Introduction
Gastrointestinal stromal tumor (GIST) which arises
from interstitial cells of Cajal (ICC) or Cajal-like precursor
cells (1) is the most common
mesenchymal neoplasm of the gastrointestinal tract (2). Activating mutations in KIT and
platelet-derived growth factor receptor α (PDGFRA) have been
identified in ~85 and 10% of GISTs, respectively (3). Although inhibitors of KIT and PDGFRA
such as imatinib mesylate (Gleevec; Novartis Pharmaceuticals) have
increased progression-free survival in the majority of metastatic
or recurrent GIST cases and have led to an unparalleled success in
molecular-targeted therapy (4),
<3% of patients achieve complete remission (5). Moreover, resistance to tyrosine kinase
inhibitors is also a vital issue. Hence, KIT inhibition alone is
unlikely to cure GISTs. Further studies must focus on other
therapeutic targets and intracellular pathways.
ETV1, known as ETS-related protein 81 (ER81),
encodes a member of the well known ETS family of transcription
factors. It has been defined as an oncoprotein overexpressed in
breast cancer (6,7), prostatic carcinoma (8,9),
melanoma (10), Ewing sarcoma
(11) as well as GISTs, and plays a
key role in neoplastic formation, development and progression. ChiP
(12) confirmed the observation
that ETV1 is a lineage-specific survival factor which cooperates
with KIT in GISTs. This observation showed that strong ETV1
expression and constitutively activated KIT are implicated in
hyperplasia ICC set which may give rise to GISTs. Furthermore, ETV1
directly regulates ICC- and GIST-specific transcription. Notably,
activated KIT can prolong ETV1 protein stability through
KIT-independent signaling. Together these results demonstrate that
ETV1 is essential for the growth of GIST cells and activated KIT
may collaborate with ETV1 to promote tumorigenesis (5).
ETV1 is a unique transcription factor specific to
GISTs that has been reported to date. In addition, it has been
thought that downstream Ras/Raf/MAPK signaling cascade is reactive
when KIT/PDGFRA kinases are activated by its ligand stem cell
factor (SCF) in GISTs (13,14). These data raise the intriguing
possibility that ETV1 cooperates with cellular signaling pathways
to promote tumorigenesis. In the present study, we aimed to explore
the relationship between ETV1 and cellular signaling pathways which
may provide novel molecular-targeted therapies for GISTs.
Materials and methods
Patients and clinical tissue samples
Tumor tissue samples and corresponding
tumor-adjacent normal samples were collected from patients with
suspected GISTs who received surgical resection from May 2012 to
September 2013 at The First Affiliated Hospital, College of
Medicine, Zhejiang University, Hangzhou, China. The study protocol
was approved by The First Affiliated Hospital Ethics Committee.
Written informed consent statements were obtained from all the
participants involved in the present study before surgery. Patients
who had received any preoperative chemotherapy, molecular-targeted
therapies such as imatinib mesylate or not diagnosed as having
GISTs were excluded. The diagnosis of GIST was based on hematoxylin
and eosin staining for histopathological appearance which was
compatible with GIST and was confirmed by positive
immunohistochemical staining for c-KIT. A total of 72 patients were
included in the present study. Another 10 cases of other sarcoma
types such as leiomyoma were utilized as the control group. Tissue
specimens were placed into liquid nitrogen and store at −80°C until
RNA and protein extraction.
Western blot analysis
The total protein was lysed from 72 pairs of tissue
samples using cell lysis buffer (Cell Signaling, Beverly, MA, USA)
according to the manufacturer’s protocol. Equal amounts of protein
were applied to 8–12% gels and subjected to SDS-PAGE. The samples
were then transferred to polyvinylidene fluoride (PVDF) membranes
(Bio-Rad, Hercules, CA, USA) for 1 h. Membranes were blocked for 2
h at room temperature with 5% dry milk in TBST and incubated
overnight at 4°C with primary antibodies against c-KIT (1:2,000);
phospho-c-KIT (Tyr703) (1:2,000); c-Raf (1:1,000); phospho-c-Raf
(Ser338) (1:1,000); MEK1/2 (1:2,000); phospho-MEK1/2 (Ser217/221
for MEK1 and Ser222/226 for MEK2) (1:2,000); ERK1/2 (1:2,000);
phospho-ERK1/2 (Thr202/Tyr204 for ERK1 and Thr183/Tyr185 for ERK2)
(1:2,000); Smad2/3 (1:2,000); caspase-3 (1:1,000); Bcl-2 (1:2,000);
Bax (1:2,000); E-cadherin (1:2,000); vimentin (1:2,000); β-catenin
(1:1,000); GSK-3β (1:1,000); Dvl2 (1:1,000); GAPDH (1:5,000) (Cell
Signaling) and ETV1 (1:1,000) (Abcam, Cambridge, UK). Membranes
were washed in Tris-buffered saline with Tween-20 (TBST) and
incubated with 1:5,000 anti-rabbit horseradish peroxidase
(HRP)-conjugated secondary antibodies for 1 h at room temperature
and washed again. Immune complexes were detected with an enhanced
chemiluminescence reagent (Millipore, Billerica, MA, USA) and
acquired in the linear range of the scanner and analyzed using
Quantity One software (Bio-Rad).
Immunohistochemistry
Formalin-fixed, paraffin-embedded 4-μm sections of a
GIST tissue microarray (TMA) (GIST 801–802) were purchased from
Alinabio (Xi’an, China). TMA sections with 156 cases were
deparaffinized in 100% xylene and re-hydrated in graded ethanol
solutions. Endogenous peroxidase activity was blocked using 0.3%
hydrogen peroxide for 20 min at room temperature. Heat-activated
antigen retrieval was performed in sodium citrate buffer (pH 6.0).
After blocking with 3% bovine serum albumin (BSA), TMA sections
were incubated with a primary anti-ETV1 antibody diluted 1:100 or
anti-KIT antibody diluted 1:200 in TBS containing 1% BSA overnight
in a humidified chamber at 4°C. After washing (5 min ×3) in
phosphate-buffered saline (PBS), the sections were incubated with
anti-rabbit horseradish peroxidase-conjugated antibody at room
temperature. Diaminobenzidine (Vector Laboratories, Burlingame, CA,
USA) was used as the chromogen, and color development was stopped
by dipping the slides in distilled water. The nuclei were then
counterstained with hematoxylin (Sigma). Blinded evaluations of the
ETV1 immunostaining and independent observation were carried out
simultaneously by two experienced pathologists. The
immunohistological score calculated was the sum of the percentage
of the positive area (negative, 0 point; <25%, 1 point; 26–50%,
2 points; 51–100%, 3 points) and the staining intensity (negative,
0; weak, 1; moderate, 2 and strong, 3). Five horizons were randomly
selected and the average score in each case represented the final
positive staining points: 0–2 points for each case was assigned as
negative; 3–4 points was regarded as weak/moderate and 5–6 points
was assigned as strong.
Quantitative real-time PCR
The total RNA of 72 frozen tissue specimens was
extracted using TRIzol reagent (Invitrogen, San Diego, CA, USA)
according to the manufacturer’s instructions. Total RNA (1 μg) was
reverse-transcribed using PrimeScript™ RT reagent kit with gDNA
Eraser (Perfect Real-Time) (Takara, Dalian, China). DNA products
were amplified with SYBR® Premix Ex Taq™ (Tli RNaseH
Plus) (Takara). The primer sequences were as follows: KIT, forward
5′-AATGGCACGGTTGAATGTAAG-3′ and reverse 5′-GGA
TGGATTTGCTCTTTGTTGT-3′; ETV1, forward 5′-GCA GTCAAGAACAGCCCTTTA-3′
and reverse 5′-TCAGGTT TCGGTGTATGAGTTG-3′; GAPDH, forward 5′-AGAAG
GCTGGGGCTCATTTG-3′ and reverse 5′-AGGGGCCATC CACAGTCTTC-3′.
Thermocycling conditions were 10 min at 95°C as the initial
denaturation step, followed by 40 cycles at 95°C for 30 sec and
60°C for 34 sec. Reactions were carried out in an Applied
Biosystems 7500 Real-Time PCR System. Gene expression levels were
measured using the threshold cycle (Ct) value, and relative
fold-expression changes were normalized to the amplification of
glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Calculation of
the gene copy number was carried out using the ΔΔCt method.
Statistics
Statistical analyses were conducted using the SPSS
Software Package version 16.0 for Windows (SPSS, Chicago, IL, USA).
Two-tailed paired sample t-test and independent sample t-test were
utilized to determine differences between the tumor group and the
uninvolved normal group as well as the control group. The
relationship between expression levels of ETV1 and
clinicopathological factors was examined using χ2 test
or Fisher’s exact test in cross tables. A P-value <0.05 was
considered to indicate a statistically significant result.
Results
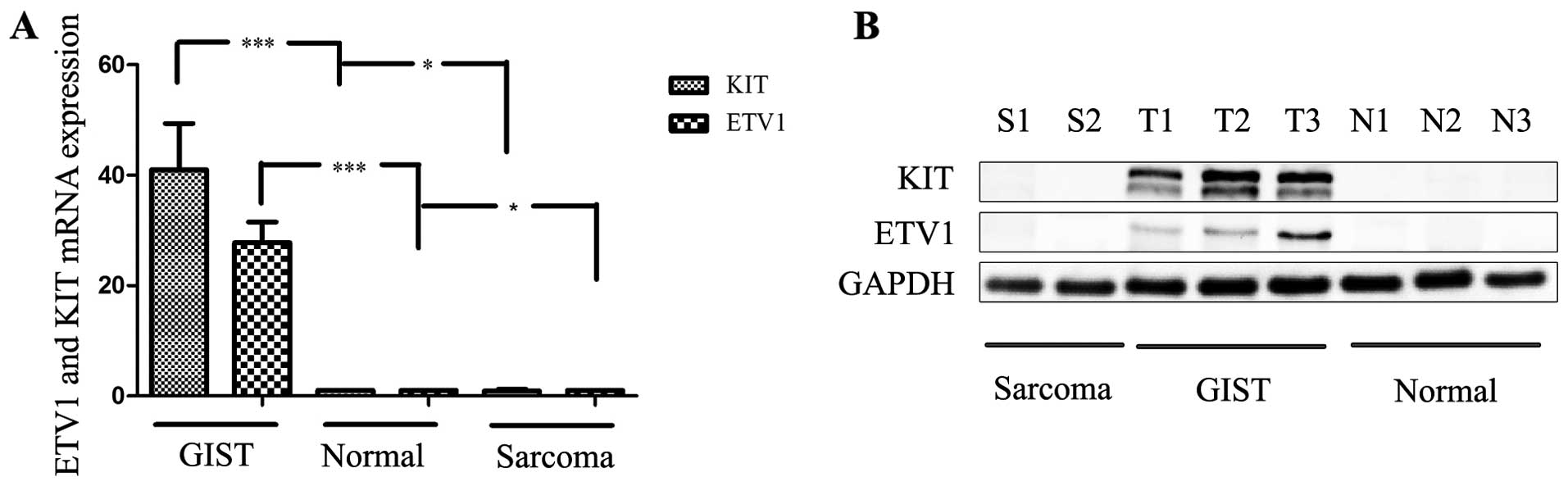
In the first step, semi-quantitative RT-PCR analysis
of 72 paired GIST patient tissue samples confirmed that ETV1 mRNA
expression was significantly upregulated in the clinical tissue
specimens when compared with that in the corresponding
tumor-adjacent normal samples (P<0.05) (Fig. 1A); 55 patients (76.4%) were
considered to have positive ETV1 expression.
Next, we determined the ETV1 protein expression in
the same 72 patient tissue samples and found that ETV1 was
upregulated in 65.3% (47/72) of these cases (P<0.05). No
expression of ETV1 was detected in the other sarcomas. The ratio of
the protein expression levels in the image was measured by
densitometry using Quantity One software, which revealed that the
pattern of expression of the ETV1 protein was similar to the
semi-quantitative RT-PCR results (Fig.
1B).
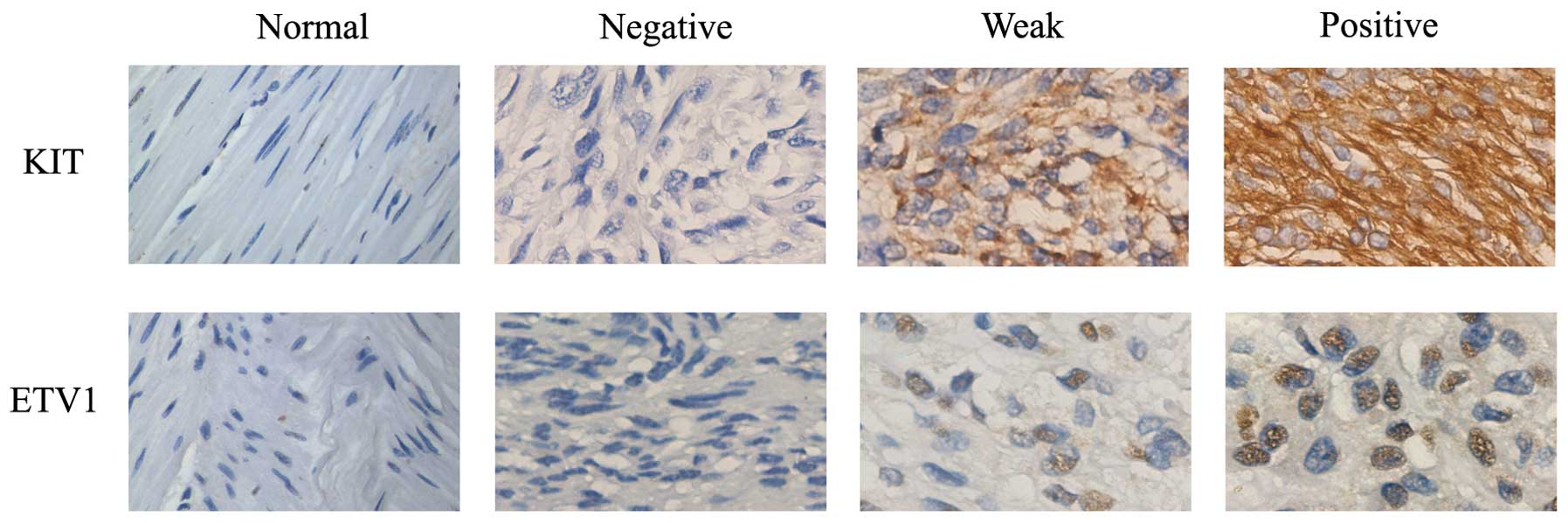
In addition, immunohistochemical staining revealed
that ETV1 was strongly expressed in the GIST TMA sections which
contained 156 cases. ETV1 was diffusely positive in the nuclei of
the tumor cells. Among the cases examined, 108 patients (69.2%)
were considered to be positive for ETV1. As shown in Fig. 2, no expression of ETV1 was observed
in the adjacent normal tissues. We analyzed the relationship
between the expression of ETV1 and patient age, gender and tumor
location (Table I). The data showed
a strong correlation between ETV1 and KIT expression (P<0.05).
Moreover, GISTs were prone to localize in the stomach (P<0.05).
Table II summarizes the levels of
ETV1 and KIT and their frequencies in the subtypes of the tumor
samples. First, high frequencies of ETV1 and KIT were mainly found
in the GIST tumors and were significantly higher in the GIST tumor
samples with low risk or intermediate risk. ETV1-positive cases
were identified in 67.3% (72/107 cases) of the low-risk group and
in 71.0% (22/31 cases) of the intermediate-risk group,
respectively. The frequencies were still lower than that noted for
KIT, which was identified in 87.0% (93/107) and 90.3% (28/31) of
the cases in the corresponding groups. Second, in the GIST with
high-risk, the frequency of ETV1 positivity was higher when
compared to that for KIT expression. ETV1 positivity was identified
in 50.0% (9/18) of the cases and only 38.9% (7/18) of the cases
demonstrated KIT positivity. We then analyzed the relationship
between ETV1 and KIT (Table III).
Similarly, ETV1 was associated with KIT expression in the
GISTs.
 | Table IClinical characteristics of the 156
patients. |
Table I
Clinical characteristics of the 156
patients.
| Factor | No. | ETV1 (+) | ETV1 (−) | P-value |
|---|
| KIT (+) | 133 | 88 | 45 | <0.05 |
| KIT (−) | 23 | 20 | 3 | |
| Age (years) | 50.9±11.9 | 48.6±10.0 | 50.2±10.8 | 0.534 |
| Gender |
| Male | 89 | 63 | 26 | 0.628 |
| Female | 67 | 45 | 22 | |
| Location |
| Stomach (gastric
cardia) | 65 | 37 | 28 | <0.05 |
| Small intestine | 48 | 36 | 12 | 0.298 |
| Colon | 11 | 10 | 1 | 0.174 |
| Rectum | 17 | 12 | 5 | 0.898 |
| Other location | 15 | 13 | 2 | 0.151 |
| Risk
classification |
| Low | 107 | 72 | 35 | 0.438 |
| Intermediate | 31 | 22 | 9 | 0.815 |
| High | 18 | 14 | 4 | 0.404 |
| Table IIFrequency of oncogenic ETV1
expression in comparison with KIT expression in the GIST tumor
samples with different risk levels. |
Table II
Frequency of oncogenic ETV1
expression in comparison with KIT expression in the GIST tumor
samples with different risk levels.
| | | Immunoreactive to
ETV1 | | | Immunoreactive to
KIT | | |
|---|
| | |
| | |
| | |
|---|
| GIST | Average age | Case no. | Ngt (0–2) | Wk/Md (3–4) | OEPT (5–6) | Expression rate
(%) | Overexpression rate
(%) | Ngt (0–2) | Wk/Md (3–4) | OEPT (5–6) | Expression rate
(%) | Overexpression rate
(%) |
|---|
| Low-risk | 48.3±9.8 | 107 | 35 (0.3±0.4) | 36 (3.7±0.5) | 36 (5.7±0.5) | 67.3% (72/107) | 33.6% (36/107) | 14 (0.8±0.5) | 16 (3.6±0.5) | 77 (5.6±0.5) | 87.0% (93/107) | 72.0% (77/107) |
| Intermediate
risk | 54.3±12.3 | 31 | 9 (0.2±0.4) | 6 (3.3±0.5) | 16 (5.3±0.4) | 71.0% (22/31) | 51.6% (16/31) | 3 (0.7±0.6) | 3 (3.3±0.6) | 25 (5.4±0.5) | 90.3% (28/31) | 80.6% (25/31) |
| High-risk | 48.0±9.2 | 18 | 4 (0) | 5 (3.8±0.4) | 9 (5.6±0.5) | 77.8% (14/18) | 50.0% (9/18) | 6 (0.7±0.5) | 5 (3.6±0.5) | 7 (5.3±0.4) | 66.7% (12/18) | 38.9% (7/18) |
| Total | 50.9±11.9 | 156 | 48 (0.2±0.4) | 47 (3.7±0.5) | 61 (5.5±0.5) | 69.2%
(108/156) | 39.1% (61/156) | 23 (0.7±0.5) | 24 (3.5±0.5) | 109 (5.7±0.5) | 85.2%
(133/156) | 69.9%
(109/156) |
| Table IIIETV1 is associated with KIT
expression in GIST tumor samples with high-risk. |
Table III
ETV1 is associated with KIT
expression in GIST tumor samples with high-risk.
| Immunoreactive to
KIT |
|---|
|
|
|---|
| Levels of ETV1
expression | Negative n (%) | Positive n (%) | Total n (%) |
|---|
| Low-risk |
| Negative | 4 (3.7) | 31 (29.0) | 35 (32.7) |
| Weak/moderate | 3 (2.8) | 33 (30.8) | 36 (33.6) |
|
Overexpression | 7 (6.5) | 29 (27.1) | 36 (33.6) |
| Total | 14 (13.1) | 93 (87.0) | 107 (100) |
| Intermediate
risk |
| Negative | 0 (0.0) | 9 (29.0) | 9 (29.0) |
| Weak/moderate | 0 (0.0) | 6 (19.4) | 6 (19.4) |
|
Overexpression | 3 (9.7) | 13 (41.9) | 16 (51.6) |
| Total | 3 (9.7) | 28 (90.3) | 31 (100) |
| High-risk |
| Negative | 0 (0.0) | 4 (22.2) | 4 (22.2) |
| Weak/moderate | 2 (11.1) | 3 (16.7) | 5 (27.8) |
|
Overexpression | 4 (22.2) | 5 (27.8) | 9 (50.0) |
| Total | 6 (21.4) | 12 (83.3)a | 18 (100) |
ETV1 was found to be expressed at a significantly
higher level in GISTs than that in the normal tissues and acts as a
unique transcription factor which is essential to the growth of
GIST cells. However, to date, the mechanism of how it works is not
clear. To identify which signaling pathway may be associated with
ETV1 driving GIST oncogenesis, we detected the relative levels of
phosphorylation of kinases and their protein substrates including
Raf/MEK/extracellular signal-regulated kinase (ERK1/2),
Bcl-2/Bax/caspase-3 and cadherin/β-catenin signaling cascades in
the 72 paired tissue samples.
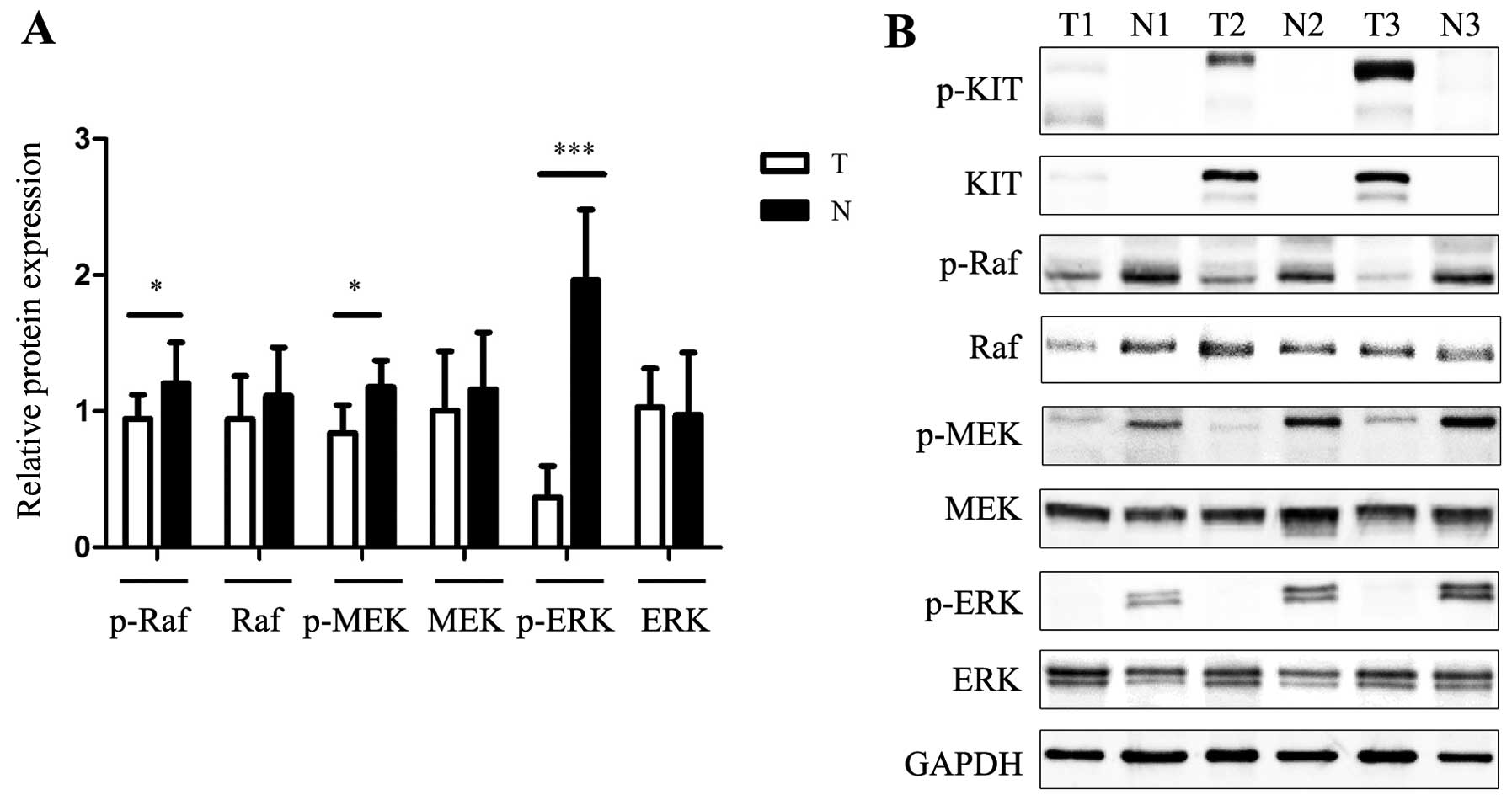
The Raf/MEK/ERK pathway often takes part in
controlling cell survival, cell cycle progression and
differentiation. Notably, the present study showed opposite
results. The phosphorylation levels of ERK1/2 and MEK in the GISTs
were significantly downregulated whereas total protein levels were
unchanged. Phosphorylation of MEK and ERK was significantly higher
in the normal group than that in the tumor group (Fig. 3) We hypothesized that this
phenomenon was due to innate tumor-suppressive responses, which
were elicited as a protection against cancer.
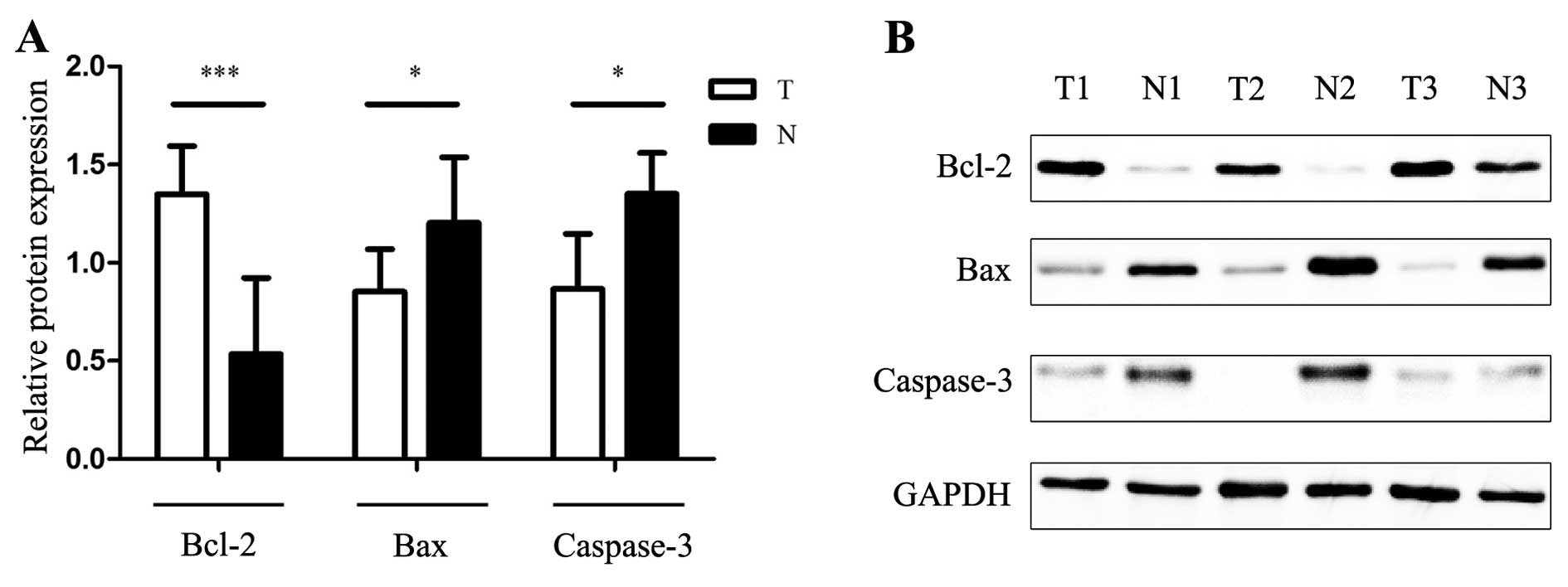
As shown in Fig. 4,
Bax/Bcl-2 and caspase-3 protein were detected in a matching
approach. Prominent expression of Bcl-2 in the GIST tissues was
investigated, and the expression of Bax and caspase-3 was much
higher in the uninvolved normal group when compared with the tumor
group. Accumulating evidence suggests that overexpression of
anti-apoptotic protein Bcl-2 and underexpression of pro-apoptotic
protein Bax prevented tumor cells from undergoing apoptosis in
response to a variety of stimuli and promoted tumorigenesis.
A previous study revealed that convergence of Wnt,
β-catenin and cadherin pathways may control cell-cell adhesion and
determine the cell fate. Cadherin may also act as a negative
regulator of β-catenin signaling as cadherin binds β-catenin
forming a cadherin-catenin complex and thereby sequesters it from
the nucleus (15). A recent study
investigated the tight junction between E-cadherin and the prostate
tumor suppressor SPDEF, the latest discovered transcription factor
of the ETS family (16). In the
present study, we investigated whether activated ETS family member
ETV1 was correlated with the wnt-β-catenin-cadherin signaling
pathway in the tumor progression of GISTs. In a similar experiment,
we observed that rarely was E-cadherin expression found in the GIST
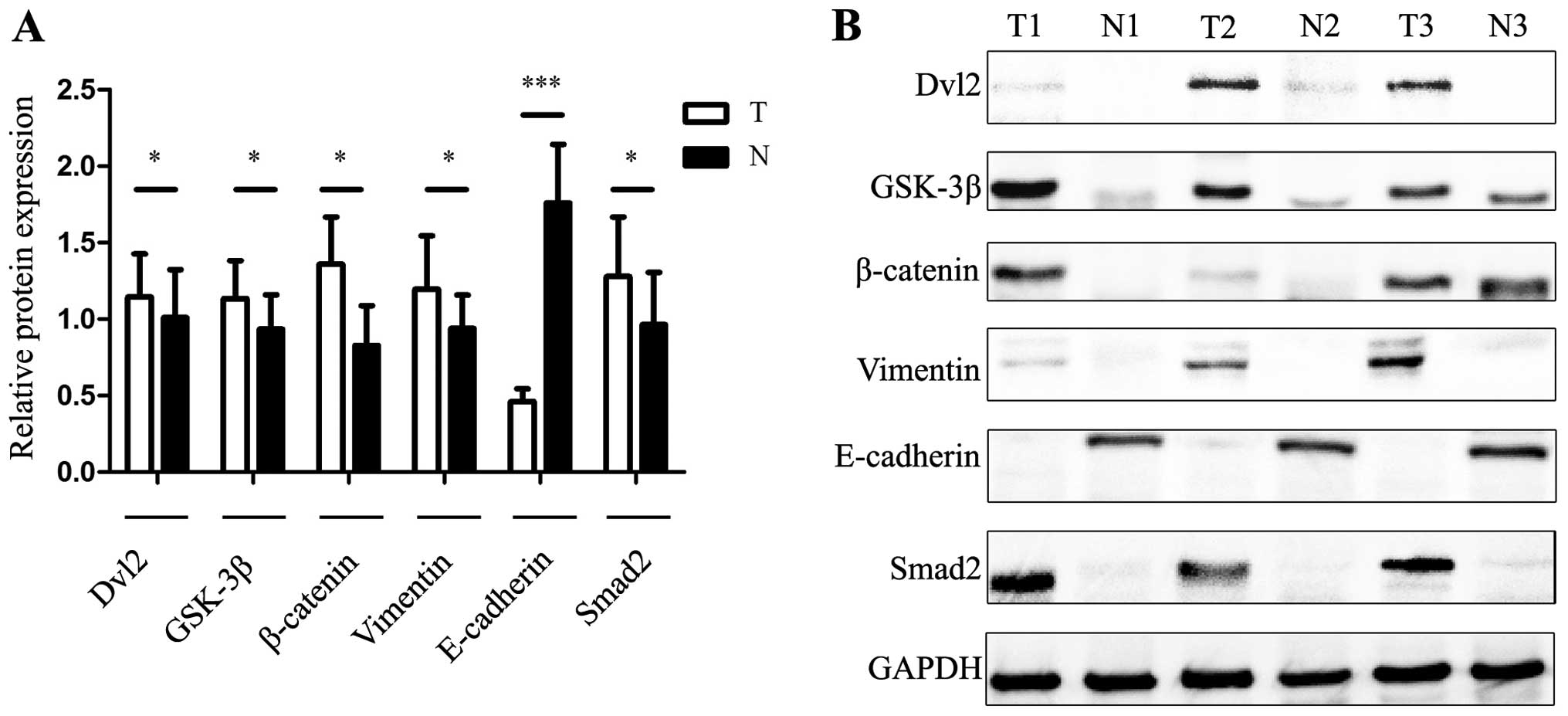
group when compared with the non-tumor group. Nevertheless,
significant high levels of β-catenin and Dvl2 were detected
(Fig. 5). These results point to a
potential role of the wnt-β-catenin-E-cadherin pathway in
ETV1-regulated GIST tumorigenesis.
Discussion
GISTs are clinically and histologically
heterogeneous neoplasms that are driven by oncogenic KIT or PDGFRA
mutations (17). Our findings
demonstrated that prominent ETV1 expression was noted in the
majority of GISTs both at the mRNA and protein levels. IHC analysis
confirmed that ETV1 was strongly expressed in the GISTs and
revealed that ETV1 was correlated with KIT expression. Fig. 2 and Tables II and III show that the frequencies of ETV1 and
KIT were high in the GISTs. ETV1-positive cases were identified in
67.3% (72/107) of the cases in the low-risk group and 71.0% (22/31)
of the cases in the intermediate-risk group, respectively. The
frequency of KIT was higher in 87.0% (93/107) of the low-risk cases
and in 90.3% (28/31) of the cases in the intermediate-risk group,
respectively. As to the GIST tumor samples with high-risk, ETV1
expression was significantly higher. ETV1 positivity was identified
in 50.0% (9/18) of the cases and only 38.9% (7/18) of the cases
showed KIT positivity. Taken together, ETV1 was strongly expressed
in the GISTs which can aid in the diagnosis of GISTs particularly
when KIT is negative in the high-risk group.
Novel prognostic markers have been successively
found in GISTs, yet the action mode of relevant factors has
received little attention. Thus, we aimed to explore how ETV1
integrates with cellular upstream or downstream signaling molecules
in the tumorigenesis of GIST. In light of our findings from ChiP
(12), we inferred that ETV1 can be
stabilized through KIT-independent MAPK signaling. Furthermore, the
ERK pathway induced by Ras/Raf mutations participates in cell
proliferation, differentiation and cell migration in a variety of
cancer types (18). However, in our
cohort, lower expression of the phosphorylation of the ERK pathway
was noted in tumor tissues than that in the tumor-adjacent normal
tissues, since the total protein levels were not significantly
altered. Immunohistochemical results reported by Schoppmann et
al (19) demonstrated that only
29.1% of patients showed pMEK1/2 overexpression and 86.3% of
patients showed upregulation of PEBP1, known as a Raf kinase
inhibitory protein in GIST. Downregulation of p-ERK expression was
also reported in colorectal cancer in a previous study (20). Aberrant Raf/MEK/ERK signaling can be
involved as follows. First, ERK signaling is rarely detected,
suggesting that the Raf/MEK/ERK pathway does not provide a growth
advantage or significant inflammation reaction. It should be noted
that ERK inactivation promotes a less differentiated phenotype
(20). Our studies did not
investigate the expression at different stages, thus we cannot
exclude that possibility. However, normal tissues, which can be
referred as premalignant lesions to some extent (21,22),
sustained activation of the Raf/MEK/ERK pathway eliciting
senescence-like growth arrest responses compared with the tumor
tissues. This phenomenon can be interpreted as a physiologic
fail-safe antitumorigenic mechanism (23). Furthermore, it is noteworthy that
Hollenhorst et al (9)
observed that ETS proteins activated a MEK/ERK-regulated gene
expression program in the absence of ERK action. ETS/AP-1-binding
sites in the PLAU enhancers can act as response elements for the
Ras/MAPK signaling pathway. In RWPE-1 cells in the presence of the
MEK inhibitor U0126 or lacking supplements, PLAU expression was
elevated by oncogenic ETS proteins ETV1 and ERG. Thus,
overexpression of oncogenic ETS proteins can replace Ras/MAPK
pathway activation in prostate cells. ETV1 can activate targeted
gene expression when the Ras/MAPK pathway is off and superactivates
when the pathway is on. We hypothesized that ETV1 may serve the
same function in GIST, promoting cell proliferation in the absence
of the Ras/Raf/MEK/ERK pathway. Based on the findings, the activity
of the system was dynamic so that we could not draw a definite
conclusion or take this phenomenon as direct proof that the
signaling was downregulated. Further study on the specific
mechanism is warranted.
Cancer is both a consequence of uncontrolled
proliferation, as well as disturbed differentiation (24,25).
Thus, we detected Bcl-2/Bax/caspase-3 expression levels. Bcl-2 is
an integral membrane protein located mainly on the outer membrane
of mitochondria (26). Previous
studies have found the involvement of Bcl-2 expression in GIST
(27,28). In the pre-imatinib era, Bcl-2
expression was associated with worse prognosis. In contrast,
Steinert et al (27)
analyzed data from 81 GIST patients and demonstrated that higher
levels of Bcl-2 were associated with improved PFS in patients who
were subsequently treated with imatinib. In the present study,
Bcl-2 expression was found in the majority of GISTs. The caspase
family also plays a key role in the initiation and execution of
programmed cell death through both the intrinsic and extrinsic
signaling pathways. Western blot analysis indicated that GIST
tissues antagonized apoptosis by elevating anti-apoptotic Bcl-2
activity, whereas the normal tissues were prone to prominent
apoptosis. Although its ability to promote proliferation and
tumorigenesis, inflammatory factors such as Akt and ERK could not
protect against ROS-mediated cell death but rather sensitized cells
to this cell death (29). ROS are
natural by-products of oxidative energy metabolism which play a
critical role in determining the lifespan of mammalian cells and
induce severe damage such as DNA lesions and protein oxidation. Bax
is phosphorylated and translocated to the mitochondrial outer
membrane. Subsequently release of cytochrome c from the
mitochondrial inter-membrane space into the cytosol occurs. The
mitochondrial caspase cascade is activated (30,31).
Collectively, these data suggest that an anti-apoptotic effect is
obvious in GIST.
Importantly, our results also indicated that the
wnt-β-catenin-E-cadherin signaling pathway may be involved in GIST
progression. ETS family member, SPDEF, is overexpressed in
aggressive PC3 prostate cancer cells, leading to increased
E-cadherin expression, an adhesion molecule with a crucial role in
preventing metastatic spread (16).
Loss of E-cadherin has been regarded as a central event in the
switch to epithelial-mesenchymal transition and tumor metastasis
(32). In the GIST group, we
detected vimentin instead of E-cadherin and confirmed the component
of the tumor arising from mesenchymal tissue. Loss of E-cadherin
makes it difficult for β-catenin to combine with. In addition, our
data suggest that Wnt/β-catenin also existed in the GISTs.
Activation of Wnt/ β-catenin signaling triggers Dvl2 activity and
GSK-3β inhibition, thereby phosphorylation of β-catenin is reduced.
On the basis of our research, we hypothesized that loss of
E-cadherin and reduced phosphorylation of β-catenin resulted in
accumulation of cytoplasmic β-catenin, which became available to
bind the TCF/LEF family of transcription factors, induce target
gene expression and promote GIST pathogenesis. ETV1 and SPDEF
appear to be two mutual antagonistic transcription factors in the
ETS family demonstrated in prostate cancer (9). It is plausible that a similar
mechanism may also operate in GISTs and whether ETV1 interacts with
Wnt/β-catenin/ E-cadherin remains unclear.
In conclusion, our investigation revealed that ETV1
was upregulated at a higher rate in GISTs and was correlated with
KIT expression, which is consistent with previous research. Limited
by the patient number and follow-up time, we could not obtain the
long-term outcome data of patients and analyze the prognostic value
of ETV1 in GIST. Several studies have carried out relevant
research, demonstrating that there is no correlation with clinical
outcome (19,33,34).
The prognostic significance is still controversial and warrants
detailed study. Novel molecular pathways were found activated in
GISTs, raising the possibility of an interaction between them in
the tumorigenesis in GISTs. Nevertheless, more specific mechanisms
still need to be explored.
Acknowledgements
This study was supported by the Scientific Research
Foundation of the Ministry of Health of China (WKJ2011-2-002). We
also thank the patients of the Department of Oncology and
Gastrointestinal Surgery at The First Affiliated Hospital of
Zhejiang University.
References
|
1
|
Corless CL, Barnett CM and Heinrich MC:
Gastrointestinal stromal tumours: origin and molecular oncology.
Nat Rev Cancer. 11:865–878. 2011.PubMed/NCBI
|
|
2
|
Rubin BP, Heinrich MC and Corless CL:
Gastrointestinal stromal tumour. Lancet. 369:1731–1741. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Downs-Kelly E and Rubin BP:
Gastrointestinal stromal tumors: molecular mechanisms and targeted
therapies. Patholog Res Int. 2011:7085962011.PubMed/NCBI
|
|
4
|
Blay JY: A decade of tyrosine kinase
inhibitor therapy: historical and current perspectives on targeted
therapy for GIST. Cancer Treat Rev. 37:373–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rubin BP: Bioinformatic mining of gene
expression datasets identifies ETV1 as a critical regulator of
oncogenesis in gastrointestinal stromal tumors. Cancer Cell.
18:407–408. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen Y, Zou H, Yang LY, Li Y, Wang L, Hao
Y and Yang JL: ER81-shRNA inhibits growth of triple-negative human
breast cancer cell line MDA-MB-231 in vivo and in vitro. Asian Pac
J Cancer Prev. 13:2385–2392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shin S, Bosc DG, Ingle JN, Spelsberg TC
and Janknecht R: Rcl is a novel ETV1/ER81 target gene upregulated
in breast tumors. J Cell Biochem. 105:866–874. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baena E, Shao Z, Linn DE, et al: ETV1
directs androgen metabolism and confers aggressive prostate cancer
in targeted mice and patients. Genes Dev. 27:683–698. 2013.
View Article : Google Scholar
|
|
9
|
Hollenhorst PC, Ferris MW, Hull MA, Chae
H, Kim S and Graves BJ: Oncogenic ETS proteins mimic activated
RAS/MAPK signaling in prostate cells. Genes Dev. 25:2147–2157.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jané-Valbuena J, Widlund HR, Perner S, et
al: An oncogenic role for ETV1 in melanoma. Cancer Res.
70:2075–2084. 2010.
|
|
11
|
Oh S, Shin S and Janknecht R: ETV1, 4 and
5: an oncogenic subfamily of ETS transcription factors. Biochim
Biophys Acta. 1826:1–12. 2012.PubMed/NCBI
|
|
12
|
Chi P, Chen Y, Zhang L, et al: ETV1 is a
lineage survival factor that cooperates with KIT in
gastrointestinal stromal tumours. Nature. 467:849–853. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duensing A, Medeiros F, McConarty B, et
al: Mechanisms of oncogenic KIT signal transduction in primary
gastrointestinal stromal tumors (GISTs). Oncogene. 23:3999–4006.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Duensing S and Duensing A: Targeted
therapies of gastrointestinal stromal tumors (GIST) - the next
frontiers. Biochem Pharmacol. 80:575–583. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nelson WJ and Nusse R: Convergence of Wnt,
β-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
|
|
16
|
Pal M, Koul S and Koul HK: The
transcription factor sterile alpha motif (SAM) pointed
domain-containing ETS transcription factor (SPDEF) is required for
E-cadherin expression in prostate cancer cells. J Biol Chem.
288:12222–12231. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heinrich MC and Corless CL: Cancer:
oncogenes in context. Nature. 467:796–797. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schoppmann SF, Beer A, Nirtl N,
Ba-Ssalamah A, Brodowicz T, Streubel B and Birner P: Downregulation
of phosphatidylethanolamine binding protein 1 associates with
clinical risk factors in gastrointestinal stromal tumors, but not
with activation of the RAF-1-MEK-ETV1 pathway. Cancer Lett.
335:26–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gulmann C, Sheehan KM, Conroy RM, et al:
Quantitative cell signalling analysis reveals down-regulation of
MAPK pathway activation in colorectal cancer. J Pathol.
218:514–519. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Braig M, Lee S, Loddenkemper C, et al:
Oncogene-induced senescence as an initial barrier in lymphoma
development. Nature. 436:660–665. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu PK, Hong SK, Veeranki S, Karkhanis M,
Starenki D, Plaza JA and Park JI: A mortalin/HSPA9-mediated switch
in tumor-suppressive signaling of Raf/MEK/extracellular
signal-regulated kinase. Mol Cell Biol. 33:4051–4067. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mooi WJ and Peeper DS: Oncogene-induced
cell senescence - halting on the road to cancer. N Engl J Med.
355:1037–1046. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Klonisch T, Wiechec E, Hombach-Klonisch S,
Ande SR, Wesselborg S, Schulze-Osthoff K and Los M: Cancer stem
cell markers in common cancers - therapeutic implications. Trends
Mol Med. 14:450–460. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghavami S, Hashemi M, Ande SR, et al:
Apoptosis and cancer: mutations within caspase genes. J Med Genet.
46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu J, Cui H, Peng X, et al: Dietary high
fluorine induces apoptosis and alters Bcl-2, Bax, and caspase-3
protein expression in the cecal tonsil lymphocytes of broilers.
Biol Trace Elem Res. 52:25–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Steinert DM, Oyarzo M, Wang X, et al:
Expression of Bcl-2 in gastrointestinal stromal tumors: correlation
with progression-free survival in 81 patients treated with imatinib
mesylate. Cancer. 106:1617–1623. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Q and Kou YW: Study of the
expressions of p53 and bcl-2 genes, the telomerase activity and
apoptosis in GIST patients. World J Gastroenterol. 13:2626–2628.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nogueira V, Park Y, Chen CC, et al: Akt
determines replicative senescence and oxidative or oncogenic
premature senescence and sensitizes cells to oxidative apoptosis.
Cancer Cell. 14:458–470. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Corazza N, Jakob S, Schaer C, et al: TRAIL
receptor-mediated JNK activation and Bim phosphorylation critically
regulate Fas-mediated liver damage and lethality. J Clin Invest.
116:2493–2499. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013.PubMed/NCBI
|
|
33
|
Birner P, Beer A, Vinatzer U, et al:
MAPKAP kinase 2 overexpression influences prognosis in
gastrointestinal stromal tumors and associates with copy number
variations on chromosome 1 and expression of p38 MAP kinase and
ETV1. Clin Cancer Res. 18:1879–1887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kubota D, Yoshida A, Tsuda H, et al: Gene
expression network analysis of ETV1 reveals KCTD10 as a novel
prognostic biomarker in gastrointestinal stromal tumor. PLoS One.
8:e738962013. View Article : Google Scholar : PubMed/NCBI
|