Introduction
Cancer stem cells have the phenotype of a high level
of resistance to a range of anticancer agents, including platinum
compounds. Maintenance of cancer stem cells is dependent on the
Hedgehog (Hh) pathway. The Hh role in platinum resistance is
related in part to the positive transcription factor Gli1 and its
role in the regulation of c-jun and ERCC1 (1,2). The
knockdown of Gli1 using shRNA results in a series of events: the
c-jun phosphorylation pattern changes from the anti-apoptotic
Ser63/73 to the pro-apoptotic Thr91/93; c-jun-dependent DNA repair
genes are not upregulated; cisplatin-DNA adduct repair is markedly
reduced; and cells are sensitized to cisplatin by a factor of six
(1).
Cellular resistance to cisplatin is multifactorial
(3–5). One of the factors that controls the
level of cisplatin resistance is altered cellular accumulation of
drug (4–7). Cellular accumulation of cisplatin is
regulated by several processes, including passive diffusion,
carrier-mediated uptake and carrier-mediated efflux. In the
excellent review by Hall et al (8), the five known mechanisms of cisplatin
uptake and the four known mechanisms of cisplatin efflux are
summarized. Uptake of cisplatin is mediated by: i) passive
diffusion; ii) a protein gate, linked to the Na+
K+ -ATPase pump; iii) fluid phase endocytosis; iv)
organic cation transporter (OCT) 1–3 proteins; and v) copper
transporter CTR1 and CTR2 proteins. Efflux is mediated by: i)
melanosomes; ii) ATP7B dependent vesicles; iii) ATP7A protein; and
iv) MRP1–5 proteins.
Uptake of cisplatin is mediated by proteins that
have multiple transport functions. Specifically OCT1, OCT2 and OCT3
function as transporters of organic cations, monoamine
neurotransmitters, xenobiotics and various drugs. OCT1, OCT2 and
OCT3 transport cisplatin and oxaliplatin. Uptake of cisplatin is
also mediated by two copper transporters: CTR1 and CTR2. CTR1 and
CTR2 normally function for copper influx. Both of these CTR
transporters have been shown to transport cisplatin and
carboplatin, but CTR1 is additionally involved in the transport of
oxaliplatin.
Efflux of cisplatin occurs through the ATPase copper
transporters ATP7A and ATP7B. The ATP7A and ATP7B transporters are
both involved in copper sequestration and efflux and transport both
cisplatin and carboplatin.
In our examination of the molecular sequences of the
genes discussed above, we found potential Gli-binding sites (GBS)
in five of these genes: OCT1, OCT2, OCT3, CTR1 and ATP7B. These
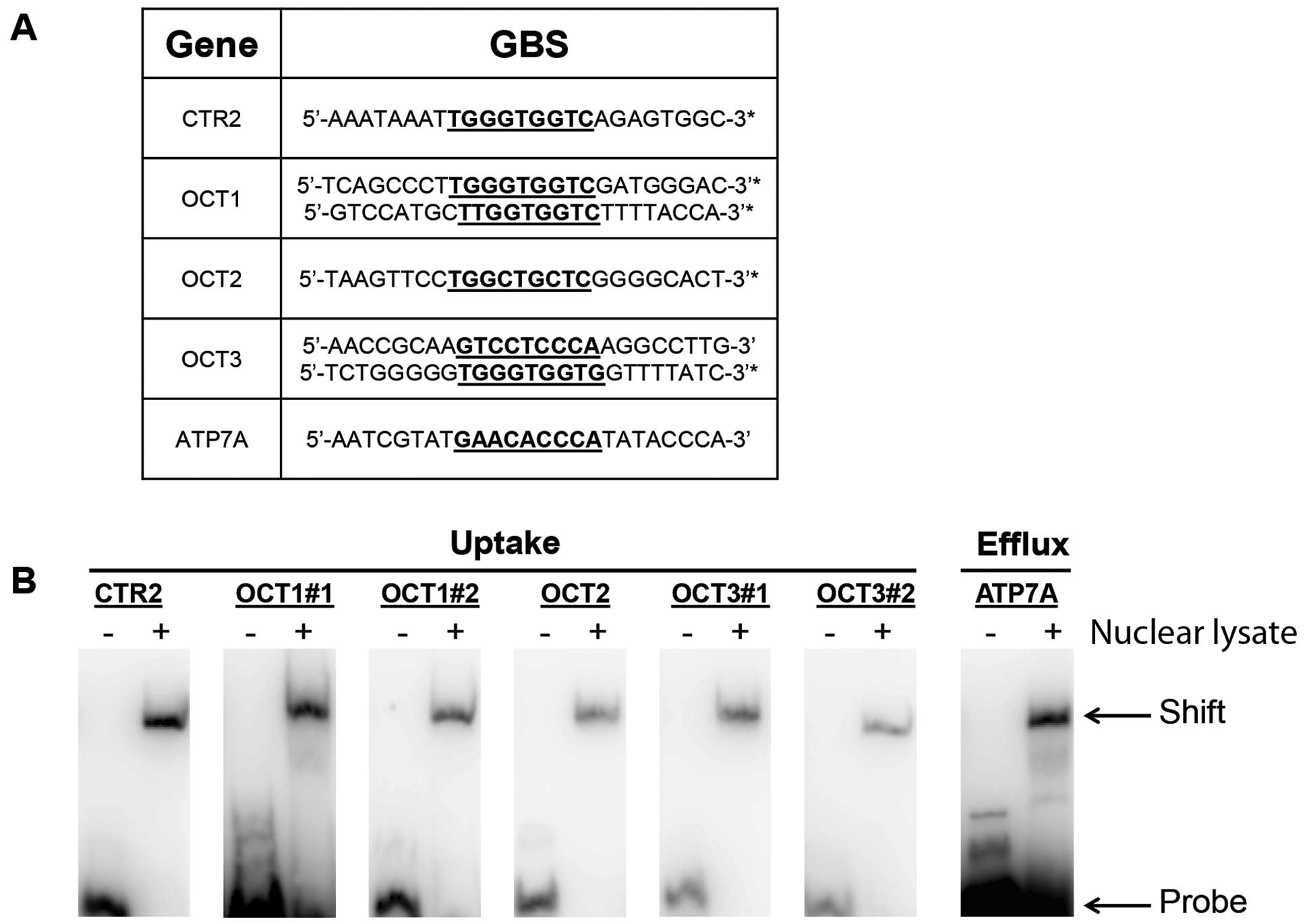
potential Gli binding sites are given in Fig. 2A. In the present study, we explore
whether inhibition of Gli1 has any effect on total cellular
accumulation of cisplatin in A2780-CP70 cisplatin-resistant human
ovarian cancer cells. We also examined whether there is any
evidence that the Gli- binding sites for these genes, would be
recognized by nuclear lysates from this cell line. Our results
suggest that Gli1 may play a strong role in modulating total
cellular accumulation of cisplatin in these cells, through altered
drug uptake and altered drug efflux.
Materials and methods
Cells
The cisplatin resistant A2780-CP70 ovarian cancer
cells were used in all experiments. Cells were retrieved from a
frozen stock and experiments were performed between passages 5 and
30. Cells were cultured in RPMI-1640 media (Gibco/Invitrogen,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum
(Gibco), L-glutamine (Gibco), insulin (Sigma-Aldrich, St. Louis,
MO, USA) and penicillin/streptomycin (Gibco). During active
experiments, cells were carried in media without
penicillin/streptomycin.
Whole cell platinum analysis
Cells were treated under two experimental
conditions. One set of cells were treated with anti-Gli1 shRNA at
an IC20 dose for 24 h, prior to treatment with
cisplatin. Gli1 was targeted for degradation using shRNA specific
for Gli1 (1). Control cells were
treated with scrambled shRNA at the same micromolar dose for 24 h,
prior to cisplatin treatment.
A2780-CP70 cells were seeded at 2×106 in
a 10-cm2 dish. The following morning, cells were
transfected with Gli1 or scrambled shRNA using Lipofectamine
according to the manufacturer’s instructions (Invitrogen).
Twenty-four hours later cells were treated with 30 μM cisplatin for
1 h, the IC50 dose when cisplatin is used alone.
Cisplatin-containing media was then removed and plates were washed
with cold PBS. The zero hour time point was immediately after the
1-h cisplatin dose. Cells were harvested by trypsinization and
collected.
For the 12-h time-point, cisplatin-containing media
was removed after the 1-h drug exposure. Cells were washed with PBS
and fresh cisplatin-free media was then added. Cells were collected
by trypsinization 12 h later. Cells were harvested by
centrifugation and counted by trypan blue dye exclusion assays. The
collected cell pellets were stored at −80°C until analysis.
Cell pellets were prepared for atomic absorption
spectroscopy (AAS) analysis by wet ashing as previously described
(9). Briefly, cell pellets were
treated with 0.5 ml nitric acid and incubated in a water bath at
100°C for 5 min. Polypropylene 15 ml conical tubes were used.
Samples were cooled to room temperature under running water.
Hydrogen peroxide, 0.5 ml of 30%, was added to the tube and
re-submerged into the water bath at 100°C for 5 min. Samples were
again cooled to room temperature before analysis by AAS.
Platinum in each sample was measured by AAS using a
Perkin Elmer 600 AAnalyst AAS machine with Zeeman background
correction and a platinum lamp (Perkin-Elmer, Walthan, MA, USA).
Platinum standards were used to generate a standard curve. Total
platinum per 1×106 cells was determined based on the
cell counts and the total platinum in each sample. Total cellular
platinum per million cells at 0 h, was compared with the total
platinum level per million cells at 12 h.
Binding site search
Genomic sequences containing 10 kB upstream of the
translation start site were obtained from Ensembl (www.ensembl.org) for the following transporter genes:
ATP7A (ATP7A-001), ATP7B (ATPB-001), CTR1 (SLC31A1-001), CTR2
(SLC31A2-001), OCT1 (SLC22A1-001), OCT2 (SLC22A2-001) and OCT3
(SLC22A3-001). The following known Gli-binding sites were used in
the search parameters: GAGCAGCCA, GACGACCCC, GGCCCCCCA, GACCGCCCC,
AACCAACCCC, GTCCTCCCA, GACCAC CCA (10), GAACACCCA, CACCACCCA and GACCACCAA
(11). A pairwise BLAST (basic
local alignment search tool, bl2seq, http://blast.ncbi.nlm.nih.gov/Blast.cgi) was performed
to compare the promoter region with each GBS. The parameters of the
pairwise BLAST were kept at the default setting except for the word
size was changed from 11 to 7. Activator protein 1 (AP1) binding
sites were searched in each transporter promoter using the
following sequence TGAGTCA (12,13)
and keeping in the same pairwise BLAST search parameters.
Electrophoretic mobility shift assays
(EMSAs)
The putative Gli-binding sites found in the
promoters in the listed transporters were synthesized as DNA
oligonucleotides and the reverse compliment containing a 5′ biotin
were obtained for the following transporters: ATP7A
(AATCGTATGAACACC
CATATACCCA), CTR2 (AAATAAATTGGGTGGTCAGA GTGGC), OCT1
(TCAGCCCTTGGGTGGTCGATGGGAC,
GTCCATGCTTGGTGGTCTTTTACCA), OCT2
(TAAGTTC CTGGCTGCTCGGGGCACT) and OCT3
(AACCGCAAG
TCCTCCCAAGGCCTTG, TCTGGGGGTGGGTGGTGG TTTTATC)
(Integrated DNA Technologies, Coralville, IA, USA). Each
oligonucleotide was resuspended in Milli-Q-water at a final
concentration of 100 μM. Double stranded DNA (dsDNA) probes were
generated by adding 1 μM of the forward and reverse compliment
oligonucleotides in annealing buffer (10 mM Tris, 1 mM EDTA, 50 mM
NaCl, pH 8.0) and heated to 95°C and cooled to room temperature at
a rate of 1°C/min.
Nuclear lysate was prepared from A2780-CP70 as
previously described (1). Protein
concentration was determined using BCA kit (Thermo Pierce,
Rockford, IL, USA). The EMSA DNA-binding reaction was carried out
using LightShift Chemiluminescent EMSA kit (Thermo Pierce). Each
reaction consisted of 20 fmol of biotin labeled dsDNA, 1 μg poly
(dI-dC), 20 μg nuclear lysate in a 20 μl of reaction buffer (40 mM
HEPES, 25 mM KCl, 10 mM MgCl2, 10 mM ZnSO4,
500 μM EDTA, 10% glycerol, pH 7.8). The DNA-binding reaction was
incubated on ice for 30 min and 5 μl of loading buffer was added.
Samples were separated on a 6% native polyacrylamide gel, and
transferred to a positively charged PVDF membrane (BrightStar Plus;
Ambion, Austin, TX, USA). The dsDNA biotin probe was visualized
using the reagents of the kit.
Results
Anti-Gli1 shRNA reduces cisplatin influx
and shuts down cisplatin efflux
We first assessed total cellular platinum in these
cells, after pretreatment with anti-Gli1 shRNA for 24 h, followed
by cisplatin treatment for 1 h. This was compared to pretreatment
for 24 h with scrambled shRNA control followed by cisplatin
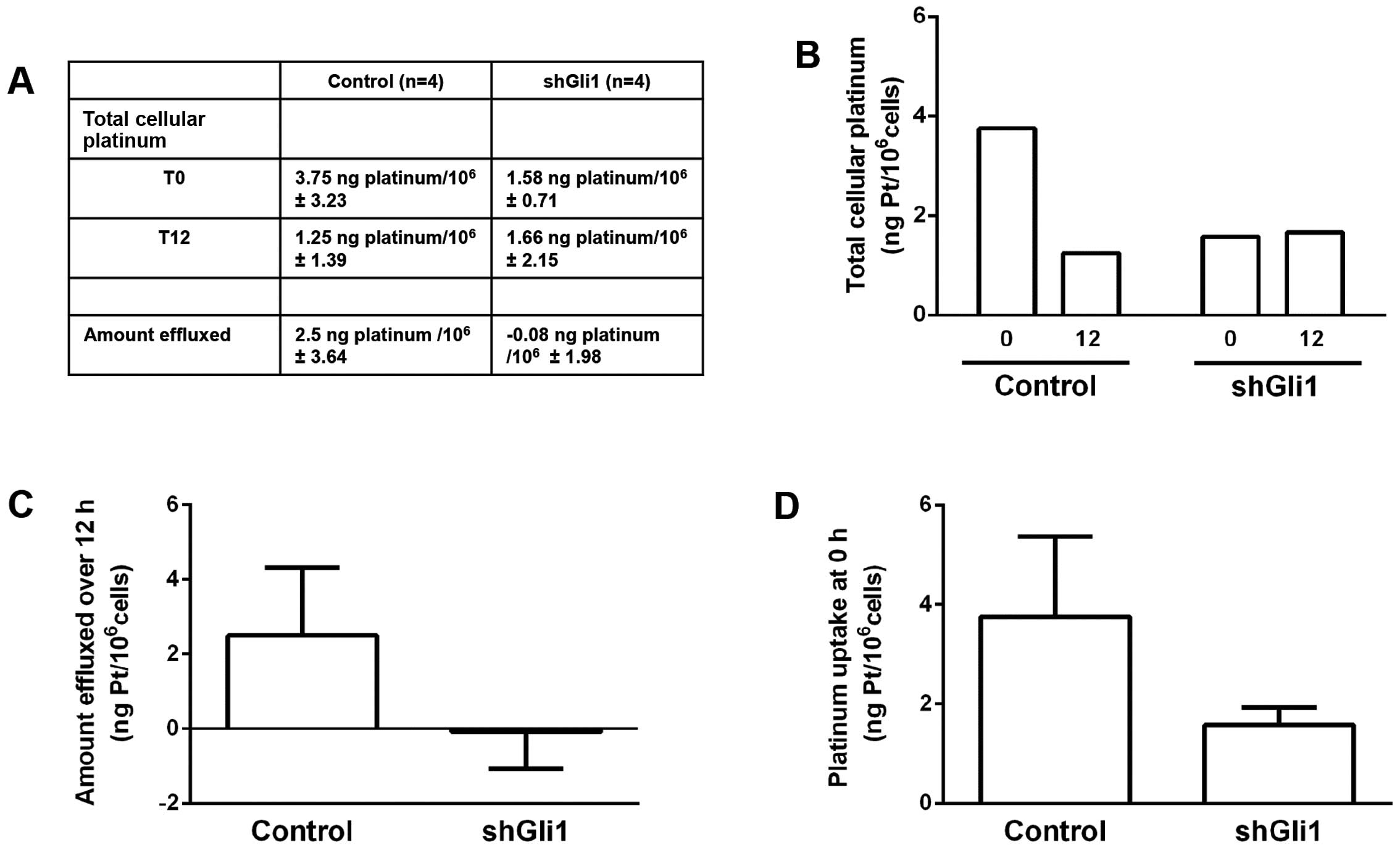
treatment for 1 h, at the same cisplatin dose. As shown in Fig. 1A and B, under control conditions,
cells accumulated 3.75 ng Pt/106 cells, after a 1 h
cisplatin exposure. Total cellular platinum was also measured 12 h
after cisplatin was removed from cells. Total cellular platinum was
reduced to 1.25 ng Pt/106 cells, indicating that these
cells effluxed 2.5 ng Pt/106 cells over this 12-h
period. When cells were treated with anti-Gli1 shRNA, the platinum
level at 0 h was 1.58 ng Pt/106 cells, which was
essentially unchanged 12 h later, measured at 1.66 ng
Pt/106 cells. This indicates that under control
conditions, 67% of total cellular platinum was removed over 12 h
(3.75 down to 1.25 ng). Furthermore, when cells were pretreated
with anti-Gli1 shRNA, these cells were unable to efflux platinum
over the same 12-h period. Fig. 1C
graphically shows the differences with respect to total drug
effluxed during the 12-h experiment.
Fig. 1D, graphically
shows the differences between conditions with respect to the
initial levels of total cellular platinum. When cells were
pretreated with scrambled shRNA total cellular drug was 3.75 vs.
1.58 units when pretreated with anti-Gli1 shRNA; a 58% reduction in
drug uptake. This suggests that drug uptake was greatly reduced by
the inhibition of Gli1.
Nuclear lysate recognizes the GBS of each
of five platinum transport genes
We studied seven of 12 known platinum transport
genes, for the possibility of harboring one or more Gli-binding
sites in their 5′UTR. The genes we studied were CTR1, CTR2, ATP7A,
ATP7B, OCT1, OCT2 and OCT3. Approximately 10 kB upstream of each
gene was obtained and searched for the binding sites using BLAST
(http://blast.ncbi.nlm.nih.gov/Blast.cgi). For two of
these seven genes, there was no indication of a GBS in the 5′UTR;
CTR1 and ATP7B. For five genes that contained a GBS in the 5′UTR,
those sequences are listed in Fig.
2A. Two of these genes, OCT1 and OCT3, have two GBS sites each,
which are given in Fig. 2A.
We next assessed nuclear lysates from A2780-CP70
cells, for the ability to bind the synthesized Gli-binding sites
for each of the platinum transporters listed in Fig. 2A. In Fig. 2B, we show the EMSAs for each GBS for
each gene. The EMSA is run with the synthetic GBS mixed with
nuclear lysate. The negative control, GBS alone, is run to check
for gross abnormalities of the probe.
CTR2, OCT1, OCT2 and OCT3, are platinum influx
proteins/genes. In each case the GBS probe demonstrated the
predicted shift within the nuclear lysate lane. ATP7A is a platinum
efflux protein/gene. In this case as well, the GBS probe
demonstrated the predicted shift. Therefore, for each of the five
genes studied, and a total of seven GBS probes, the nuclear lysate
of these cells generated the predicted shift. This indicates that a
GBS-binding protein exists in the nuclear lysate of these cells for
each of these five platinum transport genes. As shown in Fig. 1, when Gli1 is disrupted by the use
of anti-Gli1 shRNA, cellular influx of cisplatin is reduced and
cellular efflux of cisplatin is abrogated.
Comparing cellular accumulation and DNA
repair in the cells
We have extended the studies we previously performed
with respect to the repair of platinum-DNA adducts. This is
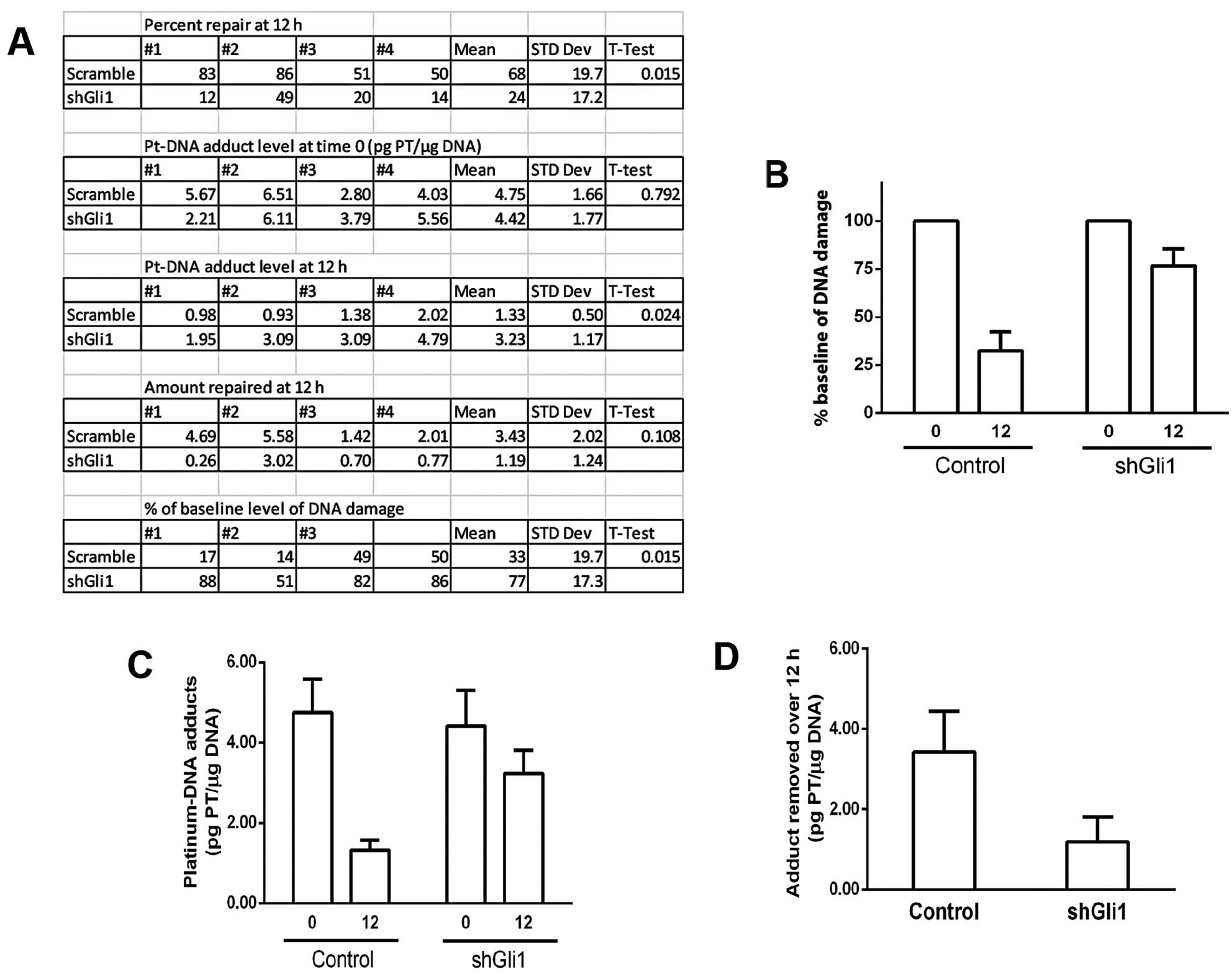
summarized in Fig. 3. As we have
reported (1), the repair of
platinum-DNA damage is greatly inhibited by using anti-Gli1 shRNA
in these cells. Fig. 3A, gives the
raw data from n=4. Fig. 3A–C show
that under control conditions, ~68% repair occurs for platinum-DNA
damage over 12 h. When cells are treated with anti-Gli1 shRNA, ~24%
repair occurs (P=0.015). Fig. 3A and
D, show that the absolute amount of DNA adduct repaired over
the 12-h time frame, is ~3-fold greater under control conditions,
as compared to when Gli1 is inhibited (3.43 vs. 1.19 units).
We next sought to compare these two experimental
conditions, in a manner where total cellular drug and total DNA
damage could be assessed concurrently, at 0 h and at 12 h. This is
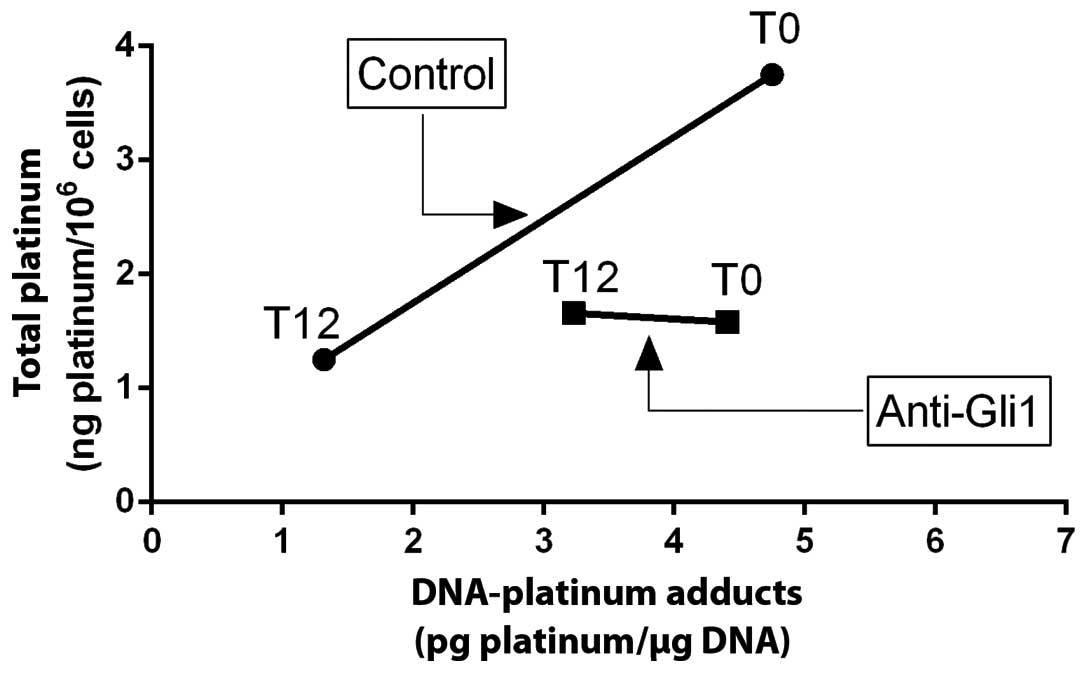
graphically shown in Fig. 4. Here,
total cellular platinum is plotted on the vertical axis, and total
DNA damage is assessed on the horizontal axis. Cells treated under
control conditions are plotted at 0 h and at 12 h. Cells treated
with anti-Gli1 shRNA are plotted at 0 h and at 12 h.
Fig. 4 shows is that
at time zero (immediately after the 1-h exposure to cisplatin),
cells treated with anti-Gli1 shRNA had less than half the total
cellular platinum than control cells treated at the same cisplatin
dose. This illustrates that inhibition of Gli1, in this manner,
greatly inhibits cisplatin uptake in these cells. Also of interest,
is that at this IC50 cisplatin dose, the total amount of
DNA damage experienced by these cells is almost exactly the same,
~4.5 units. This is consistent with previous reports of the
relationships between DNA damage levels and cisplatin
IC50 values in other cell lines (5,14).
Also shown in Fig.
4, under control conditions, reductions in total cellular
levels of drug and removal of DNA damage, appear to be
phenotypically linked. In contrast, in cells where Gli1 is
inhibited, DNA repair continues to a limited degree although
cellular efflux of drug is frozen. Previous studies from this group
suggest that in these cells, DNA repair and cellular accumulation
of drug may be linked processes (5). It appears from Fig. 4, that this linkage can be disrupted
through inhibition of Gli1.
Discussion
Cellular resistance to cisplatin has been associated
with the phenotypic linkage of DNA repair and cellular accumulation
of drug, since the mid-1980s. Eastman was the first to show in a
cellular system, that for levels of cisplatin resistance to
~10-fold over baseline, these two molecular processes go
hand-in-hand (15–17). The consensus belief is that this
level of resistance was probably the range that was clinically
relevant to human patients (18).
Parker and colleagues were the first to show that in human ovarian
cancer cells, levels of resistance up to ~13-fold over baseline
were associated with phenotypic linkage of DNA repair and cellular
accumulation of drug (5). This was
also observed in human T lymphocyte cell lines (19). Masuda et al (20) and Ferry and colleagues (21) confirmed in a series of studies in
human ovarian cancer cells, that at this level of resistance, DNA
repair was very important. They also showed that at levels of
resistance in the range above 30-fold over baseline, cytosolic
inactivation of drug appeared to become the predominant molecular
mechanism of cisplatin resistance (22,23).
In the present study we report on the A2780-CP70
cell line, where these two processes are linked. Here, we show that
Gli1 appears to be an important common molecular entity that
controls cellular uptake of drug and cellular efflux of drug.
Previously we showed that Gli1 is critical to platinum-DNA
repair.
Enhanced DNA repair is associated with cisplatin
resistance. The involvement of Gli1 in DNA damage and repair
pathways has been reported by several groups (1,24,25).
Mazumdar and colleagues demonstrated that inhibition of Gli1 using
small molecule inhibitors induces double strand DNA breaks (DSBs)
coupled with reduced DNA repair in colon cancer (26,27).
Leonard and colleagues demonstrated increased expression of Gli1 in
HEK293 cells resulted in the inability to activate Chk1 after
ionizing radiation (25).
Our group has identified the importance of Gli1 in
the regulation of nucleotide excision repair and base excision
repair in response to platinum DNA damage (1). Gli1 shRNA treated cells exhibited a
switch in the c-jun phosphorylation cascade, from a pro-survival to
a pro-apoptotic pattern. In response to cisplatin treatment, the
upregulation of DNA repair genes by c-jun is blocked and cells are
unable to repair platinum-DNA lesions.
Our laboratory identified the isoform of Gli1, the
130-kDa isoform, responsible for upregulating c-jun (28). Gli1 has 5 known isoforms:
full-length, two mRNA splice variants, and two post-translational
truncations of the full-length protein. The 130-kDa Gli1 isoform is
generated by an N-terminal truncation of the full-length protein
resulting in the removal of the N-terminal Degron domain and the
Sufu-binding domain. Ovarian cancer specimens express higher levels
of the 130-kDa isoform in comparison to non-cancer specimens.
Additionally, the 130-kDa isoform was expressed at higher levels in
the cisplatin resistant cell lines A2780-CP70 and A2780-CIS when
compared with the parental cell line A2780. This opens the
possibility that the Gli1 130-kDa isoform may play a role in both
components of cisplatin drug resistance; i.e., DNA repair and
cellular accumulation of drug.
There are a large number of genes/proteins
associated with platinum transport across cellular membranes. This
has been reviewed by Hall et al (8). Many of these genes have been shown to
be highly conserved, with analogs that exist in grapes (29), rice (30), barley (31) and a range of vertebrates. In humans,
these genes tend to be highly expressed in kidney, in the inner
ear, and in various tissues throughout the body (8). In the present study, we focused on
those transporters where there is a Gli-binding site in the 5′UTR
of that specific gene. For these transporters (Fig. 2), there exists the possibility that
Gli1 may have direct impact on the regulation of those genes.
We further analyzed the 5′UTR of these genes for
binding sites for other regulatory proteins. For the transporters
we examined, we could find binding sites for either: Gli, c-jun or
AP1 (data not shown). We have previously demonstrated that Gli1
effects changes in cisplatin-DNA adduct repair through its
influence on c-jun (1). c-jun
heterodimerizes with c-fos to form the transcriptionally active AP1
(32). This suggests that when Gli1
is inhibited by using anti-Gli1 shRNA, there may also be a strong
indirect effect on the regulation of some cisplatin transporters,
through Gli1′s modulation of c-jun or AP1.
Acknowledgements
The present research was supported by the Intramural
Research Program of the NIH, National Institute on Minority Health
and Health Disparities.
References
|
1
|
Kudo K, Gavin E, Das S, Amable L, Shevde
LA and Reed E: Inhibition of Gli1 results in altered c-Jun
activation, inhibition of cisplatin-induced upregulation of ERCC1,
XPD and XRCC1, and inhibition of platinum-DNA adduct repair.
Oncogene. 31:4718–4724. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Steg AD, Bevis KS, Katre AA, et al: Stem
cell pathways contribute to clinical chemoresistance in ovarian
cancer. Clin Cancer Res. 18:869–881. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ulasov IV, Nandi S, Dey M, Sonabend AM and
Lesniak MS: Inhibition of Sonic hedgehog and Notch pathways
enhances sensitivity of CD133+glioma stem cells to
temozolomide therapy. Mol Med. 17:103–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reed E: Cisplatin, carboplatin and
oxaliplatin. Cancer Chemotherapy and Biotherapy: Principles and
Practice. 4th edition. Chabner BA and Longo DL: Lippincott Williams
and Wilkins; Philadelphia: pp. 332–343. 2006
|
|
5
|
Parker RJ, Eastman A, Bostick-Bruton F and
Reed E: Acquired cisplatin resistance in human ovarian cancer cells
is associated with enhanced repair of cisplatin-DNA lesions and
reduced drug accumulation. J Clin Invest. 87:772–777. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reed E: Cisplatin and platinum analogs.
Cancer Principles and Practice of Oncology. 8th edition. DeVita VT,
Rosenberg SA and Lawrence T: Lippincott Williams and Wilkins;
Philadelphia: pp. 419–426. 2008
|
|
7
|
Reed E and Chabner BA: Platinum analogues.
Cancer Chemotherapy and Biotherapy: Principles and Practice. 5th
edition. Chabner BA and Longo DL: Lippincott Williams and Wilkins;
Philadelphia: pp. 310–322. 2011
|
|
8
|
Hall MD, Okabe M, Shen DW, Liang XJ and
Gottesman MM: The role of cellular accumulation in determining
sensitivity to platinum-based chemotherapy. Annu Rev Pharmacol
Toxicol. 48:495–535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reed E, Sauerhoff S and Poirier MC:
Quantitation of platinum-DNA binding in human tissues following
therapeutic levels of drug exposure: a novel use of graphite
furnace spectrometry. Atomic Spectroscopy. 9:93–95. 1988.
|
|
10
|
Laner-Plamberger S, Kaser A, Paulischta M,
Hauser-Kronberger C, Eichberger T and Frischauf AM: Cooperation
between GLI and JUN enhances transcription of JUN and selected GLI
target genes. Oncogene. 28:1639–1651. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu H and Lo HW: The human
glioma-associated oncogene homolog 1 (GLI1) family of transcription
factors in gene regulation and diseases. Curr Genomics. 11:238–245.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hess J, Angel P and Schorpp-Kistner M:
AP-1 subunits: quarrel and harmony among siblings. J Cell Sci.
117:5965–5973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stein B, Angel P, van Dam H, et al:
Ultraviolet-radiation induced c-jun gene transcription: two AP-1
like binding sites mediate the response. Photochem Photobiol.
55:409–415. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee KB, Parker RJ, Bohr V, Cornelison T
and Reed E: Cisplatin sensitivity/resistance in UV repair-deficient
Chinese hamster ovary cells of complementation groups 1 and 3.
Carcinogenesis. 14:2177–2180. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richon VM, Schulte N and Eastman A:
Multiple mechanisms of resistance to
cis-diamminedichloroplatinum(II) in murine leukemia L1210 cells.
Cancer Res. 47:2056–2061. 1987.PubMed/NCBI
|
|
16
|
Eastman A and Schulte N: Enhanced DNA
repair as a mechanism of resistance to
cis-diamminedichloroplatinum(II). Biochemistry. 27:4730–4734. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eastman A: Mechanisms of resistance to
cisplatin. Cancer Treat Res. 57:233–249. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reed E: Cisplatin. Cancer Chemotherapy and
Biological Response Modifiers. Pinedo H, Longo D and Chabner B:
Elsevier Science; Amsterdam: pp. 83–90. 1992
|
|
19
|
Dabholkar M, Parker R and Reed E:
Determinants of cisplatin sensitivity in non-malignant
non-drug-selected human T cell lines. Mutat Res. 274:45–56. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Masuda H, Ozols RF, Lai GM, Fojo A,
Rothenberg M and Hamilton TC: Increased DNA repair as a mechanism
of acquired resistance to cis-diamminedichloroplatinum (II) in
human ovarian cancer cell lines. Cancer Res. 48:5713–5716.
1988.PubMed/NCBI
|
|
21
|
Ferry KV, Hamilton TC and Johnson SW:
Increased nucleotide excision repair in cisplatin-resistant ovarian
cancer cells: role of ercc1-xpf. Biochem Pharmacol. 60:1305–1313.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Godwin AK, Meister A, O’Dwyer PJ, Huang
CS, Hamilton TC and Anderson ME: High resistance to cisplatin in
human ovarian cancer cell lines is associated with marked increase
of glutathione synthesis. Proc Natl Acad Sci USA. 89:3070–3074.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yao KS, Godwin AK, Johnson SW, Ozols RF,
O’Dwyer PJ and Hamilton TC: Evidence for altered regulation of
gamma-glutamylcysteine synthetase gene expression among
cisplatin-sensitive and cisplatin-resistant human ovarian cancer
cell lines. Cancer Res. 55:4367–4374. 1995.
|
|
24
|
Agyeman A, Mazumdar T and Houghton JA:
Regulation of DNA damage following termination of Hedgehog (HH)
survival signaling at the level of the GLI genes in human colon
cancer. Oncotarget. 3:854–868. 2012.PubMed/NCBI
|
|
25
|
Leonard JM, Ye H, Wetmore C and Karnitz
LM: Sonic Hedgehog signaling impairs ionizing radiation-induced
checkpoint activation and induces genomic instability. J Cell Biol.
183:385–391. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mazumdar T, Devecchio J, Agyeman A, Shi T
and Houghton JA: Blocking Hedgehog survival signaling at the level
of the GLI genes induces DNA damage and extensive cell death in
human colon carcinoma cells. Cancer Res. 71:5904–5914. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mazumdar T, DeVecchio J, Agyeman A, Shi T
and Houghton JA: The GLI genes as the molecular switch in
disrupting Hedgehog signaling in colon cancer. Oncotarget.
2:638–645. 2011.PubMed/NCBI
|
|
28
|
Amable L, Gavin E, Kudo K, et al: Gli1
upregulates c-jun through a specific 130 kDa isoform. Int J Oncol.
(In press).
|
|
29
|
Martins V, Hanana M, Blumwald E and Geros
H: Copper transport and compartmentation in grape cells. Plant Cell
Physiol. 53:1866–1880. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takahashi R, Bashir K, Ishimaru Y,
Nishizawa NK and Nakanishi H: The role of heavy-metal ATPases,
HMAs, in zinc and cadmium transport in rice. Plant Signal Behav.
7:1605–1607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mikkelsen MD, Pedas P, Schiller M, et al:
Barley HvHMA1 is a heavy metal pump involved in mobilizing
organellar Zn and Cu and plays a role in metal loading into grains.
PLoS One. 7:e490272012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Q, Gardner K, Zhang L, Tsang B,
Bostick-Bruton F and Reed E: Cisplatin induction of ERCC-1 mRNA
expression in A2780/CP70 human ovarian cancer cells. J Biol Chem.
273:23419–23425. 1998. View Article : Google Scholar : PubMed/NCBI
|