPK13), which had >2-fold-changes according
to the microarray assay results, were selected for further
validation by real-time qPCR. The real-time qPCR results of these
genes were generally in agreement with microarray data (Fig. 2). Although the fold-changes of these
genes determined respectively by microarray and real-time qPCR were
not completely the same, the general trends were. This was due to
some noise that was inevitable. Nevertheless, these results still
confirm our findings of differential gene expression by microarray
analysis.
Pathway analysis
We proceeded with pathway analysis based on the KEGG
database to illustrate all the available pathways containing
differentially expressed genes. The significant pathways affected
due to upregulated and downregulated genes are shown in Tables III and IV, respectively.
| Table IIIPathway analysis of upregulated
genes. |
Table III
Pathway analysis of upregulated
genes.
| PathwayID | Definition | Fisher’s
P-value | Genes |
|---|
| hsa05322 | Systemic lupus
erythematosus - Homo sapiens (human) | 7.77667E-06 |
C7//CTSG//ELANE//HIST1H2AC//HIST1H2AI//HIST1H2BB//HIST1H2BF//HIST1H2BH//HIST1H2BK//HIST1H2BL//HIST1H2BO//HIST1H3B//HIST1H3H//HIST1H4H//HIST1H4L//HIST2H2BE//HIST2H2BF |
| hsa05144 | Malaria - Homo
sapiens (human) | 0.000416632 |
CCL2//DARC//HBB//HBD//HGF//IL1B//IL8//THBS1 |
| hsa04621 | NOD-like receptor
signaling pathway - Homo sapiens (human) | 0.001132948 |
CASP5//CCL2//CXCL1//IL1B//IL8//MAPK13//MAPK9//NLRP1 |
| hsa04340 | Hedgehog signaling
pathway - Homo sapiens (human) | 0.003689235 |
BMP7//CSNK1E//CSNK1G1//PRKACB//PTCH1//SUFU//WNT4 |
| hsa05146 | Amoebiasis -
Homo sapiens (human) | 0.004635966 |
COL11A2//CTSG//CXCL1//HSPB1//IL1B//IL8//LAMC1//PRKACB//RAB5A//SERPINB13 |
| hsa04010 | MAPK signaling
pathway - Homo sapiens (human) | 0.007661671 |
CACNA1B//CACNA1E//FGF4//FGF8//FGFR3//FLNA//HSPB1//IL1B//JUN//MAPK13//MAPK8IP2//MAPK9//NTRK2//PLA2G2A//PRKACB//RAC1//RAPGEF2//RASGRP3 |
| hsa05020 | Prion diseases -
Homo sapiens (human) | 0.007782237 |
C7//EGR1//IL1B//LAMC1//PRKACB |
| hsa05120 | Epithelial cell
signaling in Helicobacter pylori infection - Homo
sapiens (human) | 0.01072619 |
CXCL1//IL8//JAM2//JUN//MAPK13//MAPK9//RAC1 |
| hsa04620 | Toll-like receptor
signaling pathway - Homo sapiens (human) | 0.01085929 |
IL1B//IL8//JUN//LY96//MAPK13//MAPK9//RAC1//SPP1//TLR6 |
| hsa04512 | ECM-receptor
interaction - THBS1 Homo sapiens (human) | 0.01106789 |
CD44//COL11A2//ITGAV//LAMC1//SPP1//SV2B//SV2C// |
| hsa05132 | Salmonella
infection - Homo sapiens (human) | 0.01264349 |
CXCL1//FLNA//IL1B//IL8//JUN//MAPK13//MAPK9//RAC1 |
| hsa05217 | Basal cell
carcinoma - Homo sapiens (human) | 0.01355671 |
FZD4//FZD5//FZD7//PTCH1//SUFU//WNT4 |
| hsa05200 | Pathways in cancer
- Homo sapiens (human) | 0.01360474 |
CCND1//CYCS//FGF4//FGF8//FGFR3//FZD4//FZD5//FZD7//HGF//IL8//ITGAV//JUN//LAMC1//MAPK9//PTCH1//RAC1//RARA//RUNX1//SUFU//WNT4 |
| hsa04310 | Wnt signaling
pathway - Homo sapiens (human) | 0.01989438 |
CCND1//CSNK1E//FZD4//FZD5//FZD7//JUN//MAPK9//PPP2R5B//PRKACB//RAC1//WNT4 |
| hsa04962 |
Vasopressin-regulated water reabsorption -
Homo sapiens (human) | 0.02006348 |
ARHGDIB//DYNLL2//PRKACB//RAB11A//RAB5A |
| hsa04510 | Focal adhesion -
Homo sapiens (human) | 0.02786726 |
CAPN2//CCND1//COL11A2//FLNA//HGF//ITGAV//JUN//LAMC1//MAPK9//PXN//RAC1//SPP1//THBS1 |
| hsa04210 | Apoptosis - Homo
sapiens (human) | 0.03465665 |
ATM//CAPN2//CYCS//IL1B//IL1RAP//PRKACB//TNFRSF10C |
| hsa04722 | Neurotrophin
signaling pathway - Homo sapiens (human) | 0.03905394 |
ARHGDIB//JUN//MAPK13//MAPK9//NTRK2//PDK1//RAC1//SH2B2//YWHAE |
| hsa05014 | Amyotrophic lateral
sclerosis (ALS) - Homo sapiens (human) | 0.04095384 |
ALS2//CYCS//MAPK13//RAB5A//RAC1 |
| hsa04964 | Proximal tubule
bicarbonate reclamation - Homo sapiens (human) | 0.0480162 |
ATP1B1//GLS//SLC38A3 |
| hsa05133 | Pertussis - Homo
sapiens (human) | 0.0495752 |
IL1B//IL8//JUN//LY96//MAPK13//MAPK9 |
| Table IVPathway analysis of downregulated
genes. |
Table IV
Pathway analysis of downregulated
genes.
| PathwayID | Definition | Fisher’s
P-value | Genes |
|---|
| hsa03040 | Spliceosome -
Homo sapiens (human) | 3.71589E-08 |
ACIN1//BUD31//CHERP//CTNNBL1//DDX39B//DHX38//HNRNPA1L2//HNRNPA3//HNRNPC//HNRNPK//HSPA8//ISY1//LSM4//NHP2L1//PPIE//PQBP1//PRPF31//PRPF6//PRPF8//RBMX//SF3B2//SF3B4//SNRNP40//SNRNP70//SNRPA//SNRPB//SNRPC//SRSF3//SRSF9//TCERG1//THOC1//THOC3//THOC4//TRA2B |
| hsa03013 | RNA transport -
Homo sapiens (human) | 2.51665E-05 |
AAAS//ACIN1//DDX39B//EEF1A1//EIF2B4//EIF2B5//EIF3C//EIF3F//EIF3G//EIF4A1//EIF4B//EIF4G1//GEMIN8//NUP188//NUP210//NUP214//NUP62//NUP93//NXF1//POM121//PRMT5//RNPS1//SEC13//SNUPN//TACC3//THOC1//THOC3//THOC4//THOC5//THOC6//WIBG//XPO5 |
| hsa03410 | Base excision
repair - Homo sapiens (human) | 0.000263071 |
APEX1//FEN1//HMGB1//LIG1//MPG//NTHL1//OGG1//PARP1//POLD2//POLE//XRCC1 |
| hsa00520 | Amino sugar and
nucleotide sugar metabolism - Homo sapiens (human) | 0.00053131 |
GALE//GALK1//GALT//GMPPA//GMPPB//GNPDA1//GNPNAT1//HEXA//NAGK//NPL//PMM1//PMM2//TSTA3 |
| hsa05221 | Acute myeloid
leukemia - Homo sapiens (human) | 0.001151056 |
AKT2//ARAF//CEBPA//GRB2//HRAS//IKBKG//MAP2K2//PIK3CD//RAF1//RELA//RPS6KB1//STAT3//STAT5A//ZBTB16 |
| hsa04142 | Lysosome - Homo
sapiens (human) | 0.001392349 |
ABCA2//AP1M1//AP4B1//ATP6AP1//ATP6V0D1//CLTCL1//CTSA//CTSC//CTSD//CTSL1//DNASE2//GALNS//GBA//GGA1//GNPTG//HEXA//IGF2R//LAMP2//MAN2B1//MCOLN1//NAGLU//PPT1//PSAP |
| hsa04141 | Protein processing
in endoplasmic reticulum - Homo sapiens (human) | 0.001583112 |
AMFR//BAK1//CANX//CAPN1//DDIT3//DNAJC5G//EDEM2//ERP29//HSP90AA1//HSP90AB1//HSPA8//LMAN1L//LMAN2//MAN1B1//NSFL1C//P4HB//PDIA4//PPP1R15A//PRKCSH//RAD23B//RPN1//RRBP1//SEC13//SEC61A1//SSR2//STUB1//UBE2J2//UBQLN4//UBXN6 |
| hsa03050 | Proteasome -
Homo sapiens (human) | 0.003429877 |
PSMA7//PSMB9//PSMC3//PSMC4//PSMC5//PSMD13//PSMD2//PSMD3//PSMD8//PSME1//PSMF1 |
| hsa05213 | Endometrial cancer
- Homo sapiens (human) | 0.003806897 |
AKT2//ARAF//AXIN1//ELK1//FOXO3//GRB2//HRAS//ILK//MAP2K2//PDPK1//PIK3CD//RAF1 |
| hsa04666 | Fc γ R-mediated
phagocytosis - Homo sapiens (human) | 0.004610906 |
AKT2//ARPC1B//CFL1//DNM2//GSN//HCK//LAT//NCF1//PIK3CD//PIP5K1C//PRKCD//PTPRC//RAC2//RAF1//RPS6KB1//VASP//WAS//WASF2 |
| hsa04662 | B cell receptor
signaling pathway - Homo sapiens (human) | 0.005562133 |
AKT2//CD79B//CD81//GRB2//HRAS//IFITM1//IKBKG//LILRB3//MAP2K2//NFKBIB//PIK3CD//PTPN6//RAC2//RAF1//RELA |
| hsa00310 | Lysine degradation
- Homo sapiens (human) | 0.006858597 |
DOT1L//EHMT2//GCDH//GLT25D1//HADHA//MLL4//PLOD1//SETD1B//SETD2//SUV39H1//WHSC1 |
| hsa04910 | Insulin signaling
pathway - Homo sapiens (human) | 0.007757841 |
AKT2//ARAF//CALML3//ELK1//EXOC7//FLOT1//FLOT2//GRB2//HRAS//MAP2K2//MKNK2//PCK2//PDPK1//PHKG2//PIK3CD//PPP1CA//PRKAG1//PRKAR1B//PYGB//RAF1//RPS6//RPS6KB1//TSC2 |
| hsa05130 | Pathogenic
Escherichia coli infection - Homo sapiens
(human) | 0.008284432 |
ABL1//ARHGEF2//ARPC1B//TUBA1A//TUBA1C//TUBA3C//TUBA4A//TUBB//TUBB2C//TUBB8//WAS//YWHAZ |
| hsa04150 | mTOR signaling
pathway - Homo sapiens (human) | 0.0108534 |
AKT2//DDIT4//EIF4B//MLST8//PDPK1//PIK3CD//RPS6//RPS6KA1//RPS6KB1//TSC2//ULK3 |
| hsa04722 | Neurotrophin
signaling pathway - Homo sapiens (human) | 0.01165631 |
ABL1//AKT2//ARHGDIA//CALML3//CAMK2G//FOXO3//GRB2//HRAS//IRAK1//MAP2K2//MAPKAPK2//NFKBIB//PIK3CD//PRKCD//RAF1//RELA//RPS6KA1//RPS6KA4//YWHAE//YWHAZ//ZNF274 |
| hsa00511 | Other glycan
degradation - Homo sapiens (human) | 0.02057153 |
GBA//HEXA//MAN2B1//MAN2B2//MAN2C1 |
| hsa03030 | DNA replication -
Homo sapiens (human) | 0.0211445 |
FEN1//LIG1//MCM5//MCM7//POLA2//POLD2//POLE//RFC2 |
| hsa00480 | Glutathione
metabolism - Homo sapiens (human) | 0.02164977 |
G6PD//GGT1//GPX2//GPX4//GSS//GSTK1//GSTZ1//IDH2//PGD//SRM |
| hsa05140 | Leishmaniasis -
Homo sapiens (human) | 0.02422244 |
C3//CYBA//ELK1//IRAK1//ITGA4//ITGB2//NCF1//NCF4//NFKBIB//PTPN6//RELA//TAB1//TGFB1 |
| hsa05220 | Chronic myeloid
leukemia - Homo sapiens (human) | 0.02422244 |
ABL1//AKT2//ARAF//CTBP1//GRB2//HRAS//IKBKG//MAP2K2//PIK3CD//RAF1//RELA//STAT5A//TGFB1 |
| hsa03015 | mRNA surveillance
pathway - Homo sapiens (human) | 0.03338131 |
ACIN1//CPSF1//DDX39B//NXF1//PABPN1//PPP2R1A//PPP2R3B//PPP2R5D//RNGTT//RNPS1//SMG5//SMG6//THOC4//WIBG |
| hsa03420 | Nucleotide excision
repair - Homo sapiens (human) | 0.04168682 |
DDB2//ERCC1//ERCC2//GTF2H4//LIG1//POLD2//POLE//RAD23B//RFC2 |
Discussion
Nucleostemin (NS) is a protein mainly expressed on
stem and tumor cells, and it is important to maintain early
embryonic development and tumor cell proliferation (1,3,4). Most
studies consider that NS could inhibit the antineoplastic function
of p53 through combining with it (1), but some researchers still consider
that NS could also play its function independent of p53 (10–15).
However, the specific mechanism of this p53-independent pathway has
yet to be elucidated.
In this study, we used lentivirus-mediated RNA
interference and DNA microarray technology to investigate the
differences of gene expressing profiling after knocking down NS in
HL-60 cells, which was p53-null, in order to further explore the
specific mechanism of NS p53-independent pathway. Our study showed
that knockdown of NS in HL-60 cells could modulate the expression
of extensive genes. Next, we proceeded to pathway analysis to
explore how NS plays its function independent of p53. In all the
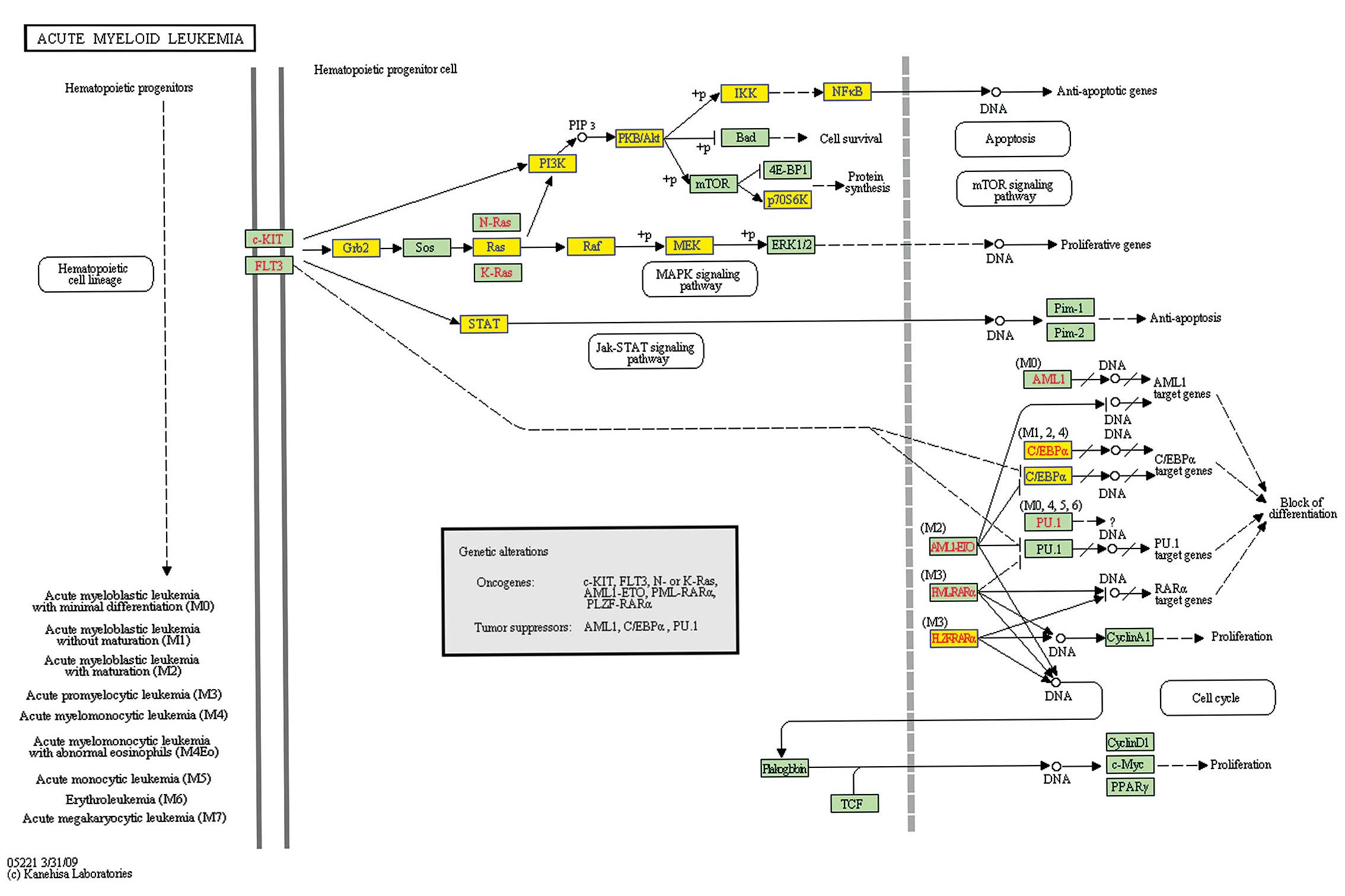
pathways which changed significantly, the acute myeloid leukemia
pathway (Pathway ID: has05221) warranted further attention, since
HL-60 was an acute myeloid leukemia cell line. The PI3K-AKT,
JAK-STAT and MAPK pathways are three main pathways included in
acute myeloid leukemia (Fig.
3).
The PI3K-AKT pathway is one of the most important
cellular signal transduction pathways involved in the regulation of
proliferation. It plays its role of inhibiting apoptosis and
promoting proliferation in cells through impacting the activated
state of a variety of downstream effectors, and is closely related
to the occurrence of many human tumors. After PI3K is activated by
upstream molecules, it phosphorylates PI (4,5) P2 on plasma
membrane to PI (3,4,5) P3, the latter could translocate AKT to
plasma membrane to activate it. Next, the activated AKT could
activate or inhibit its downstream target proteins such as Bad,
Caspase 9, NF-κB, Forkhead, mTOR, Par-4 and P21 through
phosphorylation, which mediate cell growth and promote cell
survival induced by insulin and types of growth factors; it is an
important antiapoptotic factor (16,17).
In addition, AKT could regulate the activity of IKK, cause the
nuclear translocation of NF-κB, and then promote the transcription
of NF-κB-dependent survival genes to facilitate cell survival
(18); also, the inhibition of
NF-κB could promote cell apoptosis (19). In this research, after knocking down
NS expression in HL-60 cells, the key point in the PI3K-AKT pathway
including PI3K (PIK3CD), AKT2, IKK (IκBKG) and NF-κB were all
downregulated (Fig. 3). This
suggested that the inhibition of the PI3K/AKT/NF-κB pathway may
participate in the apoptosis caused by knocking down NS in HL-60
cells.
The continuous activation of the JAK-STAT pathway is
widespread in leukemia cells; the activated key point STAT could
induce abnormal expression of genes closely related to cell
proliferation, differentiation and apoptosis, and then promote cell
proliferation and inhibit apoptosis through various pathways
(20,21). STAT family members are closely
related to tumors, especially STAT3 and STAT5A. Some studies
reported that inhibition of STAT5A could block the growth of
prostate cancer cells (22);
inhibition of STAT3 could promote the apoptosis of bladder cancer
cells (23); high-dose
methylprednisone induced HL-60 cell apoptosis, and this process was
accompanied by downregulation of STAT5A protein (24). In the present study, STAT3 and
STAT5A were both downregulated after the inhibition of NS,
indicating that downregulation of STAT may be another reason for
cell apoptosis after knocking down NS in HL-60 cells.
Mitogen-activated protein kinase (MAPK) is a type of
serine-threonine protein kinase existing in cells and plays an
important role in cell signal transduction. MAPK pathways mainly
include the RAS-RAF-MEK-ERK1/2, the JNK and the P38MAPK pathway
(25). A previous study reported
that the RAS-RAF-MEK-ERK1/2 pathway may have an important impact on
the proliferation and differentiation of melanoma cells (26); inhibiting this pathway could be an
effective approach for antitumor therapy (27–29).
Some antineoplastic drugs could induce cell apoptosis mainly
through the activation of JNK and P38MAPK pathway (30–32).
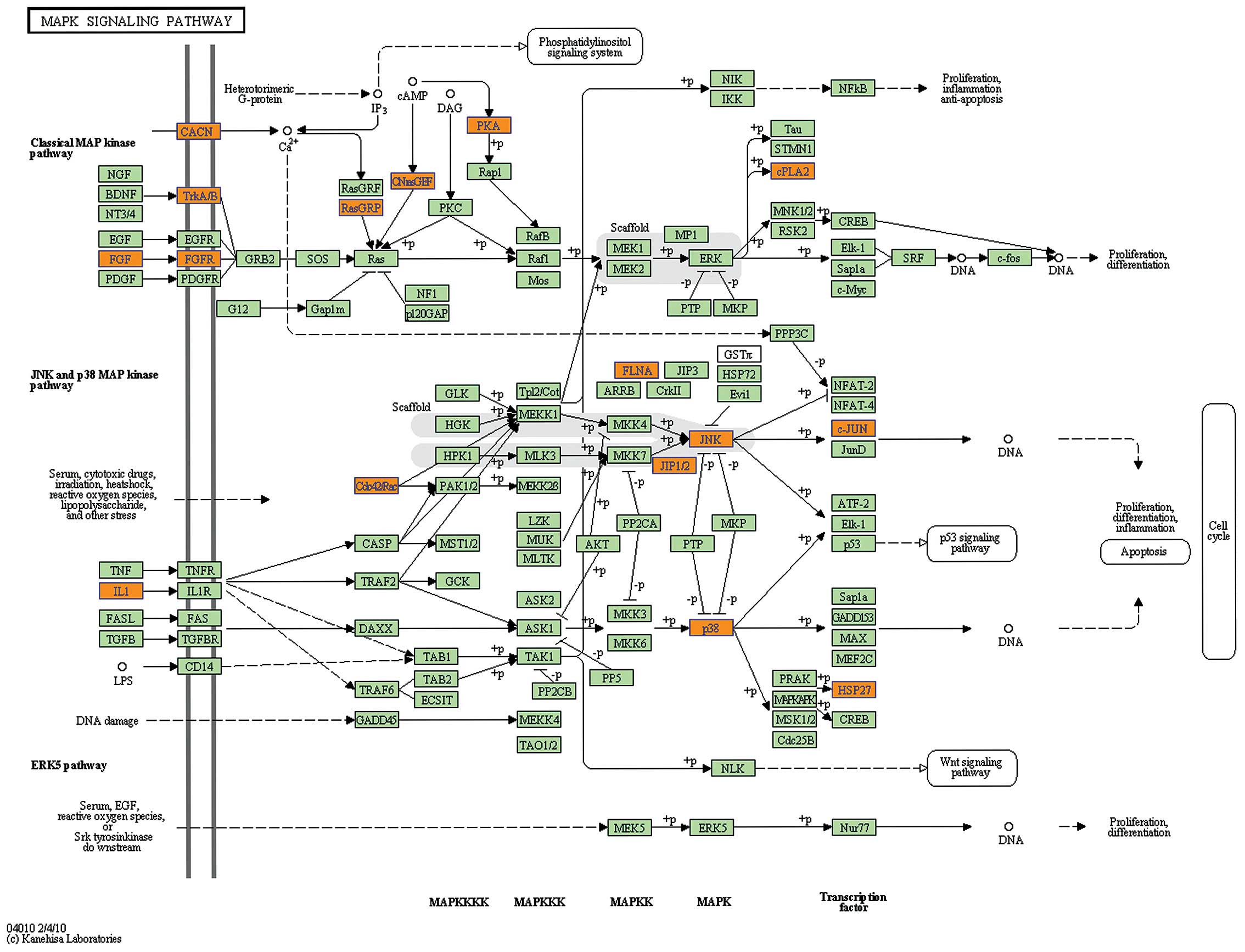
Herein, we found genes involved in the RAS-RAF-MEK-ERK1/2 pathway,
including GRB2, RAS (HRAS), RAF (RAF1), MEK2 (MAP2K2), were all
downregulated (Fig. 3), while JNK
(MAPK9) and P38 (MAPK13) were upregulated (Fig. 4). This suggested that the inhibition
of RAS-RAF-MEK-ERK1/2 pathway and activation of JNK as well as
P38MAPK pathway may also involved in HL-60 cell apoptosis caused by
knocking down the expression of NS.
The pathway analysis of downregulated genes showed
that spliceosome (Pathway ID: hsa03040) and RNA transport (Pathway
ID: hsa03013) were much more significant. Spliceosome plays a
critical role in processing pre-mRNA and folding mRNA. The data of
our research showed that many genes participated in the formation
of spliceosome, therefore it was speculated that knocking down NS
may influence the cellular gene expression through preventing RNA
processing and transport. Romanova et al presented a similar
viewpoint, that knocking down NS had an impact on the processing of
rRNA, folding and stabilization of other RNAs (33,34).
In conclusion, we detected the changes of gene
expression profiles in HL-60 cells which were p53-null after
knocking down NS expression, in order to explore how NS plays its
function without the existence of p53. The DNA microarray data
showed a large number of genes were differentially expressed.
Pathway analysis indicated that after NS was inhibited in HL-60
cells, especially inhibition of the PI3K-AKT pathway, the JAK-STAT
pathway and RAS-RAF-MEK-ERK1/2, and the activation of JNK pathway
as well as P38MAPK pathway may be involved in the cell apoptosis
caused by knocking down NS. This study provides insight into
exploring the functional mechanism of NS independent of p53, and
lays the foundations for the search of new effective therapeutic
targets of p53-null leukemia and even p53-null tumors.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (No. 81271911) and the Key
Projects of Medical Science and Technology of Henan Province (No.
201002006). The authors thank Professor Guoqiang Zhao, Basic
Medical School of Zhengzhou University, for his valuable advice on
transfection and RNA-interference technology. We also acknowledge
the Kangcheng company in Shanghai, China, for DNA microarray
technical help.
References
|
1
|
Tsai RY and McKay RD: A nucleolar
mechanism controlling cell proliferation in stem cells and cancer
cells. Genes Dev. 16:2991–3003. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu SJ, Cai ZW, Liu YJ, Dong MY, Sun LQ,
Hu GF, et al: Role of nucleostemin in growth regulation of gastric
cancer, liver cancer and other malignancies. World J Gastroenterol.
10:1246–1249. 2004.PubMed/NCBI
|
|
3
|
Liu RL, Xu Y, Zhang ZH, Wang M, Sun JT, Qi
SY, et al: Expression of nucleostemin in prostate cancer tissues
and its clinical significance. Zhonghua Nan Ke Xue. 14:418–422.
2008.(In Chinese).
|
|
4
|
Zhang GY, Yin L, Li SL, Xing WY, Zhao QM,
Le XP, et al: Expression of nucleostemin mRNA and protein in the
esophageal squamous cell carcinoma. Zhonghua Zhong Liu Za Zhi.
30:125–128. 2008.(In Chinese).
|
|
5
|
Sijin L, Ziwei C, Yajun L, Meiyu D,
Hongwei Z, Guofa H, et al: The effect of knocking-down nucleostemin
gene expression on the in vitro proliferation and in vivo
tumorigenesis of HeLa cells. J Exp Clin Cancer Res. 23:529–538.
2004.PubMed/NCBI
|
|
6
|
Ma H and Pederson T: Depletion of the
nucleolar protein nucleostemin causes G1 cell cycle arrest via the
p53 pathway. Mol Biol Cell. 18:2630–2635. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu RL, Zhang ZH, Zhao WM, Wang M, Qi SY,
Li J, et al: Expression of nucleostemin in prostate cancer and its
effect on the proliferation of PC-3 cells. Chin Med J (Engl).
121:299–304. 2008.PubMed/NCBI
|
|
8
|
Hsu JK, Lin T and Tsai RY: Nucleostemin
prevents telomere damage by promoting PML-IV recruitment to
SUMOylated TRF1. J Cell Biol. 197:613–624. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yue BH, Yu LN and Wang YY: Effects of
silent nucleostemin gene expression on apoptosis of HL-60 cells in
vitro. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 17:319–323. 2009.(In
Chinese).
|
|
10
|
Beekman C, Nichane M, De Clercq S, Maetens
M, Floss T, Wurst W, et al: Evolutionarily conserved role of
nucleostemin: controlling proliferation of stem/progenitor cells
during early vertebrate development. Mol Cell Biol. 26:9291–9301.
2006. View Article : Google Scholar
|
|
11
|
Jafamejad SM, Mowla SJ and Matin MM:
Knocking-down the expression of nucleostemin significantly
decreases rate of proliferation of rat bone marrow stromal stem
cells in an apparently p53-independent manner. Cell Prolif.
41:28–35. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nikpour P, Mowla SJ, Jafarnejad SM,
Fischer U and Schulz WA: Differential effects of Nucleostemin
suppression on cell cycle arrest and apoptosis in the bladder
cancer cell lines 5637 and SW1710. Cell Prolif. 42:762–769. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu R, Zhang Z and Xu Y: Downregulation of
nucleostemin causes G1 cell cycle arrest via a p53-independent
pathway in prostate cancer PC-3 cells. Urol Int. 85:221–227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zwolinska AK, Heagle WA, Beekman C, Sedivy
JM and Marine JC: Suppression of Myc oncogenic activity by
nucleostemin haploinsufficiency. Oncogene. 31:3311–3321. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Paridaen JT, Janson E, Utami KH, Pereboom
TC, Essers PB, van Rooijen C, et al: The nucleolar GTP-binding
proteins Gnl2 and nucleostemin are required for retinal
neurogenesis in developing zebrafish. Dev Biol. 355:286–301. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Osaki M, Oshimura M and Ito H: PI3K-Akt
pathway: its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vandermoere F, El Yazidi-Belkoura I,
Adriaenssens E, Lemoine J and Hondermarck H: The antiapoptotic
effect of fibroblast growth factor-2 is mediated through nuclear
factor-kappaB activation induced via interaction between Akt and
IkappaB kinase-beta in breast cancer cells. Oncogene. 24:5482–5491.
2005. View Article : Google Scholar
|
|
19
|
Zanotto-Filho A, Delgado-Cañedo A,
Schröder R, Becker M, Klamt F and Moreira JC: The pharmacological
NFkappaB inhibitors BAY117082 and MG132 induce cell arrest and
apoptosis in leukemia cells through ROS-mitochondria pathway
activation. Cancer Lett. 288:192–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105
|
|
21
|
Haura EB, Turkson J and Jove R: Mechanisms
of disease: Insights into the emerging role of signal transducers
and activators of transcription in cancer. Nat Clin Pract Oncol.
2:315–324. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dagvadorj A, Kirken RA, Leiby B, Karras J
and Nevalainen MT: Transcription factor signal transducer and
activator of transcription 5 promotes growth of human prostate
cancer cells in vivo. Clin Cancer Res. 14:1317–1324. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen CL, Cen L, Kohout J, Hutzen B, Chan
C, Hsieh FC, et al: Signal transducer and activator of
transcription 3 activation is associated with bladder cancer cell
growth and survival. Mol Cancer. 7:782008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaymaz BT, Selvi N, Saydam G, Sahin F and
Kosova B: Methylprednisolone induces apoptosis by interacting with
the JAK/STAT pathway in HL-60 and K-562 leukemic cells. Hematology.
17:93–99. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Raman M, Chen W and Cobb MH: Differential
regulation and properties of MAPKs. Oncogene. 26:3100–3112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Busca R, Abbe P, Mantoux F, Aberdam E,
Peyssonnaux C, Eychène A, et al: Ras mediates the cAMP-dependent
activation of extracellular signal-regulated kinases (ERKs) in
melanocytes. EMBO J. 19:2900–2910. 2000. View Article : Google Scholar
|
|
27
|
Huang D, Ding Y, Luo WM, Bender S, Qian
CN, Kort E, et al: Inhibition of MAPK kinase signaling pathways
suppressed renal cell carcinoma growth and angiogenesis in vivo.
Cancer Res. 68:81–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ji Z, Flaherty KT and Tsao H: Targeting
the RAS pathway in melanoma. Trends Mol Med. 18:27–35. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Daouti S, Wang H, Li WH, Higgins B,
Kolinsky K, Packman K, et al: Characterization of a novel
mitogen-activated protein kinase kinase 1/2 inhibitor with a unique
mechanism of action for cancer therapy. Cancer Res. 69:1924–1932.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Su JC, Lin KL, Chien CM, Lu CM, Chen YL,
Chang LS and Lin SR: Novel indoloquinoline derivative, IQDMA,
induces G(2)/M phase arrest and apoptosis in A549 cells through
JNK/p38 MAPK signaling activation. Life Sci. 85:505–516. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brosseau CM, Pirianov G and Colston KW:
Involvement of stress activated protein kinases (JNK and p38) in
1,25 dihydroxyvitamin D3-induced breast cell death. Steroids.
75:1082–1088. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Uehara N, Kanematsu S, Miki H, Yoshizawa K
and Tsubura A: Requirement of p38 MAPK for a cell-death pathway
triggered by vorinostat in MDA-MB-231 human breast cancer cells.
Cancer Lett. 315:112–121. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Romanova L, Grand A, Zhang L, Rayner S,
Katoku-Kikyo N, Kellner S, et al: Critical role of nucleostemin in
pre-rRNA processing. J Biol Chem. 284:4968–4977. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Romanova L, Kellner S, Katoku-Kikyo N and
Kikyo N: Novel role of nucleostemin in the maintenance of nucleolar
architecture and integrity of small nucleolar ribonucleoproteins
and the telomerase complex. J Biol Chem. 284:26685–26694. 2009.
View Article : Google Scholar : PubMed/NCBI
|